

Abstract
Parkinson's disease (PD) is the second most common neurodegenerative disease in developed countries. The core motor symptoms are attributable to the degeneration of dopamine (DA) neurons in the substantia nigra pars compacta (SNc). Why these neurons, and other restricted sets of non-dopamine neuron, succumb in PD is not clear. One potential clue has come from the observation that the engagement of L-type Ca2+ channels during autonomous pacemaking elevates the sensitivity of SNc DA neurons to mitochondrial toxins used to create animal models of PD, suggesting that Ca2+ entry is a factor in their selective vulnerability. Epidemiological data also supports a linkage between L-type Ca2+ channels and the risk of developing PD. This review examines the hypothesis that the primary factor driving neurodegenerative changes in PD is the metabolic stress created by sustained Ca2+ entry, particularly in the face of genetic or environmental factors that compromise oxidative defenses or proteostatic competence.
PD is a disabling neurodegenerative disorder that is strongly associated with aging, increasing exponentially in incidence above the age of 65 [1, 2]. The incidence of PD is expected to rise dramatically worldwide in the next 25 years with the extension of life expectancy by improved health care [3]. Although there are signs of distributed neuropathology (as judged by Lewy body formation) [4], the motor symptoms of PD, including bradykinesia, rigidity, and resting tremor, are clearly linked to the degeneration and death of SNc DA neurons [5, 6]. Although there are pathological changes in other select regions of the brains [4], the efficacy of the clinical gold-standard treatment of L-DOPA – a DA precursor – is testament to the centrality of DA neurons in the motor symptoms of PD.
What causes SNc DA neurons to die in PD?
The mechanisms responsible for the preferential loss of DA neurons in PD have been debated for decades. A widely held theory implicates DA itself, suggesting that oxidation of cytosolic DA (and its metabolites) leads to the production of cytotoxic free radicals [7, 8]. However, there are reasons to doubt this type of cellular stress alone is responsible for the loss of DA neurons in PD. For example, there is considerable regional variability in the vulnerability of DA neurons in PD, with some being devoid of pathological markers [9-13]. Moreover, L-DOPA administration (which relieves symptoms by elevating DA levels in PD patients) does not appear to accelerate disease progression [14], suggesting that DA is not a significant source of reactive oxidative stress, at least in the short term. Sulzer and colleagues have recently reported that Ca2+ entry through L-type channels stimulates DA metabolism in SNc DA neurons, pushing cytosolic DA concentrations into a toxic range with L-DOPA loading [15]. For this mechanism to be relevant to selective vulnerability, one would have to posit that modest elevations in cytosolic DA over decades leads to an accumulation of cellular defects that ultimately produces cell death. Although plausible, the hypothesis is not readily testable. It does suggest that treating patients in the early stages of the disease with direct acting agonists, rather than L-DOPA, should lead to a slower progression of the disease. That said, the frank death or phenotypic decline of a variety of non-dopaminergic neurons in PD argues that DA itself is not likely to be the principal culprit in the disease.
If not DA, then what? In recent years, attention has turned to the role of mitochondrial dysfunction in PD [16-18]. In addition to the ability of several toxins that target mitochondria to create a parkinsonian phenotype [19, 20], compelling evidence for mitochondrial involvement in PD comes from the study of human PD patients. In postmortem tissue samples of the SNc from sporadic PD patients, there is a substantial decrease in the activity of mitochondrial NADH ubiquinone reductase, referred to as complex I of the electron transport chain (ETC) [21]; this deficit is specific to PD patients [22] and appears to reflect oxidative damage to complex I [23]. Oxidative damage to other cellular components such as lipids, proteins and DNA also has been found in the SNc of PD brains [24]. The source of this oxidative stress is likely to be mitochrondrial. Reactive oxygen species (ROS) and other radicals are generated by inefficiencies in the ETC, which is responsible for creating the electrochemical gradient across the inner mitochondrial membrane that drives ATP synthase and the conversion of adenosine diphosphate to ATP [25]. ROS also are thought to be responsible for the high level of somatic DNA and mitochondrial DNA mutations in SNc DA neurons [26-28]. Lastly, although deficits in the complex I activity of platelets, skeletal muscle, fibroblasts, and lymphocytes have been reported in some PD patients [29], this is not a consistent feature of the disease, arguing that mitochondrial dysfunction is regionally selective [30]. Further support for a mitochondrial link in PD comes from the rapidly expanding literature on genetic mutations associated with familial forms of PD [16].
Another organelle that has been widely linked to pathogenesis in PD is the endoplasmic reticulum (ER). The ER is an integral component of the cellular machinery responsible for the production, delivery and degradation of proteins, a process referred to as proteostasis [31]. One of the hallmarks of PD is the formation of Lewy bodies (LBs), an abnormal protein aggregate found in SNc DA neurons and elsewhere in the brain [32]. These depositions reflect a deficiency in proteostasis that is accompanied by signs of ER stress and an attempt to sequester cytotoxic proteins [33]. In part, LBs in PD must reflect the decline in proteostatic competence that accompanies normal aging [31]. What appears to distinguish PD is the presence of an additional proteostatic burden that causes an aged DA neuronal ensemble to fail en masse (see below) [18, 34, 35].
Taken together, the evidence for the involvement of mitochondria and ER in the PD pathogenesis is unequivocal. The critical question is whether the disease begins with the dysfunction of these organelles. The striking regional distribution of deficits argues against this proposition. There is no evidence that mitochondria in cortical pyramidal neurons (which show little to no sign of pathology in PD) differ in any important respect from mitochondria in SNc DA neurons. There is no evidence for substantive variation in the ER (or other proteostatic elements) between vulnerable and resistant neurons either. Furthermore, there is no evidence of selective regional expression of genes associated with familial forms of PD that would be predictive of disease progression [36]. The most straightforward conclusion to be drawn from the evidence at hand is that the cellular environment in which these organelles find themselves accelerates their decline with age, making them more vulnerable to genetic or environmental stress. What then is distinctive about the organelle environment created by SNc DA neurons?
SNc DA neurons have a distinctive physiological phenotype
SNc DA neurons have an unusual physiological phenotype. Unlike the vast majority of neurons in the brain, adult SNc DA neurons are autonomously active, generating action potentials regularly (2-4 Hz) in the absence of synaptic input [37]. This pacemaking activity is believed to be important in maintaining ambient DA levels in regions that are innervated by these neurons, particularly the striatum [38]. While most neurons rely exclusively on monovalent cation channels to drive pacemaking, SNc DA neurons also engage ion channels that allow Ca2+ to enter the cytoplasm [39-41], leading to elevated intracellular Ca2+ concentrations [42, 43]. The L-type Ca2+ channels used by SNc DA neurons in pacemaking have a distinctive Cav1.3 pore-forming subunit encoded by Cacna1d [43, 44]. Cav1.3 Ca2+ channels are relatively rare, constituting only about 10% of the all the L-type Ca2+ channels found in the brain [45]. Channels with this subunit differ from other L-type Ca2+ channels in that they open at relatively hyperpolarized potentials, allowing them to contribute to the mechanisms driving the membrane potential to spike threshold underlying autonomous pacemaking [41, 43, 46].
Until recently, it was thought that L-type Ca2+ channels were essential for pacemaking in SNc DA neurons making them less than ideal drug targets if pacemaking was necessary to maintain physiologically important levels of DA in target structures [47, 48]. This inference was based upon the ability of L-type channel antagonists (dihydropyridines) to halt pacemaking. However, we know now that at the concentrations of dihydropyridine (DHP) necessary to stop pacemaking, other ion channels are being antagonized, complicating the interpretation of previous studies. This discovery was made possible by the integration of traditional patch clamp electrophysiology and two photon laser scanning microscopy in brain slices. The combination of approaches allowed dendritic Ca2+ oscillations to be monitored with high temporal resolution at the same time as somatic voltage, revealing that at lower, channel-specific DHP concentrations, pacemaking continues in SNc DA neurons, unaltered in rate and regularity, even when dendritic L-type Ca2+ channels were effectively antagonized (Fig. 1) [46]. The ability of SNc DA neurons to continue pacemaking under these circumstances reflects the robustness of the multi-channel pacemaking mechanism and that Cav1.3 Ca2+ channels play a supportive, but not necessary role. In addition to this role, Cav1.3 Ca2+ channels participate in the postsynaptic response to activation of glutamatergic synapses and burst spiking in response to reward prediction errors [M. Bevan, unpublished observations]. However, as with pacemaking, this role is supportive, rather than necessary.
Figure 1. Low concentrations of DHPs suppress dendritic Ca2+ oscillations but do not slow pacemaking.
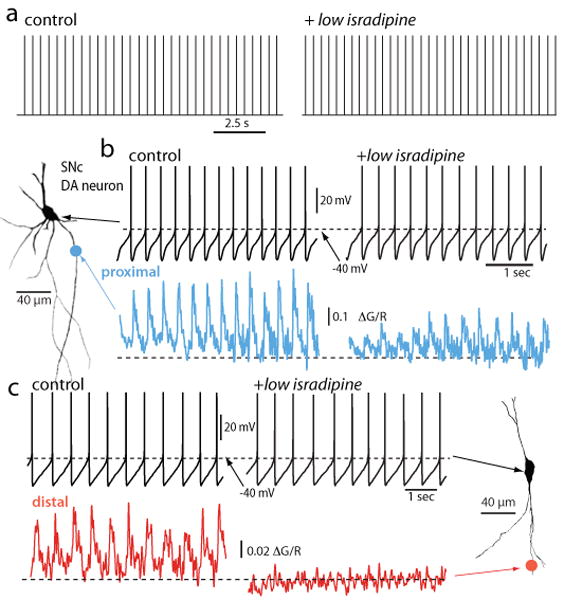
a. Digitized cell-attached patch recordings from an SNc DA neuron before and after application of isradipine (5 μM). The median discharge rate before isradipine application was 2.2 Hz and 2.4 Hz after (p>0.05, n=4). b. Whole cell recording from the cell shown to the left (projection image) before and after isradipine (5 μM) application; there was no significant change in discharge rate in this cell or in 10 others. At the bottom, 2PLSM measurements of Fluo-4 fluorescence (G) at a proximal dendritic location (∼40 μm from the soma) normalized by the fluorescence of the red Alexa dye used to image the cell. c. Somatic recording during imaging at a more distal dendritic location (∼120-200 μm from the soma). Note the complete elimination of the spike associated dendritic Ca2+ transient at the distal imaging site. Similar results were obtained in 6 other neurons. From [46]
The sustained engagement of Cav1.3 Ca2+ channels during pacemaking comes at an obvious metabolic cost to SNc DA neurons. Because of its involvement in cellular processes ranging from the regulation of enzyme activity to programmed cell death, Ca2+ is under very tight homeostatic control, with a cytosolic set point near 100 nM – 10,000 times lower than the concentration of Ca2+ in the extracellular space [49-51]. Ca2+ entering neurons is rapidly sequestered or pumped back across the steep plasma membrane concentration gradient; this process requires energy stored in ATP or in ion gradients that are maintained with ATP-dependent pumps. In most neurons, Ca2+ channel opening is a rare event, occurring primarily during very brief action potentials. This makes the task and the metabolic cost to the cell readily manageable. But in SNc DA neurons, where Cav1.3 Ca2+ channels are open much of the time, the magnitude and the spatial extent of Ca2+ influx are much larger [42].
Another distinctive feature of SNc DA neurons, and many of the other neurons that succumb in PD (e.g., locus ceruleus neurons), is their enormous axonal field. Recent anatomical work has estimated that a typical SNc DA neuron has mean axonal length of 470,000 μm [52]. Furthermore, each axon supports ∼370,000 synapses, orders of magnitude higher than the number supported by cortical pyramidal neurons for example [53]. Although the oxidative stress experienced by each of these terminals might very well be normal (this has yet to be determined), it could be that the proteostatic burden created by maintaining this extraordinary axonal tree creates a substantial metabolic load in and of itself. Moreover, the need to supply this axonal field with mitochondria to maintain ionic gradients and exocytosis could create an axonal sink for these organelles, potentially depriving the somatic region of an adequate mitochondrial complement. In fact, mitochondrial density in the somatodendritic region of SNc DA neurons appears to be abnormally low [54]. Asking a depleted mitochondrial complement in the somatodendritic region to meet the challenge posed by sustained Ca2+ entry and this large axonal tree should lead to a sustained elevation in oxidative phosphorylation and the production of superoxide and ROS.
Is PD a manifestation of Ca2+-accelerated aging?
One of the oldest and most popular theories of aging is that it is a direct consequence of accumulated mtDNA and organelle damage produced by ROS and related reactive molecules generated by the ETC in the course of oxidative phosphorylation [55, 56]. A corollary of this hypothesis is that the rate of aging is directly related to metabolic rate. There is no obvious reason not to extend this organismal postulate to individual cells. The reliance of SNc DA neurons on a metabolically expensive strategy to generate autonomous activity that taxes mitochondria should mean that they age more rapidly than other types of neuron. Is then PD simply a reflection of accelerated aging in neurons that rely too heavily upon Ca2+ channels to do their business? Age is undoubtedly the single strongest risk factor for PD [57, 58]. Stereological estimates of normal aging related cell death in humans argue that SNc DA neurons at a higher risk than other neurons in the absence of environmental toxins or pathogens, as they are lost at a significantly higher rate than many other types of neurons (some of which show no appreciable loss over a 6-7 decade span) [59]. In mammals with significantly shorter life-spans, loss of SNc DA neurons with age has not been seen reliably, but there is a clear decline in phenotypic markers with age that matches that seen in PD, as well as an increased susceptibility to toxins [60-65]. Taken together, these studies make a case that SNc DA neurons age more rapidly than the vast majority of the neurons in the brain.
This ‘wear and tear’ theory suggests that PD is first and foremost a consequence of aging. Why then do some people become symptomatic in their 50's, others in their 60's or 70's or not at all in an 80 or 90 year life? Genetic factors certainly could account for a large part of this variation [8, 36, 57]. These factors could increase the rate at which vulnerable neurons age by compromising mitochondrial function or Ca2+ homeostasis. Several of the genetic mutations associated with early onset forms of PD are directly linked to mitochondrial dysfunction [16]. Unfortunately, animal models of these forms of PD have failed to recapitulate the disease. For example, mutations of DJ-1 (PARK7) are associated with an early-onset form of PD. It has been putatively categorized as an atypical mitochondrial “peroxiredoxin-like peroxidase,” decreasing the accumulation of hydrogen peroxide and damage due to ETC superoxide generation [66]. But, how this happens is not clear. DJ-1 is redox sensitive, giving it the capacity to signal oxidative challenges and potentially coordinate a variety of mitochondrial oxidative defense mechanisms [67]. Although deleting DJ-1 increases the sensitivity of cells generally to heroic oxidative challenge, it remains to be determined how it compromises SNc DA neurons under more physiological conditions. Studies of DJ-1 knockouts have not revealed any loss of SNc DA neurons or a fundamental shift in their physiology, only a modest deficit in DA release during burst stimulation that is difficult to tie to an increased vulnerability [68]. But there have been no studies in these mice of mitochondrial physiology in mature SNc DA neurons where Ca2+ influx during pacemaking has fully developed. This could prove to be critical to an understanding of the mechanisms that could accelerate the loss of these cells over decades.
Two other genes linked to familial PD have unequivocal linkages to mitochondria. One is Parkin (PARK2). Fruit flies with functional deletions of Parkin have fragmented and apoptotic mitochrondria [69]; knockout mice have a less dramatic but a clear mitochondrial phenotype (including decreased mitochondrial (respiratory) function, decreased metabolic drive, and increased lipid and protein phosphorylation) [70]. Another is PTEN-induced putative kinase 1 or PINK1 (PARK6). Its deletion leads to an identical phenotype in Drosophila as does Parkin deletion – fragmented cristae and apoptotic mitochondria; this phenotype can be rescued by Parkin over-expression, suggesting involvement in some common biochemical pathway [71, 72]. Although found both in cytosolic and mitochrondrial preparations, PINK1 has an N-terminus mitochondrial targeting sequence [73]. PINK1 deletion also compromises DA release during burst stimulation, like DJ-1 [74]. It is not difficult to infer that loss of function mutations in either of these genes should have their biggest impact in a cell type that had a high basal level of metabolic activity, creating a mitochondrial oxidative stress. But to date, this has not been convincingly demonstrated.
Can the Ca2+-mediated cellular aging hypothesis account for the vulnerability of other cell types in PD? Other regions of the brain that have cell loss paralleling that of the SNc are the locus ceruleus (LC) and hypothalamic tuberomamillary nucleus [32, 75]. The neurons of the LC and the tuberomamillary neurons are similar to SNc DA neurons in several respects. Like SNc DA neurons, both LC and tuberomamillary neurons are autonomous pacemakers that depend upon L-type Ca2+ channels [76-78]. In contrast, DA neurons in the VTA do not rely upon L-type Ca2+ channels for pacemaking and are relatively intact in PD patients and in animal models of PD [11, 43, 79-81]. DA neurons in the olfactory bulb also are autonomous pacemakers and rely upon Ca2+ channels (although not L-type channels) [81]. While olfactory deficits have been associated with PD [82], there is no obvious loss of olfactory bulb DA neurons [83]. Although this would seem to run counter to the Ca2+ hypothesis, this could simply be a consequence of the capacity of this region for adult neurogenesis [84].
Can PD be prevented?
If PD is a consequence of Ca2+-accelerated aging in SNc DA neurons (and in those neurons with a similar phenotype), then reducing Ca2+ flux should delay the onset of PD symptoms as well as slow its progression. This might be possible with orally deliverable, DHP L-type channel antagonists shown to be safe in humans [43]. Adult SNc DA neurons readily compensate for the antagonism of L-type Cav1.3 Ca2+ channels and continue pacemaking at a normal rate [46]. More importantly, although the impact on mitochondrial and ER stress can only be inferred at this point, reducing Ca2+ influx during pacemaking dramatically diminishes the sensitivity of SNc DA neurons to toxins used to generate animal models of PD [43]. SNc DA neurons that express the Ca2+ binding protein calbindin also have a diminished sensitivity to PD toxins [79, 85]. Furthermore, at neuroprotective doses of an L-type channel antagonist, mice have no obvious motor, learning or cognitive deficits, suggesting that the patterned activity of SNc DA neurons is functionally unchanged [39, 46, 86].
Is there evidence that this strategy might work in humans to prevent or slow PD? Calcium channel antagonists (CCAs), including the DHPs used in animal studies, are commonly used in clinical practice to treat hypertension, creating a potential database to be mined. A case-control study of hypertensive patients found a significant reduction in the observed risk of PD with CCA use, but not with medications that reduce blood pressure in other ways [87]. More recently, a large Danish data set has been examined [88]. The authors agreed with the main conclusions of the Becker et al. study but extended their findings by showing that only DHPs that cross the blood-brain barrier (BBB) are associated with reduced PD risk (∼30%). Given the short period of treatment in many cases (∼2 years), variable dosing, and low relative affinity of DHPs for Cav1.3 Ca2+ channels (compared to Cav1.2 channels) [89-91], this is a surprisingly strong association and lends further credence to the proposition that a BBB permeable and potent Cav1.3 antagonist could be a very effective neuroprotective agent.
That said, these studies are not a substitute for a controlled clinical trial. In the absence of a selective Cav1.3 Ca2+ channel antagonist, the DHP isradipine is the most attractive drug for such a trial. Isradipine has a relatively higher affinity for Cav1.3 Ca2+ channels than the other known DHP and has good brain bioavailability [92, 93]. At the doses used to treat hypertension, isradipine has relatively minor side effects [94]. The question is whether it will prove neuroprotective at doses tolerated by the general population. Pharmacokinetic studies by our group have found that serum concentrations of isradipine achieved in mice that are protected (∼2 ng/ml) against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 6-hydroxydopamine toxicity are very close to those achieved in humans with a very well tolerated daily dose (10 mg/day, Dynacirc CR), suggesting that neuroprotection is achievable.
It is also worth considering how DHPs might be used in combination with other drugs that are being tested in clinical neuroprotection trials for PD. Although early trials with creatine, coenzyme Q10, and other antioxidant supplements have been disappointing [95], they share the hypothesis that oxidative stress exacerbates the symptoms and progression of PD. Coenzyme Q10 is an electron acceptor for complexes I and II that appears compromised in PD patients [96] and is neuroprotective in animal models of PD [97]. Creatine is a substrate for ATP production that can both improve mitochondrial efficiency and reduce oxidative stress by buffering fluctuations in cellular energy production [98]. Both approaches are aimed at improving mitochondrial function rather than attacking the source of stress on mitochondria. Rasagline or deprenyl also could prove to have neuroprotective effects by virtue not of its ability to inhibit monoamine oxidase B, but by their ability to its ability to induce the expression of antioxidant defenses [99, 100]. Because their sites of action differ within the chain of events leading to oxidative stress and mitochondrial dysfunction, a combination therapy could prove more effective than any one therapy alone.
Gaps in our understanding of the role of Ca2+ in the etiology of PD
There are a number of major gaps in our understanding of the role Ca2+ might play in the neuronal pathology seen in PD. Filling these gaps not only could provide novel therapeutic strategies for preventing or slowing the progression of the disease but could also help to create strategies for ‘successful aging’ generally. Figure 2 graphically summarizes some of the key points of the review and points of uncertainty.
Figure 2. A schematic summarizing a model of how Ca2+ entry during pacemaking in SNc DA neurons might lead to mitochondrial oxidative stress, accelerated aging and eventual cell death.
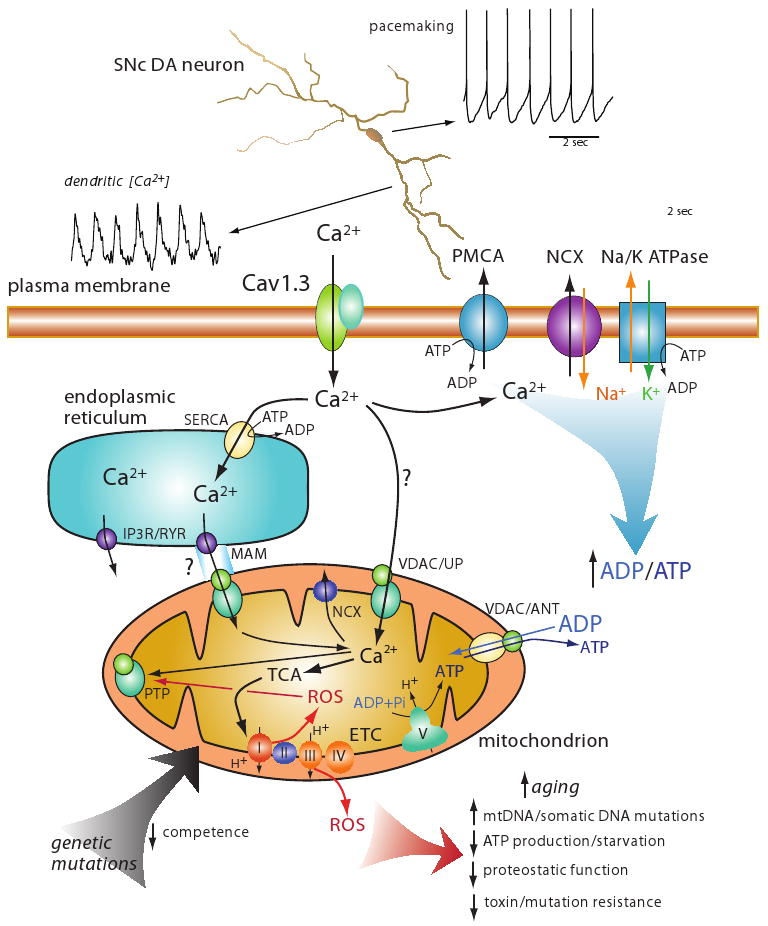
At the top is an image of a reconstructed SNc DA neuron showing the pacemaker driven somatic spiking and dendritic Ca2+ oscillations associated with pacemaking. This dendritic Ca2+ influx is attributable almost entirely to flux through Cav1.3 Ca2+ channels. At the bottom is a cartoon depicting the hypothetical mechanisms involved in elevating mitochondrial oxidative stress. Ca2+ influx through Cav1.3 channels is either sequestered in the endoplasmic reticulum by uptake through smooth endoplasmic reticulum Ca2+ (SERCA) pumps or taken up by mitochondria through channels created by voltage dependent anion channels (VDAC) and Ca2+ uniporter (UP); the extent to which this pathway is important is in question (hence the question mark on the arrow). Ca2+ could also enter mitochondria through this VDAC/UP channel at points of apposition between the ER and mitochondra where inositol trisphosphate receptors (IP3) and/or ryanodine receptors (IP3R/RYR) are positioned at specializations – mitochondrial associated membrane (MAM). This entry route has not been established in SNc DA neurons, hence the question mark. Ca2+ entering through Cav1.3 channels is moved back across the plasma membrane through either the Ca2+-ATPase (PMCA) or through a Na+/Ca2+ exchanger (NCX) that relies upon the Na+ gradient maintained by the Na/K ATPase at energetic cost; leading to conversion of ATP to ADP. ADP and ATP are exchanged by mitochondria with the VDAC and adenine nucleotide transporter (ANT). ADP stimulates oxidative phosphorylation. Ca2+ entering the mitochondrial matrix can stimulate enzymes of the tricarboxylic acid (TCA) cycle that produces reducing equivalents for the electron transport chain (ETC); complex I through V are shown at the inner mitochondrial membrane; complex V (ATP synthase) uses the electrochemical gradient to convert adenosine diphosphate (ADP) and inorganic phosphate to adenosine triphosphate (ATP). Electron movement along the ETC generates superoxide, leading to the production of reactive oxygen species (ROS) that can produce a variety of deleterious effects that can be viewed as accelerated aging (red arrow at the bottom). One other action of ROS is to promote opening of the mitochondrial permeability transition pore (mPTP); irreversible opening of the mPTP leads to release of pro-apoptetic factors into the cytosol. A basic question is whether reversible opening of the mPTP is possible under conditions of mild oxidative stress; mPTP opening and depolarization of the inner mitochondrial membrane could serve to diminish ROS production. Ca2+ is removed from mitochondrial through mitochrondrial Na+/Ca2+ exchangers (NCXs). Lastly, genetic mutations associated with Parkinson's disease, like those to DJ-1 or PINK1, might directly compromise the competence of mitochondria, leading to accelerated aging or increased oxidative stress.
One major gap is the mechanistic linkage between activity dependent Ca2+ entry into neurons and mitochondrial oxidative stress. In spite of its plausibility, there is no direct evidence that plasma membrane Ca2+ influx elevates mitochondrial oxidative phosphorylation and the production of superoxide. Limiting plasma membrane Ca2+ influx certainly diminishes the sensitivity of SNc DA neurons to mitochondrial toxins, but this effect could be indirect. The development of redox sensitive optical probes [101-103] and two photon laser scanning microscopy to allow imaging of mitochondria in situ puts this question within reach. A related, albeit more difficult question, is whether the role of mitochondria in intracellular Ca2+ buffering contributes to neuronal apoptosis in slowly progressing neurodegenerative diseases, like PD. Although mitochondria do not normally flux Ca2+ from the cytoplasm at physiological concentrations, Ca2+ released from the ER through inositol trisphosphate (IP3) or ryanodine receptors can enter mitochondria at points of apposition between the organelles, which form functional Ca2+ microdomains in which Ca2+ concentrations can rise into the micromolar range [104-106]. Through these junctions, ‘dumping’ of ER Ca2+ stores into mitochondria could trigger apoptosis in marginally competent mitochondria [107]. However, the vast majority of the studies demonstrating the existence of close interactions between mitochondria and the ER have been performed in cell lines, none have been performed in SNc dopaminergic neurons where the functional relationship between these organelles could be quite different. That said, mechanisms like this seem to be in play in Alzheimer's disease [108].
A closely related question is whether non-autonomous oxidative stress could synergize with that created by the Ca2+ phenotype. There is ample evidence that inflammation and the production of ROS and reactive nitrogen species (RNS) could accelerate the loss of SNc DA neurons and the progression of PD. Recent work has identified another possible player in translating extrinsic oxidative stress into mitochondria dysfunction. ROS mediated activation of protein kinase C beta (PKCb) phosphorylates 66-kilodalton isoform of the growth factor adapter Shc (p66shc), promoting transport into mitochondria where it alters Ca2+ responses and promotes apoptosis [109]. Because it controls lifespan [110], p66shc could be particularly important in regulating the aging of neurons, like SNc DA neurons, that are stressed by Ca2+ flux across the plasma membrane.
A second major gap in our understanding is how genetic mutations associated with PD interact with Ca2+ triggered events in SNc DA neurons. Although DA could be a factor [8], Ca2+ must be considered the prime suspect in making these mutations lethal. As outlined here, this is most likely mediated by metabolic, oxidative stress. It is easy to imagine that a negative dominant mutation of a gene like DJ-1 could exacerbate the basal oxidative stress in an SNc DA neuron and accelerate mitochondrial and proteostatic collapse, leaving neurons lacking the same basal stress largely unaffected. But, these types of interaction between mutations and cellular phenotype have not been pursued.
A third gap is our understanding is whether non-dopaminergic neurons that are vulnerable in PD share the Ca2+ phenotype. As mentioned above, there is some evidence that at least a subset of the neurons that die or functionally decline are pacemakers that engage L-type Ca2+ channels. However, the phenotypic characterization must be explored more systematically in those cell types at greatest risk (dorsal motor nucleus of the vagus, locus ceruleus, basal forebrain cholinergic neurons, dorsal raphe, etc.). Moreover, this should be done in situ (either with in vivo recording or in brain slices from adult mice) where the behavior of the neurons is as close as possible to that found in humans.
Conclusions
Ca2+ mediated cellular stress has long been thought to be important in neurodegeneration, but it usually is envisioned as a late stage consequence of organelle damage inflicted by some other challenge. The unusual reliance of SNc DA neurons on voltage-dependent L-type Ca2+ channels in autonomous pacemaking suggests that the mitochondrial stress created by sustained Ca2+ entry could be responsible for their selective vulnerability, rather than simply a late stage consequence. This hypothesis is consistent with the centrality of mitochondria in prevailing models of pathogenesis in PD. Genetic mutations and environmental challenges could easily synergize with this basal stress, hastening cellular aging and eventual death. Although plausible and consistent with regional deficits seen in normal aging and PD, the proposition that Ca2+ entry during pacemaking compromises mitochondrial function remains to be fully untested. The tools necessary to conduct this test are now becoming available. Nevertheless, given the plausibility of the link between Ca2+ and the loss of SNc DA neurons, the absence of any proven neuroprotective therapy in PD and the availability of Ca2+ channel antagonists that are well-tolerated and approved for human use, it would seem that the human experiment should proceed now in the form of clinical neuroprotection trials.
Acknowledgments
This work was supported by grants from the Hartman Foundation, USAMRMC and NIH (NS047085).
Footnotes
Disclosure: The authors have no competing financial interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology. 2004;63:1240–4. doi: 10.1212/01.wnl.0000140706.52798.be. [DOI] [PubMed] [Google Scholar]
- 2.de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, Manubens-Bertran JM, Alperovitch A, Rocca WA. Prevalence of parkinsonism and Parkinson's disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson's disease. J Neurol Neurosurg Psychiatry. 1997;62:10–5. doi: 10.1136/jnnp.62.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–6. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 5.Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–64. [PubMed] [Google Scholar]
- 6.Riederer P, Wuketich S. Time course of nigrostriatal degeneration in parkinson's disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- 7.Greenamyre JT, Hastings TG. Biomedicine. Parkinson's--divergent causes, convergent mechanisms. Science. 2004;304:1120–2. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- 8.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–50. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999;122(Pt 8):1437–48. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 10.Ito H, Goto S, Sakamoto S, Hirano A. Calbindin-D28k in the basal ganglia of patients with parkinsonism. Ann Neurol. 1992;32:543–50. doi: 10.1002/ana.410320410. [DOI] [PubMed] [Google Scholar]
- 11.Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–80. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- 12.Matzuk MM, Saper CB. Preservation of hypothalamic dopaminergic neurons in Parkinson's disease. Ann Neurol. 1985;18:552–5. doi: 10.1002/ana.410180507. [DOI] [PubMed] [Google Scholar]
- 13.Saper CB, Sorrentino DM, German DC, de Lacalle S. Medullary catecholaminergic neurons in the normal human brain and in Parkinson's disease. Ann Neurol. 1991;29:577–84. doi: 10.1002/ana.410290602. [DOI] [PubMed] [Google Scholar]
- 14.Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol. 2005;252 4:IV37–IV42. doi: 10.1007/s00415-005-4008-5. [DOI] [PubMed] [Google Scholar]
- 15.Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–29. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 17.Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson's disease: new clues. J Neurochem. 2008;107:317–28. doi: 10.1111/j.1471-4159.2008.05604.x. [DOI] [PubMed] [Google Scholar]
- 18.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–9. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 19.Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J Bioenerg Biomembr. 2004;36:375–9. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- 20.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 21.Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol. 1994;36:876–81. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- 22.Gu M, Gash MT, Cooper JM, Wenning GK, Daniel SE, Quinn NP, Marsden CD, Schapira AH. Mitochondrial respiratory chain function in multiple system atrophy. Mov Disord. 1997;12:418–22. doi: 10.1002/mds.870120323. [DOI] [PubMed] [Google Scholar]
- 23.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–64. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–9. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholls DG, Ferguson SJ. Bioenergetics3. 2002. [Google Scholar]
- 26.Soong NW, Hinton DR, Cortopassi G, Arnheim N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet. 1992;2:318–23. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- 27.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–20. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 28.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 29.Tretter L, Sipos I, Adam-Vizi V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson's disease. Neurochem Res. 2004;29:569–77. doi: 10.1023/b:nere.0000014827.94562.4b. [DOI] [PubMed] [Google Scholar]
- 30.Jenner P. Parkinson's disease, pesticides and mitochondrial dysfunction. Trends Neurosci. 2001;24:245–7. doi: 10.1016/s0166-2236(00)01789-6. [DOI] [PubMed] [Google Scholar]
- 31.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 32.Del Tredici K, Braak H. Idiopathic Parkinson's disease: staging an alpha-synucleinopathy with a predictable pathoanatomy. In: Kahle PJ, Haas C, editors. Molecular Mechanisms of Parkinson's Disease. Landes Bioscience; Georgetown, TX: 2004. pp. 1–32. [Google Scholar]
- 33.Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–8. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaushik S, Cuervo AM. Chaperone-mediated autophagy. Methods Mol Biol. 2008;445:227–44. doi: 10.1007/978-1-59745-157-4_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson's disease. Antioxid Redox Signal. 2007;9:553–61. doi: 10.1089/ars.2006.1524. [DOI] [PubMed] [Google Scholar]
- 36.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 37.Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--2. Action potential generating mechanisms and morphological correlates. Neuroscience. 1983;10:317–31. doi: 10.1016/0306-4522(83)90136-7. [DOI] [PubMed] [Google Scholar]
- 38.Romo R, Schultz W. Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements. J Neurophysiol. 1990;63:592–606. doi: 10.1152/jn.1990.63.3.592. [DOI] [PubMed] [Google Scholar]
- 39.Bonci A, Grillner P, Mercuri NB, Bernardi G. L-Type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. J Neurosci. 1998;18:6693–703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ping HX, Shepard PD. Apamin-sensitive Ca(2+)-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. Neuroreport. 1996;7:809–14. doi: 10.1097/00001756-199602290-00031. [DOI] [PubMed] [Google Scholar]
- 41.Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–56. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol. 2000;83:3084–100. doi: 10.1152/jn.2000.83.5.3084. [DOI] [PubMed] [Google Scholar]
- 43.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–6. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 44.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34:903–9. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 45.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol. 2008 doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 46.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–9. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol. 1993;466:727–47. [PMC free article] [PubMed] [Google Scholar]
- 48.Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol. 1994;113:831–8. doi: 10.1111/j.1476-5381.1994.tb17068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizzuto R. Intracellular Ca(2+) pools in neuronal signalling. Curr Opin Neurobiol. 2001;11:306–11. doi: 10.1016/s0959-4388(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 50.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 51.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–53. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arbuthnott GW, Wickens J. Space, time and dopamine. Trends Neurosci. 2007;30:62–9. doi: 10.1016/j.tins.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 54.Liang CL, Wang TT, Luby-Phelps K, German DC. Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson's disease. Exp Neurol. 2007;203:370–80. doi: 10.1016/j.expneurol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 56.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calne DB, Langston JW. Aetiology of Parkinson's disease. Lancet. 1983;2:1457–9. doi: 10.1016/s0140-6736(83)90802-4. [DOI] [PubMed] [Google Scholar]
- 58.Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991;54:388–96. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stark AK, Pakkenberg B. Histological changes of the dopaminergic nigrostriatal system in aging. Cell Tissue Res. 2004;318:81–92. doi: 10.1007/s00441-004-0972-9. [DOI] [PubMed] [Google Scholar]
- 60.McCormack AL, Di Monte DA, Delfani K, Irwin I, DeLanney LE, Langston WJ, Janson AM. Aging of the nigrostriatal system in the squirrel monkey. J Comp Neurol. 2004;471:387–95. doi: 10.1002/cne.20036. [DOI] [PubMed] [Google Scholar]
- 61.Ishikawa T, Dhawan V, Kazumata K, Chaly T, Mandel F, Neumeyer J, Margouleff C, Babchyck B, Zanzi I, Eidelberg D. Comparative nigrostriatal dopaminergic imaging with iodine-123-beta CIT-FP/SPECT and fluorine-18-FDOPA/PET. J Nucl Med. 1996;37:1760–5. [PubMed] [Google Scholar]
- 62.Backman L, Ginovart N, Dixon RA, Wahlin TB, Wahlin A, Halldin C, Farde L. Age-related cognitive deficits mediated by changes in the striatal dopamine system. Am J Psychiatry. 2000;157:635–7. doi: 10.1176/ajp.157.4.635. [DOI] [PubMed] [Google Scholar]
- 63.Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, Bakay RA, Sladek JR, Jr, Kordower JH. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis. 2007;26:56–65. doi: 10.1016/j.nbd.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanaan NM, Kordower JH, Collier TJ. Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: relevance to selective neuronal vulnerability. J Comp Neurol. 2007;502:683–700. doi: 10.1002/cne.21333. [DOI] [PubMed] [Google Scholar]
- 65.Kanaan NM, Kordower JH, Collier TJ. Age-related changes in dopamine transporters and accumulation of 3-nitrotyrosine in rhesus monkey midbrain dopamine neurons: relevance in selective neuronal vulnerability to degeneration. Eur J Neurosci. 2008;27:3205–15. doi: 10.1111/j.1460-9568.2008.06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–12. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic Biol Med. 2009;47:1354–61. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 68.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–96. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 69.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 71.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 72.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 73.Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Kruger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–8. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007;104:11441–6. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.German DC, Manaye KF, White CL, 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992;32:667–76. doi: 10.1002/ana.410320510. [DOI] [PubMed] [Google Scholar]
- 76.Williams JT, North RA, Shefner SA, Nishi S, Egan TM. Membrane properties of rat locus coeruleus neurones. Neuroscience. 1984;13:137–56. doi: 10.1016/0306-4522(84)90265-3. [DOI] [PubMed] [Google Scholar]
- 77.Stevens DR, Haas HL. Calcium-dependent prepotentials contribute to spontaneous activity in rat tuberomammillary neurons. J Physiol. 1996;493(Pt 3):747–54. doi: 10.1113/jphysiol.1996.sp021419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- 79.German DC, Manaye KF, Sonsalla PK, Brooks BA. Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci. 1992;648:42–62. doi: 10.1111/j.1749-6632.1992.tb24523.x. [DOI] [PubMed] [Google Scholar]
- 80.Belzunegui S, San Sebastian W, Garrido-Gil P, Izal-Azcarate A, Vazquez-Claverie M, Lopez B, Marcilla I, Lanciego JL, Luquin MR. The number of dopaminergic cells is increased in the olfactory bulb of monkeys chronically exposed to MPTP. Synapse. 2007;61:1006–12. doi: 10.1002/syn.20451. [DOI] [PubMed] [Google Scholar]
- 81.Pignatelli A, Kobayashi K, Okano H, Belluzzi O. Functional properties of dopaminergic neurones in the mouse olfactory bulb. J Physiol. 2005;564:501–14. doi: 10.1113/jphysiol.2005.084632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Postuma RB, Lang AE, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology. 2006;66:845–51. doi: 10.1212/01.wnl.0000203648.80727.5b. [DOI] [PubMed] [Google Scholar]
- 83.Huisman E, Uylings HB, Hoogland PV. Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson's disease patients. Mov Disord. 2008;23:1407–13. doi: 10.1002/mds.22009. [DOI] [PubMed] [Google Scholar]
- 84.Pignatelli A, Ackman JB, Vigetti D, Beltrami AP, Zucchini S, Belluzzi O. A potential reservoir of immature dopaminergic replacement neurons in the adult mammalian olfactory bulb. Pflugers Arch. 2008 doi: 10.1007/s00424-008-0535-0. [DOI] [PubMed] [Google Scholar]
- 85.Yamada T, McGeer PL, Baimbridge KG, McGeer EG. Relative sparing in Parkinson's disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990;526:303–7. doi: 10.1016/0006-8993(90)91236-a. [DOI] [PubMed] [Google Scholar]
- 86.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–76. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Becker C, Jick SS, Meier CR. Use of Statins and the Risk of Parkinson's Disease: A Retrospective Case-Control Study in the UK. Drug Saf. 2008;31:399–407. doi: 10.2165/00002018-200831050-00004. [DOI] [PubMed] [Google Scholar]
- 88.Ritz B, Qian L, Rhodes SL, Schernhammer E, Olsen J, Friis S. L-Type Calcium Channel blockers and Parkinson's Disease in Denmark. Annals Neurology. doi: 10.1002/ana.21937. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kupsch A, Sautter J, Schwarz J, Riederer P, Gerlach M, Oertel WH. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996;741:185–96. doi: 10.1016/s0006-8993(96)00917-1. [DOI] [PubMed] [Google Scholar]
- 90.Mannhold R, Rekker RF, Sonntag C, ter Laak AM, Dross K, Polymeropoulos EE. Comparative evaluation of the predictive power of calculation procedures for molecular lipophilicity. J Pharm Sci. 1995;84:1410–9. doi: 10.1002/jps.2600841206. [DOI] [PubMed] [Google Scholar]
- 91.Eisenberg MJ, Brox A, Bestawros AN. Calcium channel blockers: an update. Am J Med. 2004;116:35–43. doi: 10.1016/j.amjmed.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 92.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–6. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 93.Scholze A, Plant TD, Dolphin AC, Nurnberg B. Functional expression and characterization of a voltage-gated CaV1.3 (alpha1D) calcium channel subunit from an insulin-secreting cell line. Mol Endocrinol. 2001;15:1211–21. doi: 10.1210/mend.15.7.0666. [DOI] [PubMed] [Google Scholar]
- 94.Fitton A, Benfield P. Isradipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cardiovascular disease. Drugs. 1990;40:31–74. doi: 10.2165/00003495-199040010-00004. [DOI] [PubMed] [Google Scholar]
- 95.Hung AY, Schwarzschild MA. Clinical trials for neuroprotection in Parkinson's disease: overcoming angst and futility? Curr Opin Neurol. 2007;20:477–83. doi: 10.1097/WCO.0b013e32826388d6. [DOI] [PubMed] [Google Scholar]
- 96.Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–4. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- 97.Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998;783:109–14. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- 98.Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–9. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- 99.Magyar K, Szende B. (-)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology. 2004;25:233–42. doi: 10.1016/S0161-813X(03)00102-5. [DOI] [PubMed] [Google Scholar]
- 100.Schapira AH. Progress in neuroprotection in Parkinson's disease. Eur J Neurol. 2008;15 1:5–13. doi: 10.1111/j.1468-1331.2008.02055.x. [DOI] [PubMed] [Google Scholar]
- 101.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Desireddi JR, Farrow KN, Marks JD, Waypa GB, Schumacker PT. Hypoxia Increases ROS Signaling and Cytosolic Ca2+ in Pulmonary Artery Smooth Muscle Cells of Mouse Lungs Slices. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–53. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 104.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 105.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–21. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 107.Hajnoczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–54. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 108.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315:659–63. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- 110.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–13. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]