

SUMMARY
The embryonic pyruvate kinase M2 (PKM2) isoform is highly expressed in human cancer. In contrast to the established role of PKM2 in aerobic glycolysis or the Warburg effect1,2,3, its nonmetabolic functions remain elusive. Here we demonstrate that EGFR activation induces translocation of PKM2, but not PKM1, into the nucleus, where K433 of PKM2 binds to c-Src-phosphorylated Y333 of β-catenin. This interaction is required for both proteins to be recruited to the CCND1 promoter, leading to HDAC3 removal from the promoter, histone H3 acetylation, and cyclin D1 expression. PKM2-dependent β-catenin transactivation is instrumental in EGFR-promoted tumor cell proliferation and brain tumor development. In addition, positive correlations have been identified among c-Src activity, β-catenin Y333 phosphorylation, and PKM2 nuclear accumulation in human glioblastoma specimens. Furthermore, levels of β-catenin phosphorylation and nuclear PKM2 have been correlated with grades of glioma malignancy and prognosis. These findings reveal that EGF induces β-catenin transactivation via a mechanism distinct from that induced by Wnt/wingless4 and highlight the essential nonmetabolic functions of PKM2 in EGFR-promoted β-catenin transactivation, cell proliferation, and tumorigenesis.
Keywords: EGFR, PKM2, β-catenin, c-Src, phosphorylation, cell proliferation, metabolism, tumorigenesis
Since both EGFR activation and PKM2 expression are instrumental in tumorigenesis5,6,7, we examined whether EGFR activation regulates PKM2 functions in a subcellular compartment-dependent manner. Immunofluorescence analysis showed that EGF treatment resulted in the nuclear accumulation of PKM2 in U87/EGFR human glioblastoma (GBM) cells (Fig. 1a). In addition, expression of the constitutively active EGFRvIII mutant in U87 cells had a higher amount of nuclear PKM2 than did EGF-untreated U87/EGFR cells (Supplementary Fig. 2a). The finding that EGF induces nuclear translocation of PKM2 was further supported by cell fractionation analysis of DU145 prostate cancer cells, MDA-MB-231 breast cancer cells, and U87/EGFR cells (Supplementary Fig. 2b). In addition, PKM1 failed to translocate into the nucleus upon EGF stimulation (Supplementary Fig. 2c), indicating that EGF specifically regulates the subcellular distribution of PKM2 in multiple types of cancer cells.
Figure 1. EGF induces the PKM2–β-catenin interaction in the nucleus.
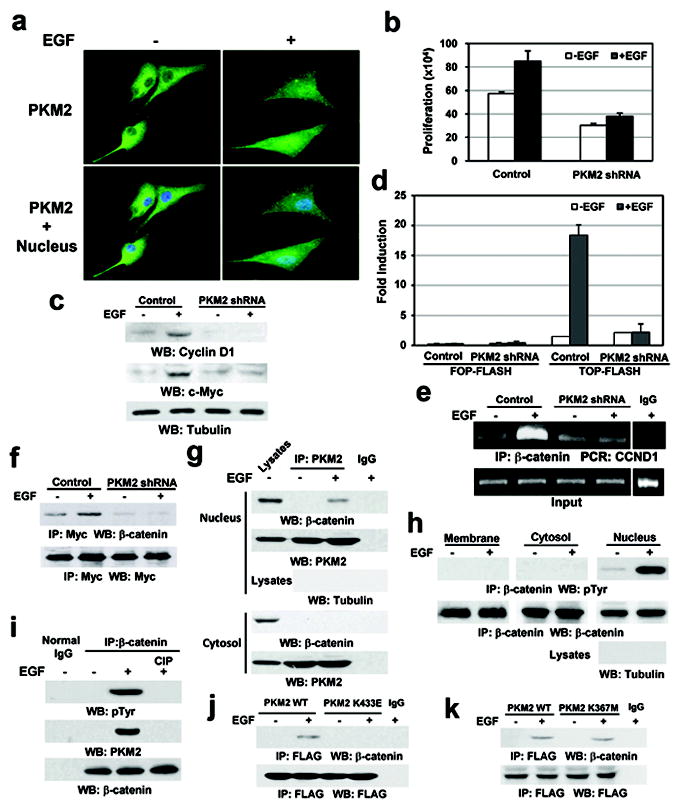
a, U87/EGFR cells were treated with or without EGF for 10 h.
b, U87/EGFR cells with or without PKM2 depletion were plated and counted 7 days after seeding. Data represent the means ± SD of three independent experiments.
c, e, U87/EGFR cells with or without PKM2 depletion were treated with or without EGF for 24 h (c) or 10 h (e).
d, U87/EGFR cells with or without PKM2 depletion were transfected with TOP-FLASH or FOP-FLASH, which was followed by EGF treatment for 10 h. Data represent the means ± SD of three independent experiments.
f, Myc-TCF4 was immunoprecipitated from PKM2-depleted or PKM2-undepleted U87/EGFR cells treated with or without EGF for 10 h.
g, h, PKM2 (g) or β-catenin (h) was immunoprecipitated from the indicated cell fractions of U87/EGFR cells treated with or without EGF for 6 h.
i, β-catenin immunoprecipitated from U87/EGFR cells with or without EGF treatment for 6 h was incubated with or without CIP (10 unit) for 30 min at 37°C followed by PBS washing for three times.
j, k, U87/EGFR cells stably expressing FLAG-tagged WT PKM2, PKM2 K433E (j), or PKM2 K367M (k) were treated with or without EGF for 6 h.
To examine whether PKM2 directly regulates gene transcription and cell proliferation, we expressed PKM2 shRNA in U87/EGFR cells (Supplementary Fig. 3a). PKM2 depletion largely reduced both basal and EGF-induced tumor cell proliferation (Fig. 1b) and blocked EGF-enhanced expression of cyclin D1 and c-Myc (Fig. 1c), which are known important regulators of cell proliferation and downstream genes of β-catenin transactivation8. To examine whether these PKM2-dependent effects were mediated by β-catenin, we performed TCF/LEF-1 luciferase reporter analyses, showing that PKM2 depletion significantly inhibited EGF-induced β-catenin transactivation (Fig. 1d). In addition, chromatin immunoprecipitation (ChIP) analyses showed that EGFR activation resulted in increased binding of β-catenin to the promoter region of CCND1 (coding for cyclin D1) (Fig. 1e) and c-myc (data not shown), which was blocked by PKM2 depletion. In addition, coimmunoprecipitation (co-IP) analyses showed that PKM2 depletion inhibited EGF-induced interaction between β-catenin and Myc-tagged TCF4 (Fig. 1f). However, PKM2 depletion failed to inhibit Wnt3a- or Wnt1- (data not shown) induced β-catenin transactivation (Supplementary Fig. 3b) and cyclin D1 expression (Supplementary Fig. 3c). In addition, Wnt3a did not induce PKM2 nuclear translocation (Supplementary Fig. 3d). These results indicate that EGF induces β-catenin transactivation via a mechanism distinct from that induced by Wnt/wingless4 and that PKM2 expression plays a pivotal role in EGF-, but not Wnt-induced β-catenin transactivation.
To examine the mechanism underlying PKM2-regulated β-catenin transactivation, we performed co-IP analyses, showing that EGF stimulation resulted in an interaction between endogenous PKM2 and β-catenin in the nuclear, but not cytosolic, fraction of U87/EGFR cells (Fig. 1g). However, an in vitro glutathione S-transferase (GST) pull-down assay showed that purified GST-β-catenin failed to bind to purified His-PKM2 (Supplementary Fig. 4). These results suggest that the interaction of these two proteins might require post-translational modifications of the proteins.
PKM2 binds to tyrosine-phosphorylated peptides, and expression of the phosphotyrosine-binding form is required for cancer cell growth9. To examine whether β-catenin is tyrosine-phosphorylated, we performed immunoblotting analyses with a phospho-Tyr antibody, showing that EGF stimulation induced Tyr phosphorylation of immunoprecipitated β-catenin in the nucleus, but not in the cytosol or at the plasma membrane (Fig. 1h). Treatment of the immunoprecipitated β-catenin with calf intestinal alkaline phosphatase (CIP) resulted in β-catenin dephosphorylation and abrogation of the PKM2–β-catenin interaction (Fig. 1i). Thus, EGF-induced Tyr-phosphorylation of β-catenin is required for the PKM2–β-catenin interaction.
PKM2 K433E mutant, which fails to bind to tyrosine-phosphorylated peptides9, had similar glycolytic enzyme activity to its WT counterpart (Supplementary Fig. 5)9. Co-IP analyses showed that EGF treatment induced the binding of β-catenin to FLAG-tagged wild-type (WT) PKM2, but not to the PKM2 K433E mutant (Fig. 1j). In contrast, a kinase-dead FLAG-PKM2 K367M10,11, acting like its WT counterpart, binds to β-catenin (Fig. 1k). These results indicate that the K433 binding residue of PKM2, but not its catalytic activity, is critical for the PKM2–β-catenin interaction.
ABL and Src have been reported to phosphorylate β-catenin12,13. Pretreatment with SU6656 (Src inhibitor) or an ABL inhibitor completely abrogated EGF-induced activation of c-Src or ABL, as shown by the reduced levels of c-Src (Y418) or ABL (Y412) phosphorylation (Supplementary Fig. 6a). However, inhibition of c-Src, but not ABL, blocked EGF-induced Tyr phosphorylation of β-catenin (Fig. 2a). In addition, deficiency of c-Src (Fig. 2b), but not of ABL (Supplementary Fig. 6b), abrogated EGF-induced β-catenin Tyr-phosphorylation and the PKM2–β-catenin interaction. These results indicate that c-Src, a downstream effector of EGFR, phosphorylates β-catenin, which is required for the PKM2–β-catenin interaction.
Figure 2. c-Src phosphorylates β-catenin at Y333 upon EGFR activation.
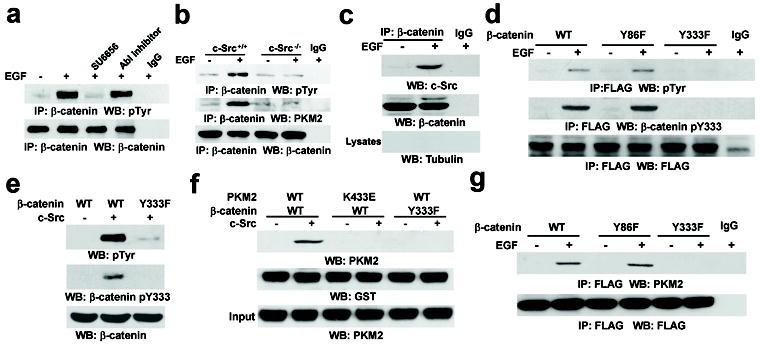
a, U87/EGFR cells were treated with SU6656 (4 μM) or an Abl inhibitor (0.2 μM) for 30 min before EGF treatment for 6 h.
b, The indicated cells were treated with or without EGF for 6 h.
c, β-catenin was immunoprecipitated from the nuclear fractions of U87/EGFR cells treated with or without EGF for 6 h.
d, g, U87/EGFR cells transiently expressing the indicated FLAG-tagged β-catenin proteins were treated with or without EGF for 6 h.
e, In vitro kinase assays were performed with purified active c-Src and purified β-catenin proteins.
f, Immobilized GST-β-catenin proteins were mixed with purified His-PKM2 proteins in the presence or absence of active c-Src.
To examine the subcellular compartment in which c-Src phosphorylates β-catenin, we conducted fractionation analyses. We found that EGF stimulation resulted in the nuclear translocation of c-Src (Supplementary Fig. 6c). In addition, co-IP analyses showed that EGF treatment induced an enhanced interaction between β-catenin and c-Src in nuclear fractions (Fig. 2c). These results, combined with the evidence that β-catenin is phosphorylated in the nucleus (Fig. 1h), strongly suggest that c-Src translocates into the nucleus and subsequently interacts with and phosphorylates β-catenin.
The Y86 residue of β-catenin has been shown to be phosphorylated by Src as well as Bcr-ABL12,13, and analysis of the amino acid sequence identified an additional potential Src phosphorylation site at Y333. Immunoblotting analysis showed that EGF stimulation resulted in tyrosine phosphorylation of FLAG-tagged WT β-catenin and β-catenin Y86F, but not β-catenin Y333F, which was further validated by immunoblotting with a phospho-β-catenin Y333 antibody (Fig. 2d). In addition, cell fractionation analysis demonstrated that EGF induced β-catenin Y333 phosphorylation primarily in the nucleus (Supplementary Fig. 6d). Furthermore, an in vitro protein kinase assay showed that active c-Src was able to phosphorylate WT β-catenin (Fig. 2e), but the β-catenin Y333F mutation largely reduced total Tyr-phosphorylation levels and completely abrogated the Y333-phosphorylation. These results reveal that c-Src binds and phosphorylates β-catenin at Y333 in vitro and in vivo.
To examine whether phosphorylation of β-catenin Y333 regulates its binding to PKM2, we performed a GST pull-down assay by mixing purified GST-β-catenin and His-PKM2 with or without purified active c-Src. Fig. 2f shows that WT PKM2 did not bind to unphosphorylated WT β-catenin. However, the presence of c-Src enabled the binding of WT PKM2 to β-catenin, which was abrogated by mutation of β-catenin at Y333 or PKM2 at K433. These in vitro results were further validated by co-IP analyses, showing that in contrast to FLAG-tagged WT β-catenin or β-catenin Y86F, FLAG-β-catenin Y333F failed to bind to endogenous PKM2 (Fig. 2g). These results indicate that c-Src-mediated β-catenin Y333 phosphorylation is required for the PKM2–β-catenin interaction.
We next examined the significance of the PKM2–β-catenin interaction in β-catenin transactivation. Fig. 3a shows that FLAG-β-catenin Y333F failed to interact with Myc-tagged TCF4 upon EGF stimulation, in contrast to its WT counterpart. ChIP analyses demonstrated that FLAG-tagged WT β-catenin bound to the CCND1 promoter region upon EGFR activation, which was abrogated by the Y333F mutation (Fig. 3b). In addition, reconstituted expression of the RNAi-resistant β-catenin (rβ-catenin) Y333F mutant, but not of WT rβ-catenin, in endogenous β-catenin-depleted U87/EGFRvIII cells failed to induce cyclin D1 and c-Myc expression (Fig. 3c). Furthermore, FLAG-PKM2 K433E and the inactive FLAG-PKM2 K367M mutant translocated into the nucleus upon EGF stimulation (Supplementary Fig. 7), but reconstituted expression of these mutants (Supplementary Fig. 8) failed to induce cyclin D1 expression as did WT rPKM2 expression (Fig. 3d). Thus, both PKM2 catalytic activity and the PKM2–β-catenin interaction are required for cyclin D1 expression upon EGFR activation.
Figure 3. The PKM2–β-catenin interaction is required for β-catenin-induced cyclin D1 expression.
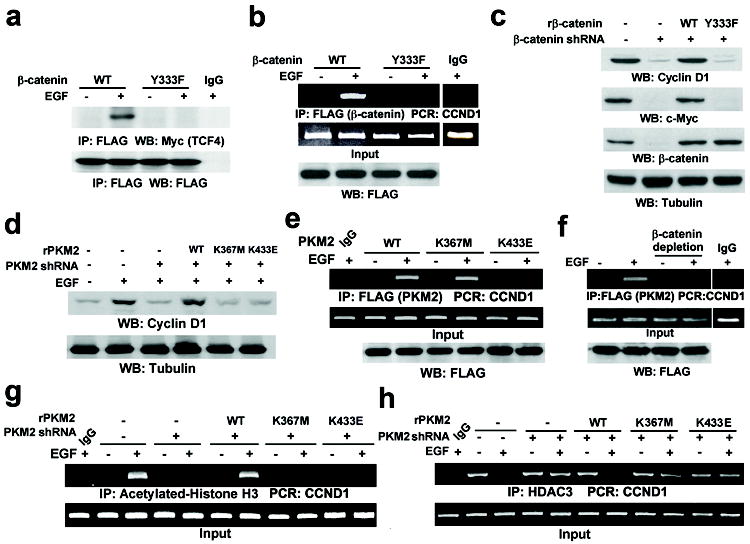
a, b, U87/EGFR cells transiently expressing FLAG-β-catenin proteins were treated with or without EGF for 10 h.
c, β-catenin was depleted in U87/EGFRvIII cells, followed by reconstituted expression of rβ-catenin.
d, g, h, U87/EGFR cells with or without depleted PKM2 and reconstituted expression of rPKM2 were treated with or without EGF for 24 h (d) or 10 h (g, h).
e, U87/EGFR cells transiently expressing the indicated FLAG-tagged PKM2 protein were treated with or without EGF for 10 h.
f, U87/EGFR cells transiently expressing FLAG-PKM2 were treated with or without EGF for 10 h. β-catenin was immunoprecipitated from the cell lysates, and the remaining supernatant was used for ChIP analyses.
To compare downstream targets of EGF and Wnt signaling, we examined the expression of other Wnt/β-catenin downstream genes: AXIN2, DKK1, and βTrCP8. Quantitative RT-PCR analysis showed that EGF treatment increased mRNA levels of DKK1, but not of AXIN2 or βTrCP, which was blocked by PKM2 depletion (Supplementary Fig. 9). These results indicate that EGF-induced and PKM2-dependent β-catenin transactivation induced transcription of a set of genes, which do not completely overlap with those induced by Wnt signaling.
The findings that PKM2 was not required for Wnt3a-induced β-catenin transactivation (Supplementary Fig. 3b) suggested that c-Src-dependent β-catenin Y333 phosphorylation is not involved in Wnt-induced signaling or cell adhesion. This assumption was supported by the results, showing that β-catenin Y333F behaved similarly to WT β-catenin in binding to APC, AXIN2, and E-cadherin and in Wnt-induced β-catenin transactivation and cellular functions (Supplementary Fig. 10, a-g). In contrast, expression of β-catenin Y333F blocked EGF- but not Wnt3a-induced cell migration (Supplementary Fig. 10h).
To investigate the mechanisms underlying PKM2- and β-catenin-dependent cyclin D1 expression, we performed ChIP analyses. Fig. 3e shows that EGF induced an enhanced binding of WT FLAG-PKM2 and FLAG-PKM2 K367M, but not of FLAG-PKM2 K433E, to the CCND1 promoter region. In addition, binding of FLAG-PKM2 to the promoter region was not detected in ChIP analyses after β-catenin had been immunodepleted from cell lysates (Fig. 3f). These results indicate that the PKM2–β-catenin interaction, but not PKM2 kinase activity, is required for both proteins to bind to the CCND1 promoter region.
We next examined whether PKM2 binding to the CCND1 promoter region regulates histone H3 acetylation, which is important for gene transcription. ChIP analyses showed that EGF treatment resulted in a significant increase of histone H3 acetylation in the CCND1 promoter region, which was blocked by PKM2 depletion (Fig. 3g). Reconstituted expression of rPKM2 K433E and the inactive rPKM2 K367M mutant failed to restore EGF-induced histone H3 acetylation, compared with the WT protein.
To further understand the mechanism underlying PKM2-regulated histone H3 acetylation, we performed ChIP analyses with antibodies against ubiquitously expressed histone deacetylase (HDAC)1, HDAC2, and HDAC314. Fig. 3h shows that HDAC3, but not HDAC1 or HDAC2 (Supplementary Fig. 11a), was prebound to the CCND1 promoter. EGF treatment resulted in the disassociation of HDAC3 from the CCND1 promoter, and this disassociation was blocked by PKM2 depletion. Furthermore, reconstituted expression of WT rPKM2, but not of rPKM2 K367M or rPKM2 K433E mutants, was able to restore EGF-induced disassociation of HDAC3 from the promoter (Fig. 3h). These results indicate that the kinase activity of PKM2 and its binding with β-catenin to the CCND1 promoter region are required for HDAC3 removal from the promoter. An additional co-IP analysis showed that WT PKM2, but not PKM2 K367M, interacted with HDAC3 upon EGF treatment (Supplementary Fig. 11b). These results suggest that PKM2 can interact with HDAC3 in an active conformation, which facilitates HDAC3 removal from the CCND1 promoter, acetylation of histone H3, and cyclin D1 expression.
To support the findings that EGF-induced and c-Src-dependent PKM2–β-catenin interaction and subsequent cyclin D1 expression are not cell line-specific, we treated GSC11 and GSC23 human primary GBM cells with EGF. Supplementary Fig. 12a shows that EGF treatment results in nuclear translocation of PKM2 and cyclin D1 expression in these cells. In addition, EGF-induced phosphorylation of β-catenin Y333 (Supplementary Fig. 12b) and its association with PKM2 (Supplementary Fig. 12c) were blocked by pretreatment with SU6656.
We next examined the significance of the PKM2–β-catenin interaction in tumor cell proliferation. Fig. 4a shows that U87/EGFRvIII cells proliferated much faster than did parental U87 cells. Expression of β-catenin shRNA (Fig. 3c) largely inhibited U87/EGFRvIII cell proliferation, which was rescued by reconstituted expression of WT rβ-catenin, but not of the rβ-catenin Y333F mutant (Fig. 4a). In addition, EGFRvIII-promoted cell proliferation was similarly inhibited by PKM2 depletion, which was rescued by the reconstituted expression of WT rPKM2, but not of the rPKM2 K433E or rPKM2 K367M mutant (Supplementary Fig. 13a and Fig. 4b).
Figure 4. The PKM2–β-catenin interaction is required for tumor development.
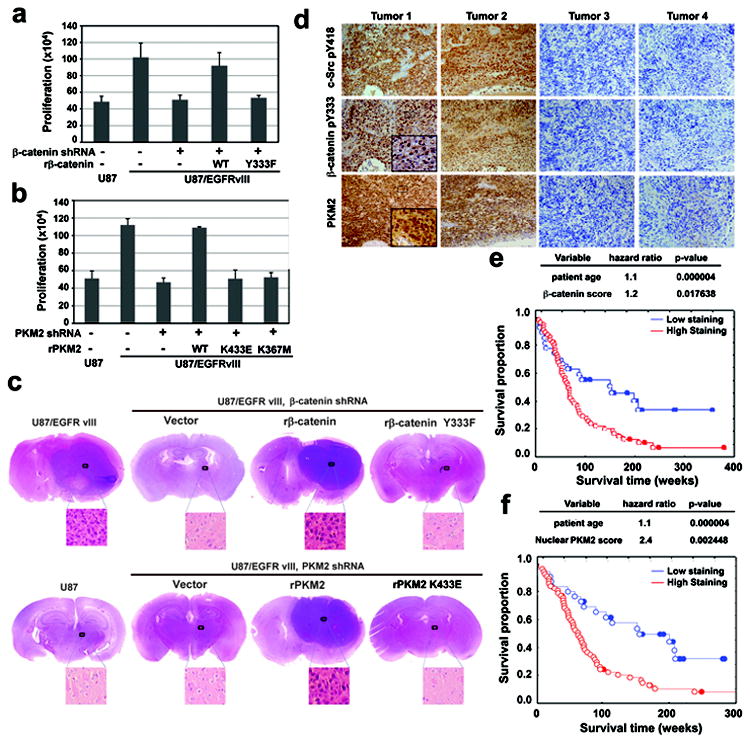
a, b, U87, U87/EGFRvIII cells with or without depleted β-catenin and reconstituted expression of rβ-catenin (a), or U87/EGFRvIII cells with or without depleted PKM2 and reconstitution of the expression of rPKM2 (b), were plated and counted 7 days after seeding. Data represent the means ± SD of three independent experiments.
c, U87 (bottom left panel), U87/EGFRvIII cells with or without depleted β-catenin and reconstituted expression of rβ-catenin (top panel), or U87/EGFRvIII cells with or without depleted PKM2 and reconstituted expression of rPKM2 (bottom right panel) were intracranially injected into athymic nude mice. After two weeks, tumor growth was examined. H&E-stained coronal brain sections show representative tumor xenografts.
d, IHC staining with the indicated antibodies was performed on 55 GBM specimens. Representative photos of four tumors are shown.
e, f, The survival time for 84 patients with low (0-5 staining scores, blue curve) versus high (6-8 staining scores, red curve) β-catenin Y333 phosphorylation (e; low, 28 patients; high, 56 patients) and nuclear PKM2 expression (f; low, 28 patients; high, 56 patients) were compared (bottom panel). The table (top panel) shows the multivariate analysis after adjustment for patient age, indicating the significance level of the association of Y133-phosphorylated β-catenin expression (e) or nuclear PKM2 expression (f) with patient survival. Empty circles represent the deceased patients, and filled circles represent the censored (alive at last clinical follow-up) patients.
β-catenin-regulated cyclin D1 expression is critical for G1-S phase transition and cell cycle progression15. Depletion of β-catenin or PKM2 resulted in an accumulation of U87/EGFRvIII cells in G0/G1 phase, which was rescued by reconstituted expression of WT rβ-catenin or WT rPKM2, but not of the rβ-catenin Y333F or rPKM2 K433E mutant (Supplementary Fig. 13b). Thus, the PKM2–β-catenin interaction is essential for cell cycle progression.
To determine the role of PKM2-dependent β-catenin transactivation in brain tumor development, we intracranially injected U87 or U87/EGFRvIII cells into athymic nude mice. U87 cells did not form a detectable tumor two weeks after injection (Fig. 4c, bottom left panel). In contrast, U87/EGFRvIII cells elicited rapid tumorigenesis (Fig. 4c, top left panel). Notably, depletion of β-catenin (top panel) or PKM2 (bottom panel) abrogated EGFRvIII-driven tumor growth, which was rescued by expression of WT rβ-catenin or WT rPKM2, but not of the rβ-catenin Y333F or rPKM2 K433E mutant. Similar results were obtained using GSC11 cells (Supplementary Fig. 14a). In addition, adding doxycycline to the drinking water 14 days after intracranial injection of GSC11 cells that expressed a tetracycline-inducible PKM2 shRNA partially depleted PKM2 expression in tumor tissues and inhibited tumor growth (Supplementary Fig. 14b). Thus, the PKM2–β-catenin interaction and PKM2 expression is instrumental in tumor growth.
To further support the role of c-Src in EGFR-induced β-catenin transactivation in vivo, we injected SU6656 intratumorally, which significantly blocked tumor growth (Supplementary Figs. 15a and 15b), reduced the phosphorylation levels of c-Src Y418 and β-catenin Y333, and inhibited cyclin D1 expression in tumor tissue (Supplementary Fig. 15c). The requirement of β-catenin transactivation in tumorigenesis was also examined by expression of Wnt1 in U87/EGFRvIII-PKM2 shRNA cells. Wnt1 expression resulted in the induction of cyclin D1 (Supplementary Fig. 16a) and largely rescued PKM2 depletion-blocked tumorigenesis (Supplementary Figs. 16b and 16c). In addition, this effect was further enhanced using Wnt1-expressing U87/EGFRvIII cells with reconstituted expression of rPKM2 K433E, which retains its catalytic activity for glycolysis. These results suggest that, while the metabolic function of PKM2 plays a critical role in aerobic glycolysis7 and tumorigenesis, brain tumor development promoted by EGFR requires PKM2-modulated β-catenin transactivation.
Analysis of publicly available microarray datasets (Affymetrix, U133) from The Cancer Genome Atlas (TCGA) and other sources16,17,18,19 revealed a correlation of c-myc and CCND1 expression with EGFR expression in GBM samples (Supplementary Fig. 17). In addition, phosphorylation levels of β-catenin Y333 correlated with phosphorylation levels of activated c-Src in seven human primary GBM cell lines (Supplementary Fig. 18).
We next performed immunohistochemical (IHC) analyses to examine c-Src activity, β-catenin Y333 phosphorylation, and PKM2 nuclear localization in serial sections of 55 human primary GBM specimens by using antibodies with validated specificities (Supplementary Fig. 19). Fig. 4d shows that the levels of c-Src Y418 phosphorylation, β-catenin Y333 phosphorylation, and nuclear PKM2 expression were correlated with each other. In addition, β-catenin Y333 phosphorylation accumulated in the nuclei of a large percentage of tumor cells (Fig. 4d, left panel). Quantification of the staining on a scale of 0 to 8.0 showed that these correlations were significant (Supplementary Fig. 20). The survival durations of 84 patients, all of whom received standard adjuvant radiotherapy after surgery, followed by treatment with an alkylating agent (temozolomide in the majority of cases), with low (0-5 staining) versus high (5.1-8 staining) β-catenin Y333 phosphorylation and nuclear PKM2 expression were compared. Patients whose tumors had low β-catenin Y333 phosphorylation or nuclear PKM2 expression had a median survival of 185.2 and 130.0 weeks, respectively. The median survival of patients decreased to 69.4 and 82.5 weeks, respectively, when their tumors showed high levels of β-catenin Y333 phosphorylation or nuclear PKM2 expression. In a Cox multivariate model, the IHC score of β-catenin phosphorylation (Fig. 4e) and nuclear PKM2 expression (Fig. 4f) were independent predictors of glioblastoma patient survival, after adjusting for the age of the patient, a relevant clinical covariate. These results support the role of PKM2 association with c-Src-phosphorylated β-catenin Y333 in the clinical behavior of human GBM and reveal a relationship between β-catenin Y333 phosphorylation/nuclear PKM2 localization and clinical aggressiveness of the tumor. These findings were further supported by IHC analyses, showing significantly lower levels of β-catenin Y333 phosphorylation in low-grade diffuse astrocytoma (30 cases) (World Health Organization [WHO] grade II; median survival time >5 years) than were present in GBM specimens (WHO grade IV)20 (Supplementary Fig. 21).
We previously showed that GSK-3β–independent transactivation of β-catenin by growth factor receptor occurs by mechanisms distinct from Wnt-dependent canonical signaling4,21-23. In this study, we describe an important and previously unknown mechanism underlying EGFR activation-induced β-catenin transactivation through interaction with PKM2, which plays a critical role in transcription of CCND1 and c-myc (Supplementary Fig. 1). The understanding that phosphorylation of β-catenin Y333 and its interaction with PKM2 are required for tumor cell proliferation and tumor development, and that the levels of β-catenin Y333 phosphorylation and nuclear PKM2 correlate with grades of glioma malignancy and prognosis, may provide a molecular basis for improved diagnosis and treatment of tumors with activated EGFR and upregulated PKM2. In summary, our findings, in combination with previous reports7,24, delineate two essential mechanisms underlying tumor development by regulation of metabolic and nonmetabolic functions of PKM2: 1) PKM2 enhances aerobic glycolysis7,24 and 2) PKM2 promotes tumor cell proliferation by binding to and transactivating Y333-phosphorylated β-catenin. Thus, PKM2 has dual roles that are essential for tumorigenesis: regulating cancer cell metabolism and gene transcription required for cell proliferation. The coordinated control of metabolism and proliferation by PKM2 is essential for tumorigenesis.
METHODS SUMMARY
Detailed methodology can be found in the Supplementary Information. In short, luciferase reporter gene assay, immunoprecipitation and immunoblotting analysis, purification of recombinant proteins, in vitro kinase assays, and cell migration assay were performed, as previously described14,21,22.
Supplementary Material
Acknowledgments
We thank Tony Hunter (The Salk Institute for Biological Studies) for ABL and c-Src knockout cells, Hans Clevers (Netherlands Institute for Developmental Biology, Hubrecht Laboratory) for the pTOP-FLASH and the pFOP-FLASH, and Yi Li (Baylor College of Medicine) for a Wnt1 lenti-vector.
This work was supported by National Cancer Institute grants 5R01CA109035 (Z.L.), 5 P50 CA127001-03, and CA16672 (Cancer Center Support Grant); a research grant (RP110252; Z. L.) from the Cancer Prevention and Research Institute of Texas (CPRIT), an American Cancer Society Research Scholar Award RSG-09-277-01-CSM (Z.L.), and a Sister Institution Network Fund from The University of Texas MD Anderson Cancer Center (Z.L.).
Footnotes
Contributions This study was conceived by Z.L. Z.L and W.Y. designed the study; W.Y., Y.X., H.J., Y.Z., and J.L. performed experiments; K.A. provided pathology assistance; W.H. and X.G. provided reagents and conceptual advice; Z.L. wrote the paper with comments from all authors.
References
- 1.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 3.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 4.Lu Z, Hunter T. Wnt-independent beta-catenin transactivation in tumor development. Cell Cycle. 2004;3:571–573. [PubMed] [Google Scholar]
- 5.Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol. 2001;21:4016–4031. doi: 10.1128/MCB.21.12.4016-4031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wykosky J, Fenton T, Furnari F, Cavenee WK. Therapeutic targeting of epidermal growth factor receptor in human cancer: successes and limitations. Chin J Cancer. 2011;30:5–12. doi: 10.5732/cjc.010.10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 8.Yochum GS, et al. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proc Natl Acad Sci U S A. 2007;104:3324–3329. doi: 10.1073/pnas.0611576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 10.Le Mellay V, et al. Regulation of glycolysis by Raf protein serine/threonine kinases. Adv Enzyme Regul. 2002;42:317–332. doi: 10.1016/s0065-2571(01)00036-x. [DOI] [PubMed] [Google Scholar]
- 11.Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Coluccia AM, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26:1456–1466. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miravet S, et al. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol. 2003;23:7391–7402. doi: 10.1128/MCB.23.20.7391-7402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia Y, et al. c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol Cell. 2007;25:219–232. doi: 10.1016/j.molcel.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 16.Freije WA, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 17.Gravendeel LA, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 18.Petalidis LP, et al. Improved grading and survival prediction of human astrocytic brain tumors by artificial neural network analysis of gene expression microarray data. Mol Cancer Ther. 2008;7:1013–1024. doi: 10.1158/1535-7163.MCT-07-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Furnari FB, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 21.Ji H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36:547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang D, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 24.Luo W, et al. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Z, et al. Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol. 1998;18:839–845. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez-Manzano C, et al. Delta-24 increases the expression and activity of topoisomerase I and enhances the antiglioma effect of irinotecan. Clin Cancer Res. 2006;12:556–562. doi: 10.1158/1078-0432.CCR-05-1892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.