

Abstract
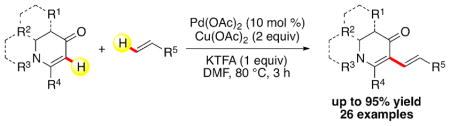
A new Pd(II)-catalyzed dehydrogenative alkenylation reaction involving two alkenes was developed. A variety of nonaromatic, cyclic enaminones were successfully coupled to primary and secondary alkenes yielding a series of unique 1,3-dienes. The generality of this transformation presents a useful strategy for directly cross-coupling alkenes and offers an attractive new approach to functionalize enaminones.
Direct C–H functionalization chemistry has seen significant progress during the last decade. 1 Cross dehydrogenative coupling reactions that use two C–H bonds to form a new C–C bond are highly sought after, because these processes do not require pre-funtionalization and as a result have high atom economy.2 The Fujiwara-Moritani reaction is a process by which aromatic substrates undergo an intermolecular dehydrogenative alkenylation. This reaction was initially developed using stoichiometric amounts of Pd(II) (Figure 1a), 3 but soon thereafter was shown to also take place with catalytic amounts of Pd(II) (Figure 1b).4 In recent years, the scope of the Fujiwara-Moritani has been expanded to a plethora of aromatic substrates.5 However, only few cases of alkenylation reactions involving two alkene C–H donors (Figure 1c) are known. They are limited to select substrates, require high Pd loading, and need long reaction time. 6 Herein, we report the development of an efficient Pd(II)-catalyzed Fujiwara-Moritani reaction, featuring nonaromatic cyclic enaminones that react with a variety of alkenes to furnish 5-alkenylated reaction products.
Figure 1.

Development of the Fujiwara-Moritani reaction.
In our quest to generate libraries for biological screening, we were particularly interested in functionalizing the cyclic enaminone nucleus I (Figure 2) because of its unique chemical and biological properties.7 Recently, we reported a Pd(II)-catalyzed direct arylation of cyclic enaminones with aryl trifluoroborates (Figure 2a). 8 Alkenyl trifluoroborates, however, did not furnish the desired alkenylated products. We speculated that the rate of transmetallation with the alkenyl reagents surpassed that of C5-palladation and as a result homocoupling depleted the alkenyl precursors and reoxidants.
Figure 2.

Pd(II)-cross coupling reactions of cyclic enaminones.
To address this problem, we examined the feasibility of a Fujiwara-Moritani reaction (Figure 2b) to access 5-alkenyl enaminone derivatives. Key to this strategy would be the use of alkenes with complementary reactivity. We envisioned that the palladated enaminone II could be orthogonally intercepted by an electron-deficient alkene to furnish product VI.
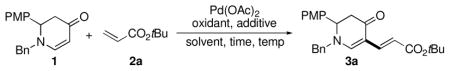
We selected enaminone 1 as the substrate and examined its reaction with tert-butyl acrylate (2a) (Table 1). Pd(OAc)2 (10 mol %) was initially selected because of its well-established reactivity.8, 9 Next, DMF was identified as the best solvent (entries 1–4). Cu(OAc)2 proved to be the most effective and economical reoxidant (entries 4–7). The incorporation of additives, KTFA specifically, notably increased the yield of the reactions (entries 8–12). Furthermore, the highest yields were observed when the reaction was run at 80 °C (entries 12– 14), and the reaction time was reduced from 24 h to 3 h (entries 12 and 15).
Table 1.
Optimization of the reaction conditionsa
 | |||||
|---|---|---|---|---|---|
| entry | solvent | reoxidant | additive | temp (°C) | 3a (%)b |
| 1 | tBuOH | Cu(OAc)2 | -- | 80 | 55 |
| 2 | DMSO | Cu(OAc)2 | -- | 80 | 53 |
| 3 | DMA | Cu(OAc)2 | -- | 80 | 70 |
| 4 | DMF | Cu(OAc)2 | -- | 80 | 78 |
| 5 | DMF | CuCl2 | -- | 80 | 0 |
| 6 | DMF | AgOAc | -- | 80 | 43 |
| 7 | DMF | PhCO3tBuc | -- | 80 | 59 |
| 8 | DMF | Cu(OAc)2 | LiBF4 | 80 | 80 |
| 9 | DMF | Cu(OAc)2 | BiCl3 | 80 | 39 |
| 10 | DMF | Cu(OAc)2 | CsOAc | 80 | 77 |
| 11 | DMF | Cu(OAc)2 | K2CO3 | 80 | 68 |
| 12 | DMF | Cu(OAc)2 | KTFA | 80 | 85 |
| 13 | DMF | Cu(OAc)2 | KTFA | 50 | 73 |
| 14 | DMF | Cu(OAc)2 | KTFA | 110 | 69 |
| 15 | DMF | Cu(OAc)2 | KTFA | 80 | 87d |
Reaction conditions unless otherwise specified: 1 (0.2 M), 2a (2 equiv), Pd(OAc)2 (10 mol %), reoxidant (2 equiv), additive (1 equiv) under N2 at 80 °C in 24 h. (PMP=para-methoxyphenyl)
1H NMR yield vs. Ph3SiMe (1 equiv) as the internal standard.
1 equiv.
Completed in 3 h. (Detailed optimization is available in the Supporting Information.)
With these optimized conditions, we were pleased to find that this direct alkenylation reaction was amenable to a variety of alkenes (Scheme 1). Acrylate esters and vinyl ketones readily reacted with 1, providing the desired dienes (3a–3e) in excellent yields. Notably, the highest yield (95%) was observed with N,N-dimethylacrylamide (3f). Phosphonates, styrene and sulfones were also viable coupling partners, yet furnished the products (3g–3i) in slightly lower yields. Acrylic acid and vinyl ethers, however, failed to afford the desired products 3j or 3k. Methyl crotonate, as a multi-substituted alkene, could also be coupled to 1 to produce a single isomer (3l), whereas α-methylene-γ-lactones afforded both the conjugated and unconjugated diene products with a greater preference for the latter (3ma:3mb=1:2.5). This preference for unconjugated dienes has been previously noted.5k, 10 Interestingly, alkenylation of cyclohexene yielded two inseparable, unconjugated dienes (3na/3nb). Mechanistically, we speculated that 3nb was generated from 3na through Pd–H insertion and immediate β-H elimination.11
Scheme 1.

Scope of alkenesa
a Conditions: 1 (0.2 M), 2 (2 equiv), Pd(OAc)2 (10 mol %), Cu(OAc)2 (2 equiv), KTFA (1 equiv) in DMF under N2 at 80 °C for 3 h. Isolated yield. b Isolated ratio. c 1H NMR ratio.
We next assessed a series of enaminones (Scheme 2). We found that this reaction could be extended to mono-and bicyclic, electronically unattenuated enaminones (5a–5g). Importantly, alkenylation of the diastereomeric substrates (5a/5b) took place without epimerization of the stereocenters and with virtually the same yields. The introduction of a C6-substituent to the cyclic enaminone, however, significantly decreased the yield (of 5h). N-H and N-Cbz enaminones also showed poor reactivity (for products 5i and 5j) which is consistent with our previous findings.8 4-Pyridone was found to furnish a mono-coupling product 5k albeit in only 7% yield. An E-enaminone was also tested, but only a trace amount of product 5l was observed.
Scheme 2.

Scope of enaminonesa
a Conditions: 4 (0.2 M), 2a (2 equiv), Pd(OAc)2 (10 mol %), Cu(OAc)2 (2 equiv), KTFA (1 equiv) in DMF under N2 at 80 °C for 3 h. Isolated yield.
To elucidate the alkenylation process, a mechanistic analysis of the initial interaction of Pd(II) with enaminone 1 was carried out by 1H NMR (in DMSO-d6 at room temperature, Figure 3). 50 mol % of Pd(OAc)2 with 1 (Figure 3b) furnished intermediate 6 at room temperature along with the same amount of acetic acid and unreacted 1. With 100 mol % of Pd(OAc)2 (Figure 3c), a complete conversion of 1 was observed after only 20 min, yielding only 6 and acetic acid (See the Supporting Information for the full spectra). 20% of product 3a was subsequently furnished from intermediate 6 when heated with acrylate 2a at an elevated temperature (140 °C) (Scheme 3).12 This suggests that the C–C bond formation is the rate-limiting step. It is worth noting that intermediate 6, however, was not detected when DMF-d7 was used as the solvent, presumably because DMF does not stabilize 6 as well as DMSO. 13 The discrepancy of their stabilizing effect as solvents was also reflected in the yield of 3a, where 78% was produced in DMF compared to 53% in DMSO (Table 1, entries 2 and 4).
Figure 3.

Detection of palladated intermediate 6 by 1H NMR: (a) Pure 1; (b) 1 with 50 mol % of Pd(OAc)2; (c) 1 with 100 mol % of Pd(OAc)2.
Scheme 3.

Reaction of 1 with acrylate 2a in DMSO-d6
Hence, we suggest the following mechanism (Figure 4).5a An electrophilic attack of Pd(II) on enaminone A followed by deprotonation forms palladated B, which then undergoes alkene insertion to afford C. Subsequent β-H elimination delivers product D. Reductive elimination and reoxidation by Cu(II) regenerates Pd(II).
Figure 4.

Suggested mechanism of dehydrogenative alkenylation.
In summary, we have developed a direct, convenient and highly atom-economic approach for the dehydrogenative coupling of nonaromatic, cyclic enaminones and simple alkenes. The generality of this transformation presents a useful strategy for directly cross-coupling alkenes and offers an attractive new approach to prepare functionalize enaminones.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM081267) for their support of our program. We thank Dr. Subhashree Francis from the Institute for Therapeutics Discovery and Development at the University of Minnesota for her mass spectrometry assistance.
Footnotes
Supporting Information Available. Experimental procedures, detailed reaction optimization data, and results from the mechanistic study, and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For reviews: Hirano K, Miura M. Synlett. 2011:294.Messaoudi S, Brion JD, Alami M. Eur J Org Chem. 2010:6495.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.McGlacken GP, Fairlamb IJS. Eur J Org Chem. 2009:4011.Kulkarni AA, Daugulis O. Synthesis. 2009:4087.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058.Ackermann L, Vicente R, Kapdi AR. Angew Chem Int Ed. 2009;48:9792. doi: 10.1002/anie.200902996.Li BJ, Yang SD, Shi ZJ. Synlett. 2008:949.Lewis JC, Bergman RG, Ellman JA. Acc Chem Res. 2008;41:1013. doi: 10.1021/ar800042p.Kakiuchi F, Kochi T. Synthesis. 2008:3013.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n.Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318. doi: 10.1021/cr068006f.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760.Dick AR, Sanford MS. Tetrahedron. 2006;62:2439.Wolfe JP, Thomas JS. Curr Org Chem. 2005;9:625.
- 2.For recent reviews: Yoo WJ, Li CJC-H. Activation. Vol. 292. Springer-Verlag; Berlin, Berlin: 2010. p. 281.Scheuermann CJ. Chem-Asian J. 2010;5:436. doi: 10.1002/asia.200900487.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.
- 3.Moritani I, Fujiwara Y. Tetrahedron Lett. 1967:1119. [Google Scholar]
- 4.(a) Fujiwara Y, Moritani I, Matsuda M, Teranishi S. Tetrahedron Lett. 1968:3863. [Google Scholar]; (b) Fujiwara Y, Noritani I, Danno S, Asano R, Teranishi SJ. Am Chem Soc. 1969;91:7166. doi: 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]
- 5.For recent examples: Le Bras J, Muzart J. Chem Rev. 2011;111:1170. doi: 10.1021/cr100209d.Kim D, Hong S. Org Lett. 2011;13:4466. doi: 10.1021/ol2018278.Chappell B, Dedman N, Wheeler S. Tetrahedron Lett. 2011;52:3223.Ye MC, Gao GL, Yu JQ. J Am Chem Soc. 2011;133:6964. doi: 10.1021/ja2021075.Garcia-Rubia A, Fernandez-Ibanez MA, Arrayas RG, Carretero JC. Chem Eur J. 2011;17:3567. doi: 10.1002/chem.201003633.Donets PA, Van der Eycken EV. Synthesis. 2011:2147.Zhang XG, Fan SL, He CY, Wan XL, Min QQ, Yang J, Jiang ZXJ. Am Chem Soc. 2010;132:4506. doi: 10.1021/ja908434e.Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512.Nishikata T, Lipshutz BH. Org Lett. 2010;12:1972. doi: 10.1021/ol100331h.Li M, Li L, Ge H. Adv Synth Catal. 2010;352:2445.Miyasaka M, Hirano K, Satoh T, Miura M. J Org Chem. 2010;75:5421. doi: 10.1021/jo101214y.Lu Y, Wang DH, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:5916. doi: 10.1021/ja101909t.Jiang HF, Feng ZN, Wang AZ, Liu XH, Chen ZW. Eur J Org Chem. 2010:1227.Zhang YH, Shi BF, Yu JQ. J Am Chem Soc. 2009;131:5072. doi: 10.1021/ja900327e.Wu JL, Cui XL, Chen LM, Jiang GJ, Wu YJ. J Am Chem Soc. 2009;131:13888. doi: 10.1021/ja902762a.Cheng D, Gallagher T. Org Lett. 2009;11:2639. doi: 10.1021/ol900627q.
- 6.(a) Hatamoto Y, Sakaguchi S, Ishii Y. Org Lett. 2004;6:4623. doi: 10.1021/ol047971u. [DOI] [PubMed] [Google Scholar]; (b) Xu YH, Lu J, Loh TP. J Am Chem Soc. 2009;131:1372. doi: 10.1021/ja8084548. [DOI] [PubMed] [Google Scholar]; (c) Xu YH, Wang WJ, Wen ZK, Hartley JJ, Loh TP. Tetrahedron Lett. 2010;51:3504. [Google Scholar]; (d) Yu HF, Jin WW, Sun CL, Chen JP, Du WM, He SB, Yu ZK. Angew Chem Int Ed. 2010;49:5792. doi: 10.1002/anie.201002737. [DOI] [PubMed] [Google Scholar]
- 7.(a) Niphakis MJ, Turunen BJ, Georg GI. J Org Chem. 2010;75:6793. doi: 10.1021/jo100907u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Edafiogho IO, Kombian SB, Ananthalakshmi KVV, Salama NN, Eddington ND, Wilson TL, Alexander MS, Jackson PL, Hanson CD, Scott KR. J Pharm Sci. 2007;96:2509. doi: 10.1002/jps.20967. [DOI] [PubMed] [Google Scholar]; (c) Negri G, Kascheres C, Kascheres AJ. J Heterocycl Chem. 2004;41:461. [Google Scholar]; (d) Elassar AZA, El-Khair AA. Tetrahedron. 2003;59:8463. [Google Scholar]
- 8.Ge H, Niphakis MJ, Georg GI. J Am Chem Soc. 2008;130:3708. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- 9.Yao Q, Kinney EP, Yang Z. J Org Chem. 2003;68:7528. doi: 10.1021/jo034646w. [DOI] [PubMed] [Google Scholar]
- 10.(a) Cai GX, Fu Y, Li YZ, Wan XB, Shi ZJ. J Am Chem Soc. 2007;129:7666. doi: 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]; (b) Garcia-Rubia A, Arrayas RG, Carretero JC. Angew Chem Int Ed. 2009;48:6511. doi: 10.1002/anie.200902802. [DOI] [PubMed] [Google Scholar]
- 11.Ozawa F, Kubo A, Matsumoto Y, Hayashi T, Nishioka E, Yanagi K, Moriguchi K. Organometallics. 1993;12:4188. [Google Scholar]
- 12.Conversion of 6 required a high temperature, which, however, led to a low yield of 3a due to its thermal instability. See the Supporting Information for more detail.
- 13.Zierkiewicz W, Privalov T. Organometallics. 2005;24:6019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.