

Abstract
Striatal spiny neurons (SPNs) associate a diverse array of cortically processed information to regulate action selection. But how this is done by SPNs is poorly understood. A key step in this process is the transition of SPNs from a hyperpolarized ‘down-state’ to a sustained, depolarized ‘up-state’. These transitions are thought to reflect a sustained synaptic barrage, involving the coordination of hundreds of pyramidal neurons. Indeed, in mice simulation of cortical input by glutamate uncaging on proximal dendritic spines produced potential changes in SPNs that tracked input time course. However, brief glutamate uncaging at spines on distal dendrites evoked somatic up-states lasting hundreds of milliseconds. These regenerative events depended upon both NMDA receptors and voltage-dependent Ca2+ channels. Moreover, they were bidirectionally regulated by dopamine receptor signaling. This capacity not only changes our model of how up-state are generated in SPNs, it has fundamental implications for the associative process underlying action selection.
Introduction
Striatal spiny neurons (SPNs) are key elements in the basal ganglia circuitry controlling contextually appropriate action selection 1,2. SPNs integrate cortically processed information about the environment, internal body state and past experience to facilitate actions that maximize positive and minimize negative outcomes. Dopamine plays a critical role in shaping the activity of SPNs, providing feedback about the outcome of choosing a particular action 3,4.
The integrative mechanisms underlying the capacity of SPNs to differentiate one context from another in the process of promoting an appropriate action are poorly defined. A potentially important clue as to how this is done comes from in vivo recordings from SPNs showing that their somatic membrane rapidly moves between a stable hyperpolarized state near the K+ equilibrium potential (‘down-state’) to a depolarized state near spike threshold (‘up-state’). The 15–20 mV transition from the down- to up-state depends upon a temporally convergent barrage of excitatory, glutamatergic synaptic input from the cerebral cortex 5. Because the connectivity between any one cortical pyramidal neuron and an individual SPN is sparse, consisting of a few en passant synapses, a successful transition has been hypothesized to require the synchronization of a great many cortical pyramidal neurons 5-7, creating a coincidence threshold that could be used to discriminate context.
Although induced by cortical synaptic input, up-states have a number of features – a stereotyped voltage trajectory and durations of hundreds of milliseconds or more – that suggest they are the product of an intrinsic bistability 5-7. However, electrophysiological studies using somatic electrodes have failed to find evidence of bistability in SPNs 5. This conclusion is consistent with recent work showing that backpropagation of somatic action potentials into dendrites is decrementing and lacks any obvious intrinsic boosting by ion channels carrying inward, depolarizing current 8,9. If up-state initiation and maintenance is dependent upon sustained synaptic depolarization, then typical up-states lasting hundreds of milliseconds must require the coordination of hundreds, if not thousands, of cortical pyramidal neurons. This hypothetical feature of synaptic integration in SPNs clearly constrains the corticostriatal network engaged in action selection and limits its pattern recognition capacity 10.
One of the limitations of the pertinent studies to date is that they have not been able to directly interrogate dendrites, particularly distal dendrites. SPN dendrites are too small to record from with electrodes. To overcome this obstacle, a combination of optical approaches – two photon laser scanning microscopy (2PLSM) and two photon laser uncaging (2PLU) of glutamate – were used in combination with somatic patch clamp methods to study SPNs in brain slices from transgenic mice in which D1 and D2 dopamine receptor expression was reported by green fluorescent protein (GFP) – allowing the identification of direct pathway (d)SPNs and indirect pathway (i)SPNs 4. In agreement with previous studies 5,8,9,11, the proximal dendritic membrane exhibited little evidence of intrinsic bistability that might contribute to state transitions. However, in distal dendrites of both populations of SPNs, uncaging glutamate at about ten adjacent spines on a single dendrite was capable of evoking a local regenerative event that produced a somatic up-state lasting hundreds of milliseconds. These up-states were dependent upon activation of NMDA receptors and voltage-dependent Ca2+ channels. Furthermore, both adenosine and dopamine receptor signaling modulated up-state duration.
Results
Somatic up-states were triggered by distal synaptic input
Previous studies have shown that SPN up-states are not a product of an intrinsic bistability in the soma and proximal dendrites 5,12. However, because of their small size, distal dendrites, which can be electrotonically remote from the soma, have not been directly examined using patch clamp techniques. To interrogate these regions, SPNs were visualized by using a somatic patch electrode containing the dye Alexa 568. This allowed both proximal (40–60 μm from the soma) and distal (>100 μm from the soma) dendrites and spines to be seen using 2PLSM. 2PLU of bath applied MNI-caged-glutamate was used to activate glutamate receptors at the heads of spines, which are the sites of corticostriatal and thalamostriatal synapses 13,14. The uncaging laser power was adjusted to produce simulated excitatory postsynaptic potentials (uEPSPs) at the soma resembling those evoked by stimulation of glutamatergic fibers (Supplementary Fig. 1). Neighboring spines were activated in rapid succession (2 ms inter-event time) to mimic convergent cortical input (Fig. 1a). When performed at distal dendrites (115 ± 6 μm from soma, n=8, Fig. 1b–d), this stimulation paradigm evoked a rapid, stereotyped somatic depolarization after which the membrane potential plateaued, extending well beyond the end of the stimulation, often lasting for hundreds of milliseconds. The somatic membrane potential during these distally evoked plateaus closely resembled that found in vivo during up-states (Fig. 1b). The threshold for evoking a sustained somatic depolarization was about 11 spines (11.9 ± 0.4, n=39) within a 20 μm stretch of distal dendrite (21 ± 1 μm, n=39, Fig. 2d) greater than 100 μm from the soma (113 ± 2 μm, n=39). The duration of the resulting membrane potential plateau was not dependent upon the direction (toward or away from the soma) of spine stimulation (Supplementary Fig. 2).
Figure 1.
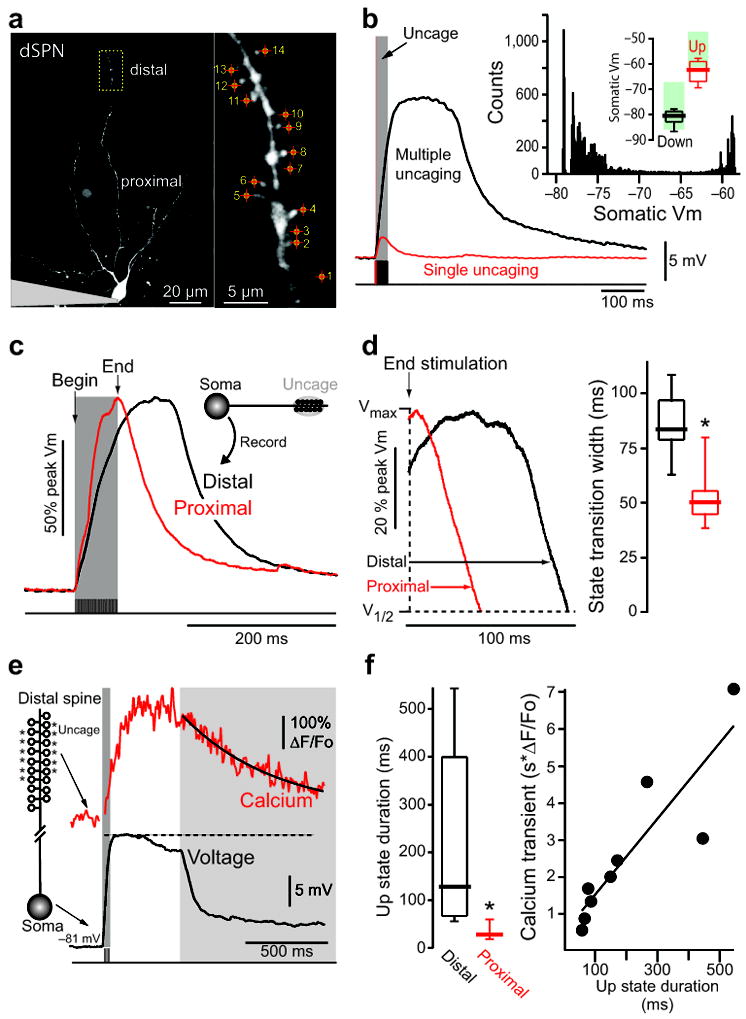
State transitions can be triggered by local stimulation of distal dendrites. (A) Low (left) and high (right) magnification maximum intensity projections (MIPs) of a dSPN filled with Alexa Fluor 568. Spines were stimulated with 1 ms uncaging pulses at 500 Hz, and are numbered in the order stimulated. (B) Somatic voltage recording of the cell shown in (A) in response to glutamate uncaging of a single spine (red), or all labeled spines in rapid succession (black). Red and black shaded regions indicate timing of uncaging pulses. An all points histogram is shown in the inset to the upper right. State membrane potentials are shown above as box plots (n=56 cells). For all box plots, lines indicate median, boxes outline upper and lower quartiles and whiskers are the upper and lower 10% values. Shaded green boxes indicate state membrane potentials reported in a previous in vivo study 5 and are shown for reference. (C–D) State transitions were evoked by uncaging at distal (black) dendritic spines; the same protocol was then used to excite a proximal dendrite (red). Somatic voltage traces aligned to the last uncaging stimulus and normalized show up-states are readily induced by distal but not proximal activation. Box plots indicate time between end of uncaging and 25% voltage fall (n=8, p=0.002, Mann-Whitney rank sum test). (E) Example spine Ca2+ transient (top) and corresponding somatic membrane potential (bottom) of an example up state in a dSPN. Ca2+ transient is measured using the low affinity Ca2+ indicator Fluo 4FF. (F) Experiment presented in 1c repeated using the Ca2+ indicator Fluo 4FF (n=9 cells; *p<0.01, Mann-Whitney rank sum test). Up-state duration is plotted against the corresponding local distal dendritic Ca2+ transient (measured at the dendritic shaft around which spines were stimulated).
Figure 2.
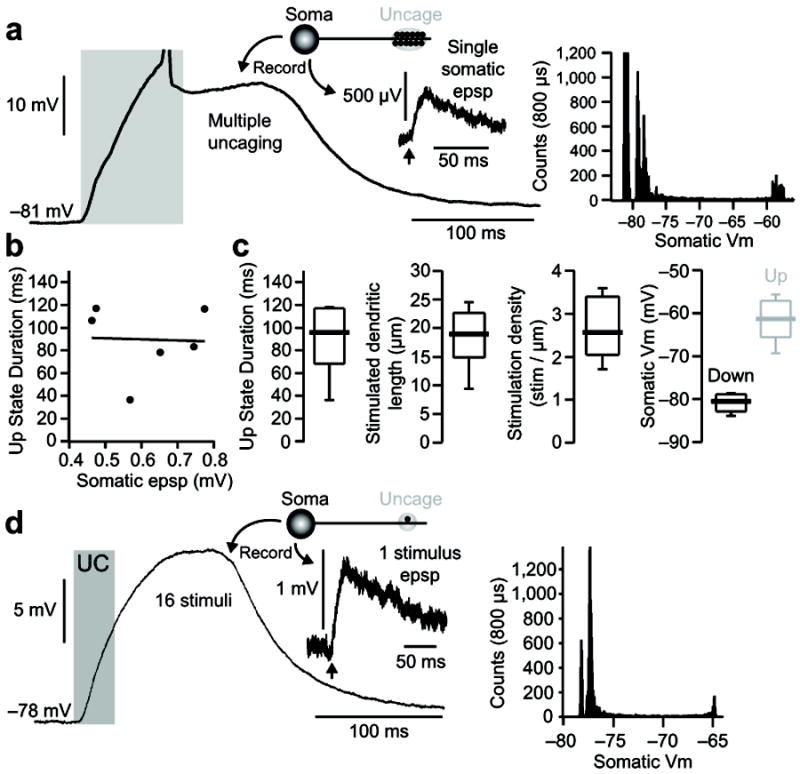
State transitions generated by proximal and distal dendritic glutamate uncaging are independent of the somatic EPSP size and recording internal used. (A) Example of a state transition induced in a dSPN: recording was made using a K-gluconate based internal: (in mM) 115 K-gluconate, 20 KCl, 10 Na-phosphocreatine, 10 HEPES, 2 Mg-ATP, and 0.3 Na-GTP)47 and Fluo 4FF; laser power was tuned to evoke small (463 μV) somatic EPSPs from a 0.5ms stimulation of a single distal dendritic spine (inset). The corresponding all points histogram for this cell is shown to the right. (B) Once a state transition is achieved, the up-state duration is not correlated with either the somatic EPSP induced by stimulation of a single distal dendritic spine or the recording internal solution used (n=6 cells; internal = KMeSo4 or K-gluconate with Fluo 4 or Fluo 4FF). (C) Though requiring a higher stimulation density, up-states induced using small amplitude (463–774 μV; n=6 cells) somatic EPSP are similar to those induced by supra-mV EPSP (i.e. figure 1). (D) In one case a modest state transition was produced by the repetitive stimulation of a single distal spine (16 stimuli, 0.5 ms duration, 500 Hz). Average somatic EPSP evoked by a single stimulus and the corresponding all points histogram are shown to the right.
Once the laser power and spine number required to induce a state transition were determined by stimulating distal dendrites, the identical laser power was then used to stimulate the same number of spines in a proximal dendrite of the same cell (49 ± 1 μm from soma, n=8). Somatic uEPSPs generated by proximal uncaging of glutamate were similar in amplitude, kinetics and evoked spine Ca2+ transients (as measured using the high affinity Ca2+ indicator Fluo 4) to those evoked distally (Supplementary Fig. 3). Although somewhat surprising, the similarity in uEPSPs was attributable to anatomical features of the SPN dendrite (Supplementary Fig. 3). This synaptic scaling translated to endogenously generated spontaneous EPSPs, as locally increasing spontaneous neurotransmitter release to distal and proximal dendrites (via local application of high osmolarity ACSF) yielded identical somatic EPSP amplitude distributions (Supplementary Fig. 4). Stimulation of clustered spines on proximal dendrites produced a rapid somatic depolarization, but unlike the situation when distal spines were stimulated, the membrane potential rapidly decayed back to the resting membrane potential when the stimulation ended (Fig. 1c,d). This relationship was observed at both room and elevated temperatures (Supplementary Fig. 5).
Though it was not feasible to directly measure dendritic voltage, changes in dendritic Ca2+ concentration produced by glutamate uncaging were measurable, providing an indirect assay of dendritic voltage 8,9. Robust Ca2+ transients were observed in distal dendritic spines and shafts following glutamate uncaging that led to up-state generation. Ca2+ transients were measured using the low affinity calcium indicator Fluo 4FF, to avoid dye saturation (Fig. 1e) 15. These experiments revealed a strong correlation between the duration of the somatic up-state and the duration of the distal Ca2+ signal (Fig. 1f).
Although uEPSPs of a millivolt or more were used to generate state transitions in the experiments described thus far, smaller, microvolt uEPSPs were also capable of triggering these events (Fig. 2). There was no correlation between uEPSP amplitude and up-state duration once a threshold number of spines had been stimulated (Fig. 2b). However, these experiments were difficult to conduct with the radially oriented dendrites of SPNs because they typically required 15 or more co-planar spines. As a consequence, subsequent studies were performed with uncaging events that produced 1–3 mV uEPSPs (e.g., Fig. 2d).
There are significant differences between dSPNs and iSPNs in the propagation of somatic spikes into dendrites 8. However, both cell types responded to distal synaptic stimulation in much the same way (Fig. 3a,b). There were no differences in up- and down-state somatic membrane potentials of the two SPNs (Fig. 3c). Nor were there any differences in the density or spatial distribution of uncaging sites required for up-state generation (Fig. 3d,e). Moreover, in neither cell type was there a relationship between the number of stimulated spines and up-state duration (Fig. 3f), just as one would expect of a non-linear process with a threshold.
Figure 3.
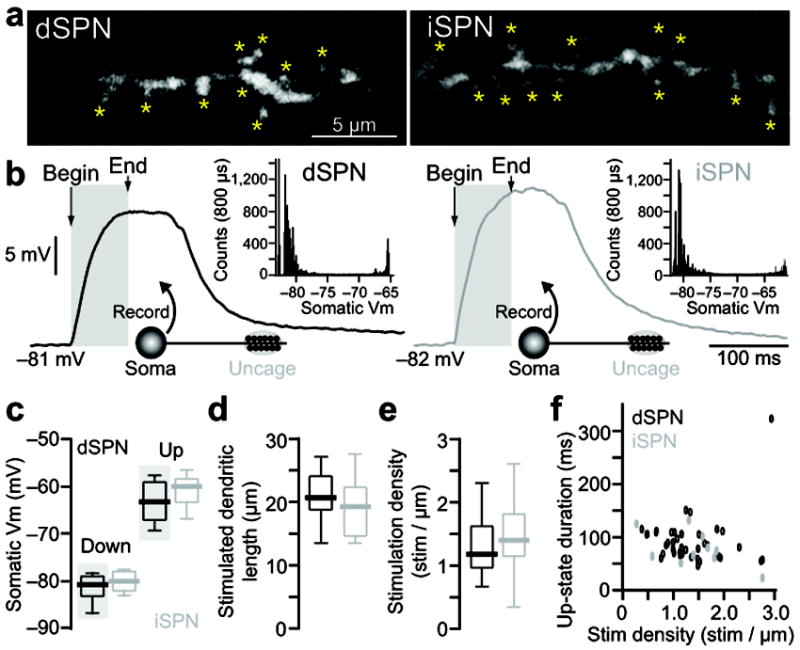
State transition generation is similar in dSPN and iSPNs. (A–B) State transitions were generated by glutamate uncaging on distal dendrites of dSPN (left) and iSPNs (right). High magnification MIP images showing the location of glutamate uncaging (A) and the resulting somatic voltage traces (B). Insets show all point histograms of membrane potentials for each trace. (C) Somatic membrane potentials of ‘up’ and ‘down’ states in dSPNs (n=56 cells) and iSPNs (n=18 cells). Data from figure 1b is shown for comparison and highlighted in green in 2c. (D–E) The length of spiny distal dendrite and the stimulation density (number of stimuli per 1 μm dendritic length) required to evoke state transitions is similar in dSPNs (n=39) and iSPNs (n=11). (F) No obvious differences were observed in the relationship between stimulus density and up-state duration in D1 (n=39) or D2 (n=11) SPNs.
Up-states required NMDA receptors and calcium channels
Because previous studies in pyramidal neurons have implicated NMDA receptors in synaptically evoked regenerative dendritic events 16,17, our working hypothesis was that the state transitions evoked by distal glutamate uncaging in SPNs had similar determinants. To test this hypothesis, the NMDA receptor antagonist ((2R)-amino-5-phosphonovaleric acid (AP5, 100 μM) was bath applied. AP5 dramatically shortened somatic up-state duration in response to glutamate uncaging at distal spines (Fig. 4a).
Figure 4.
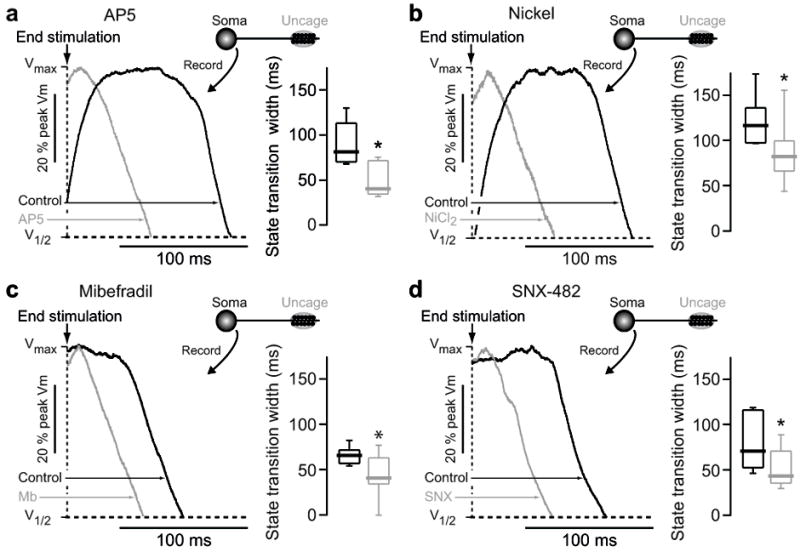
State transition generation requires NMDA receptors and voltage gated Ca2+ channels. (A) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of the NMDA receptor antagonist AP5 (100 μM). Shaded region indicates timing of glutamate uncaging. Voltage traces are normalized to maximum amplitude and overlain. Traces shown to the right are aligned to the end of the last uncaging pulse and normalized to the maximum amplitude of the state transition, as shown in figure 1d. Box plots indicate time between end of uncaging stimulation and 25% voltage fall. AP5 significantly reduced state transition duration, abolishing it in most cases (n=5, p<0.05 Mann-Whitney rank sum test). (B–D) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of (B) NiCl2 (50 μM; n=8), (C) Mibefradil (20 μM; n=8) or (D) SNX-482 (0.3 μM; n=6). Box plots indicate time between end of uncaging stimulation and 25% voltage fall. All three agents significantly reduce the somatic state transition duration (* p<0.05 Mann-Whitney rank sum test).
Voltage-dependent ion channels also are potential contributors to regenerative events in dendrites 18,19. In the apical dendrite of pyramidal neurons, voltage-dependent Na+ channels contribute to spike propagation. This role is more modest in SPNs 8 and bath application of the Nav1 Na+ channel antagonist tetrodotoxin (TTX, 1–2 μM) had no effect on up-states evoked by distal uncaging (Supplementary Fig. 6a). Voltage-dependent Ca2+ channels also contribute to dendritic electrogenesis 8,9. In SPNs, L-type Ca2+ channels prolong the depolarization produced by a brief somatic current pulse 20, and these channels are found in SPN dendrites and spines 9,21. However, the regenerative responses to uncaging glutamate were unaffected by bath application of the Cav1.2/1.3 L-type channel antagonist isradipine (5 μM; Supplementary Fig. 6b) or the mamba toxin calciseptine (0.4 μM; Supplementary Fig. 6c). Hence, while Cav1.3 L-type Ca2+ channels are certainly activated near the somatic up-state potential 9, they do not appear to be necessary for the maintenance of distally generated plateau potentials. Although L-type Ca2+ channels were not necessary, other voltage-dependent Ca2+ channels did appear to be involved as the inorganic Ca2+ channel antagonist Ni2+ (50 μM) effectively reduced plateau duration (Fig. 4b). SPNs express Cav3.2 and Cav3.3 mRNAs that code for the pore forming subunit of low-threshold T-type Ca2+ channels 22. Although not present in the somatic and proximal dendritic membrane 23, these channels are found in more distal dendrites and spines 9. Consistent with their participation in the dendritic regenerative event, the T-type channel antagonist mibefradil (20 μM) significantly decreased the duration (but not amplitude) of somatic up-states evoked by uncaging (Fig. 4c). Another voltage-dependent Ca2+ channel found in the dendrites of SPNs is the R-type Ca2+ channel Cav2.3 9,24. The tarantula toxin SNX-482 (0.3 μM), a selective R-type channel antagonist, significantly decreased the duration of somatic up-states evoked by uncaging (Fig. 4d). Thus, T-and R-type Ca2+ channels participate in the maintenance of distally initiated up-states.
The results presented thus far suggest that distal SPN dendrites are capable of regenerative events that depend upon glutamate-bound NMDA receptors. If this were true, then prolonging the opening of NMDA receptors should increase up-state duration and possibly up-state amplitude. To test this hypothesis, NMDA and glycine (10 μM) were added to the bath to sustain NMDA receptor agonism. In the absence of uncaged glutamate, this manipulation increased synaptic noise but did not significantly effect the average somatic membrane potential (Fig. 5a; Vrest = −78.9 ± 0.54 (control), −77.9 ± 0.44 mV (+NMDA), n=5, p>0.05, Mann-Whitney rank sum test). Moreover, this manipulation in and of itself did not render SPNs more responsive to somatic current injection (Fig. 5b). However, it did enhance the response to stimulation of distal synapses. The duration of up-states triggered by distal glutamate uncaging was significantly increased (Fig. 5c). In contrast, the amplitude of the somatic up-state was not significantly altered (−66.5 ± 2.9 (control), −57.9 ± 4.5 mV (+NMDA), n=5, p>0.05, Mann-Whitney rank sum test). Because bath application of NMDA could induce sustained cortical activity that might contribute to lengthening SPN up-states (Supplementary Fig. 7), these experiments were repeated in the presence of TTX (1 μM) to prevent this activity from being propagated to the striatum. The addition of TTX did not prevent bath application of NMDA from increasing up-state duration (Fig. 5d). This outcome also excludes the potential involvement of striatal interneurons and argues that increased opening of NMDA receptors was responsible for the lengthening. To provide an alternative test of this hypothesis, glutamate was repetitively uncaged at spines on distal dendrites to see if up-states could be extended. Indeed, repetitive stimulation readily extended up-state duration (Fig. 5e), suggesting that up-state duration was controlled by the time course of synaptic input and NMDA receptor activation, in agreement with previous work 5-7.
Figure 5.
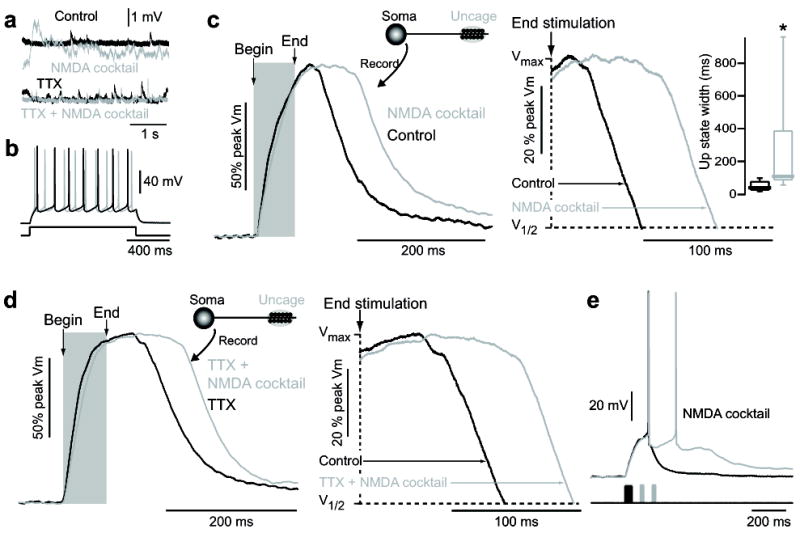
NMDA acts at distal dendritic synapses to enable state transition generation. (A) Somatic voltage recordings of spontaneous depolarizations in a dSPN before and after application of 10 μM NMDA + 10 μM glycine + 10 μM gabazine (SR95531) (“NMDA cocktail”, top) shows an increase in excitatory inputs, which is prevented by 1 μM TTX (bottom). (B) Somatic voltage of a dSPN in response to suprathreshold current injection in the presence and absence of 10 μM NMDA/glycine/gabazine. No regenerative changes in somatic voltage were observed, implying both the location of up-state duration and the NMDA receptors involved in their maintenance are located distally. (C) State transitions were evoked by glutamate uncaging on distal dendritic spines of dSPNs, then again in the presence of 10 μM NMDA/glycine/gabazine. Somatic up-state duration was significantly enhanced (n=6, p<0.05 Signed Rank test). Shaded region indicates timing of glutamate uncaging. Traces are normalized to the maximum amplitude. Box plots indicate time between end of uncaging and 25% voltage fall. (D) The increase in up-state duration in dSPNs caused by 10 μM NMDA/glycine/gabazine is not occluded by 1 μM TTX, suggesting the action of NMDA is directly on distal SPN dendrites and does not rely upon action potential dependent glutamate release (n=4). (E) A state transition was induced by stimulation of 10 distal dendritic spines (twice each) in a dSPN. The up-state could be maintained by supplemental stimulation of these same spines (once each) every 50 ms. Uncaging pulses are indicated by shaded regions.
The data presented to this point suggests that distal dendrites are capable of regenerative activity. Because these dendritic regions are not accessible to direct interrogation and manipulation, a NEURON simulation was used to better understand the phenomenon. In the model, sequential activation of ten glutamatergic synapses along a stretch of distal dendrite generated a local plateau potential (Fig. 6a,b). Moreover, the dendritic plateau depolarized the somatic membrane in a way that resembled that seen experimentally (Fig. 6b). Removal of either NMDA receptors or T- or R- type Ca2+ channels reduced the duration of the plateau potential in the dendrites and the up-state in the soma (Fig. 6c–e), as seen experimentally. Interestingly, the simulations suggested that the sub-cellular positioning of Ca2+ channels was important to plateau generation: placement of T-type channels in the dendritic shafts and R-type channels in spines was critical to the state transitions (Fig. 6d–e). Closer examination of spine and shaft voltage trajectories at the site of up-state initiation provided an explanation. In spine heads, the depolarization produced by synaptic stimulation was much greater than in the shaft, reaching levels needed for opening the high-threshold, R-type channels. Shaft placement of low-threshold, T-type channels provided a means of boosting the depolarization created by currents generated in spines.
Figure 6.

Neuron modeling supports synergistic action of NMDA receptors and T/R Ca2+ channels in up-state generation and maintenance. (A) NEURON model of a SPN containing 6 primary dendrites, 12 secondary dendrites and 24 tertiary dendrites. 4 of the tertiary dendrites are invested with spines and indicated by thick blue lines (s3°). The other 20 tertiary dendrites do not contain localized spines (ns3°). All tertiary dendrites have diameters tapered from 2.5–0.3 μm. AMPA and NMDA receptors are located in spines only; Cav3.2 channels are located in spines and tertiary dendrites, except where noted in (E); R-type channels are located only in tertiary spines, except where noted in (D). (B) Simulations were run using the same stimulation protocol described experimentally, and the resulting membrane potential is shown for the first stimulated spine, the stimulated spiny 3° dendrite, the corresponding parent 2° dendrites of a stimulated and nonstimulated 3° dendrite, and the soma. (C–E) Simulations were run in ‘control’ conditions and in the absence of (C) functional NMDA receptors, (D) R-type channels or (E) Cav3.2 T-type channels. Voltage traces are of the first stimulated spine (top), the activated tertiary dendrite (middle) and soma (bottom). (F) Simulations were run using the same protocol, gradually increasing the spine ‘leak’ conductance from 0 to 10-3 S/cm2. Increasing the leak conductance, effectively decreasing the input impedance, reduces the membrane potential amplitude and plateau duration in both the stimulated spine (top), 3° dendrite (middle) and soma (bottom). Shaded regions indicate timing of stimulation.
Given the close correspondence between the model and experimental observations, we used the model to address why dendritic plateaus were restricted to distal dendrites. One contributing factor appeared to be the distal distribution of T-type Ca2+ channels. But this alone was not enough (data not shown). Another factor could be that the high input impedance of fine distal dendrites allows synaptic input to produce the stronger local depolarization needed for a regenerative event 16. SPN dendrites taper and spine density decreases with distance from the soma 25; these features created a distance independence to uEPSPs in the dendritic regions examined here, consistent with the view that rising input impedance could be a factor (Supplemental Fig. 3). To directly test the hypothesis, a non-selective ‘leak’ conductance was placed in tertiary dendrites and the effect on the response to synaptic stimulation mapped. As the leak increased and input impedance fell, the local response lost its regenerative features and the somatic membrane potential shift became smaller and shorter (Fig. 6f).
Dopamine and adenosine receptors modulated up-states
Dopamine is a key modulator of SPN intrinsic excitability and corticostriatal glutamatergic synaptic transmission 3,4,26. As outlined above, the two principal types of SPNs are distinguished by their expression of D1 and D2 dopamine receptors 1,27. Postsynaptic NMDA receptors are major targets of these receptors 28. To determine if D1 dopamine receptor signaling could affect up-states, glutamate was uncaged on distal spines of D1 receptor-expressing dSPNs and the D1 receptor agonist ±-6-chloro-PB hydrobromide (6-CPB; 5 μM) applied. 6-CPB consistently lengthened up-states evoked in these SPNs (Fig. 7a). It was recently reported that D2 receptor signaling decreased NMDA receptor opening in dendritic spines of iSPNs 24. In agreement with this report, in D2 receptor-expressing iSPNs the D2 receptor agonist quinpirole (20 μM) consistently shortened up-states evoked by distal glutamate uncaging (Fig. 7b). This effect was independent of concomitant A2a receptor stimulation as it was found in the presence of the A2a receptor antagonist SCH58261 (200 nM). Although D1 receptors are not present in iSPNs, A2a receptors are, which like D1 receptors are positively coupled to adenylyl cyclase and activation of protein kinase A (PKA) 29. Bath application of the A2a adenosine receptor agonist CGS21680 (200 nM) consistently lengthened up-state duration in iSPNs, just as the D1 receptor agonist did in dSPNs (Fig.7c). Application of the A2a antagonist SCH58261 had no effect on up-state duration by itself, suggesting that there was little basal A2a receptor activity in our superfused brain slices (Fig. 7c).
Figure 7.
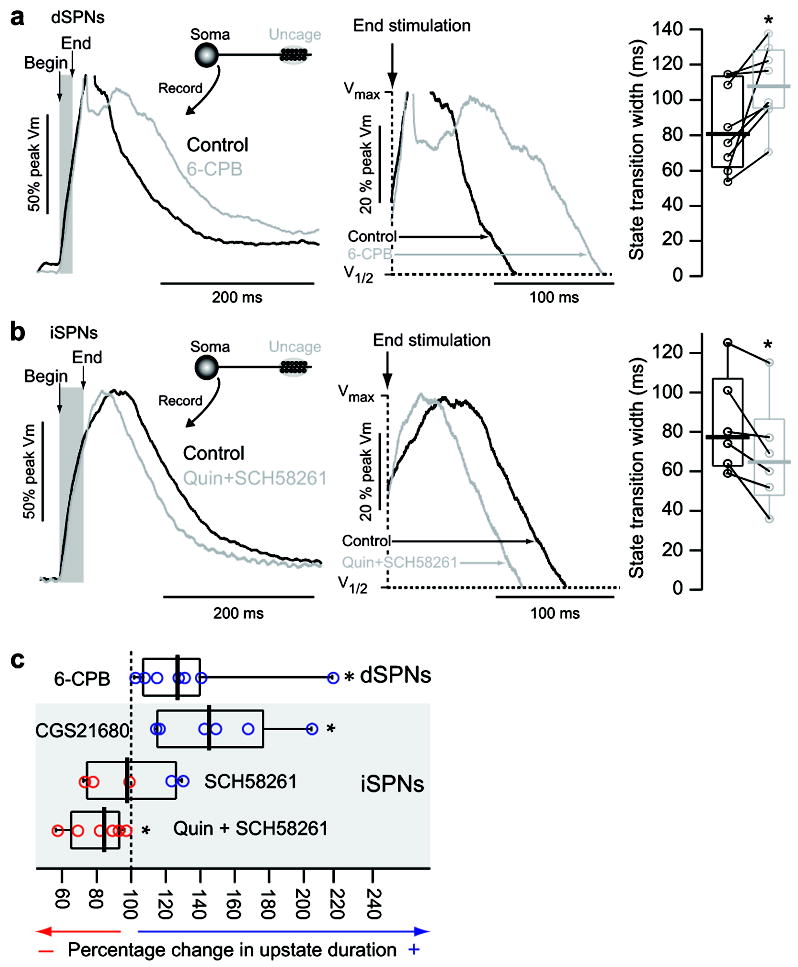
Up-state duration is modulated by dopamine and adenosine receptor signaling. (A) Example normalized traces of an up-state induced by stimulation of distal dendritic spines in a dSPN before and after application of the D1 agonist 6-CPB (5 μM). Action potentials are truncated to better display state transitions. Shaded region indicates timing of glutamate uncaging. Box plots to the right show time between end of uncaging stimulation and 25% voltage fall. 6-CPB increased up-state duration in all dSPNs examined (n=8; p=0.008 Signed rank test). Before and after data points from the same cells are connected with lines. (B) Example normalized traces of an up-state induced by stimulation of distal dendritic spines in an iSPN before and after application of the D2 agonist quinpirole (20 μM) and A2a receptor antagonist SCH58261 (200 nM). Shaded region indicates timing of glutamate uncaging. Box plots (time from end of uncaging stimulation to 25% voltage fall) to the right show that quinpirole/SCH58261 reduces up-state duration in iSPNs (n=6; p<0.05 Signed Rank Test). Before and after data points from the same cells are connected with lines. (C) Box plots showing the percent change in up-state duration in D1 and D2 SPNs by 6-CPB (from (A)), CGS21680 (200 nM; n=6, p<0.05 Signed rank test), SCH58261 (n=5, signed rank test) and quinpirole + SCH58261 (from (B)).
Discussion
Our studies revealed that uncaging glutamate on distal dendritic spines of SPNs, mimicking activation of corticostriatal synapses, triggered a local regenerative plateau potential that was effectively propagated to the soma, producing a potential change that strongly resembled an up-state in vivo. The dendritic plateau potential required the rapid activation of only about 10 neighboring synapses. Inward current through both NMDA and voltage-dependent T- and R-type Ca2+ channels contributed to the plateau, shaping its amplitude and duration. In keeping with previous work examining the modulation of SPN NMDA receptors 24,26,28, the duration of the somatic up-states was increased by activation of D1 dopamine receptors in dSPNs and shortened by activation of D2 dopamine receptors in iSPNs. Taken together, these results argue that the distal dendrites of SPNs are capable of sustaining regenerative plateau potentials that depend upon the release of glutamate and engagement of NMDA receptors. The existence of distally generated regenerative events not only reconciles the stereotyped features of state transitions in SPNs with the available physiological literature, it fundamentally shifts our model of how SPNs recognize patterns of cortical activity and use this recognition to gate action selection.
Why distal dendrites?
Synaptic stimulation of distal dendrites was much more effective in evoking dendritic plateau potentials and somatic up-states than stimulation of proximal dendrites. This difference was not attributable to a higher density of NMDA receptors at distal synapses. Rather, two other factors appeared to be important. One was the fine caliber and resulting high input impedance of distal dendritic shafts 16,25. Because these fine dendrites restrict ion flow away from the site of stimulation, more of the current flowing through synaptically located AMPA receptors is available to discharge the membrane capacitance and depolarize the neighboring membrane, leading to more effective recruitment of NMDA receptors that become unblocked by Mg2+ at depolarized potentials. Simulations of the SPN plateau potentials corroborated this inference. A similar scenario has been described in the fine basal and oblique dendrites of pyramidal neurons where NMDA receptor dependent spikes are seen 17,30,31. Because tertiary SPN dendritic diameter tapers gradually 25, the capacity to trigger plateau potentials should also increase gradually with distance from the soma, if this factor dominates the process.
Another factor that might contribute to the distal generation of plateau potentials was the investment of these dendrites with T-type Ca2+ channels. SPNs robustly express Cav3.2 and Cav3.3 mRNAs that code for a slowing gating, pore-forming subunit of the T-type Ca2+ channel 22. These channels are not present to any significant extent in the soma and proximal dendritic membrane of SPNs 23, but are present in more distal dendrites and spines 9,22. The participation of T-type channels in dendritic plateaus was inferred from the ability of the T-/L-type blocker mibefradil to shorten somatic up-states and the failure of the selective L-type antagonists isradipine and calciseptine to do the same. Simulations provided corroboration for these results showing that T-type Ca2+ channels in dendritic shafts boosted synaptically generated depolarization. A similar situation has recently been described in thalamic reticular nucleus neurons 32. These simulations also suggested the placement of R-type Ca2+channels in the spine head, rather than the dendritic shaft, was an important boosting mechanism helping to link synaptic depolarization to dendritic plateau potentials.
Are these ‘real’ up-states?
Although our studies show that temporally convergent glutamatergic input to distal dendrites can evoke somatic voltage changes that closely resemble up-states seen in vivo, it was not proven that this is how they are generated in vivo. Nevertheless, there is substantial circumstantial support for this proposition. First, SPN up-states seen in anesthetized rodents depend upon activation of corticostriatal synapses and NMDA receptors 5,33, as described here. Second, in this model, the duration of the cortical synaptic barrage dictates the duration of the up-state in vivo, as here 7,34,35. The only discrepancy is the inferred precision of this dependence. In our model, up-state duration is dependent upon locally induced regenerative events created by activation of NMDA receptors, rather than an ongoing synaptic barrage that holds the membrane potential up. Thus, the depolarized plateau potential can outlast the synaptic input by hundreds of milliseconds, depending upon glutamate clearance rates and NMDA receptor gating kinetics. A third reason for drawing a connection is that SPN up-states in vivo, as inferred from the duration of episodic spiking, are lengthened by D1 dopamine receptor activation and shortened by D2 dopamine receptor activation 36, as were up-states described here.
The most substantial discrepancy between our model and the prevailing one is the number of synaptic inputs required to drive an up-state. Rather than requiring hundreds of temporally convergent inputs to drive an up-state and hundreds more to sustain it, our data suggests that an order of magnitude fewer glutamatergic inputs are necessary because the distal dendrites of SPNs, like dendrites found in a wide variety of other neurons 37, are active and capable of regenerative activity. The restriction of the regenerative activity initiation to distal dendrites during synaptic stimulation explains why previous studies have failed to see evidence for it using somatic electrodes 5. When taken together with the stimulation densities used to evoke up-states, the high spatial convergence (approximately 20 μm of a distal dendrite) described in this study suggests a re-envisioned “synchrony threshold” for up-state induction in SPNs. Fewer highly coupled corticostriatal inputs (as mimicked by adjusting the stimulation strength to induce somatic EPSPs greater than 1 mV) are needed (range = 7–45, median = 25) to induce up-states than weakly coupled inputs (range = 16–64, median = 57). Taking coupling strength into account, and considering a corticostriatal release probability of 0.42 13, we calculate that about 59 (17–107) strongly coupled or 135 (38–152) weakly coupled synaptic units are required to evoke an up-state. Presuming active corticostriatal pyramidal neurons to fire bursts of 4–5 spikes at up to 50 Hz in vivo 38, it could take the synchronous firing of only 12–15 corticostriatal pyramidal neurons that are strongly coupled to a SPN to induce an up-state. At the other extreme it may take considerably more cortical pyramidal neurons to fire synchronously (27–34) if they are weakly coupled to the SPN. Given that there are about 30 glutamatergic cortical synapses within the requisite 20 μm stretch of distal dendrite 25, this convergence requirement does not seem difficult to reach. This number is likely an overestimate for at least two reasons. First, corticostriatal inputs show marked paired pulse inhibition, which will in effect increase the weight of subsequent spikes in a burst 13. Second, corticostriatal pyramidal neurons are not quiescent in vivo 38, which will be reflected in the SPN resting membrane potential, and potentially place the SPN in an environment even more conducive to up-state generation. Indeed, our simulations show that mimicking disperse depolarizing inputs to a SPN (by depolarizing the soma ~4 mV) lowers the number of glutamatergic inputs required to induce an up-state (Supplementary Fig. 8).
The full partnership of the post-synaptic membrane in up-state generation and the reliance upon a small number of spatially convergent inputs also helps to explain why residence time in the up-state increases in SPNs with cortical desynchronization and wakefulness 39. If the convergence requirement for up-state generation is very high, cortical desynchronization should diminish up-state transitions. However, if distal dendrites are active, cortical synchrony is not required for up-state generation. Rather, up-states could depend upon activation of neighboring corticostriatal synapses that represent meaningful patterns of cortical activity 10With wakefulness, the frequency of such meaningful cortical activity should increase, potentially resulting in a sustained up-state, as observed experimentally 39.
Implications for pattern recognition and action selection
The existence of active dendritic regions dramatically increases the pattern recognition capacity of neurons 10. This increased capacity is achieved by local interactions that confer meaning on synaptic location, as well as synaptic strength. Based upon a clusteron learning rule 10, the repeated co-activation of neighboring synapses will lead to their stabilization or strengthening as a group, providing local control of plasticity. In SPNs, longer loop feedback also appears to be provided by dopamine, as both D1 and D2 receptors modulated the duration of local plateaus in a way consistent with their global effects on synaptic plasticity 4,40. That said, this inferred form of local plasticity in distal SPN dendrites sharply contrasts with spike-timing dependent plasticity (STDP), which depends upon the temporal conjunction of local synaptic activity and a ‘global’ somatic spike that back-propagates into the dendrite 41,42. Because of this dependence, STDP must normally be limited to proximal dendrites capable of supporting bAP invasion 8. The fact that the loss of a bAP signal along an SPN dendrite is paralleled by the growth in local regenerative capacity suggests that these two forms of synaptic plasticity are, for the most part, mutually exclusive.
The ability of a small number of distal synapses to induce up-state transitions in SPNs also provides a new framework for understanding what they mean to striatal information processing. Rather than being a consequence of massed cortical activity, up-states could reflect the occurrence of a specific pattern of cortical activity that has been repeated frequently or has been paired with a psychologically meaningful event that altered striatal dopamine release. It is tempting to speculate that these ‘meaningful’ cortical patterns could represent a context that is appropriate for a particular action and that their occurrence triggers an up-state transition in a population of SPNs, making them available or responsive to some other synaptic signal that could evoke spikes. The proposition that inputs triggering spikes and those inducing up-states are different has compelling experimental support 7. In this scenario, that ‘other’ input could represent possible actions, with those that have been shaped by association with environmental contingencies being the strongest and the most likely to evoke spikes. Linking possible actions to the timing of spikes (rather than up-states) would endow activity in the striatal network with a temporal precision widely perceived as being important for movement control 43. Proximal synapses that have been shaped by processes like STDP would be the most likely to serve this purpose. In keeping with previous conjectures on this point 25, our model shows that during an up-state, the effect of a single proximal synapse is significantly enhanced as the electrotonic distance shrinks with closure of dendritic Kir2 K+ channels (Supplementary Fig. 9) 25.
If this model of cortical control of SPN activity is correct, it suggests that proximal and distal SPN synapses arise from fundamentally distinct cortical circuits – a conjecture that is not at variance with the anatomy of the corticostriatal projection 25, but one that will be difficult to test given the lack of a layered striatal cytoarchitecture. One of the few examples of dendritically segregated innervation of SPNs is that of inhibitory circuits, specifically GABAergic interneurons: parvalbumin containing FS interneurons predominantly synapse on SPN soma and proximal dendrites while somatostatin-containing PLTS interneurons predominantly synapse on more distal dendrites44. Both interneurons form feed forward corticostriatal circuits 45. The differences in subcellular targeting of these inputs might have functional consequences. For example, somatic GABAergic input could control the timing of axon initial segment spikes 45, whereas distal dendritic GABAergic input, by creating a localized shunt that decreases input resistance, could prevent up-state generation by one cortical ensemble, while leaving the capacity of another ensemble to do so intact. GABAergic modulation of state transitions need not be restricted to inputs from interneurons however, as GABAergic SPN axonal collaterals have previously been implicated in playing such a role 46.
In summary, our studies have revealed that the distal dendrites of SPNs are capable of regenerative activity that produces somatic potential changes resembling up-states in vivo. These regenerative dendritic events were dependent upon the active co-activation of NMDA receptors and voltage-dependent Ca2+ channels in small caliber dendrites having high local input impedance. The presence of active dendrites increases the pattern recognition capacity of SPNs. Moreover, it suggests that the associative functions of SPNs – linking context and appropriate action selection – are performed by the integration of two operationally distinct dendritic regions.
Methods
Brain slice preparation
Para-saggital brain slices (275 μm) were obtained from 21–30 day old BAC D1 or BAC D2 transgenic mice following procedures approved by the Northwestern University Animal Care and Use Committee. The mice were anesthetized with a mixture of ketamine (50 mg kg−1) and xylazine (4.5 mg kg−1) and perfused transcardially with 5–10 ml ice-cold artificial CSF (aCSF) containing (in mM): 124 NaCl, 3 KCl, 1 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 16.66 glucose, continuously bubbled with carbogen (95% O2 and 5% CO2). The slices were then transferred to a holding chamber where they were incubated in aCSF containing (in mM) 2 CaCl2, 1 MgCl2, at 35°C for 60 min, after which they were stored at room temperature until recording.
Electrophysiology
Patch pipettes were pulled from thick-walled borosilicate glass on a Sutter P-97 puller and fire-polished before use. Pipette resistance was typically 3–4 MΩ when filled with recording solution. The internal recording solution contained (in mM): 135 KMeSO4, 5 KCl, 10 HEPES, 2 ATP-Mg2+, 0.5 GTP-Na+, 5 phosphocreatine-tris; 5 phosphocreatine-Na+, 0.1 spermine, 0.2 Fluo-4 pentapotassium salt and 0.05 Alexa Fluor 568 hydrazide Na+ salt (Invitrogen), pH was adjusted to 7.25 with NaOH and osmolarity to 270–280 mOsm l−1. In some cases Fluo 4FF (500 μM) was used in place of Fluo 4, as indicated in the text. Slices were continuously perfused with carbogen-bubbled aCSF. Slices were transferred to a submersion-style recording chamber mounted on an Olympus BX51 upright, fixed-stage microscope. Electrophysiological recordings were obtained with a Multiclamp 700B amplifier. Stimulation and display were obtained as previously described (Day et al., 2008) using the custom-written shareware package WinFluor (John Dempster, Strathclyde University, Glasgow, Scotland, UK), which automates and synchronizes the two-photon imaging and electrophysiological protocols. The amplifier bridge circuit was adjusted to compensate for serial resistance and continuously monitored during recordings. Recordings were performed at room temperature, unless otherwise noted.
2-photon laser scanning microscopy (2PLSM)
Direct and indirect pathway SPNs were identified by somatic eGFP two-photon excited fluorescence using an Ultima Laser Scanning Microscope system (Prairie Technologies). A DODT contrast detector system was used to provide a bright-field transmission image in registration with the fluorescent images. The green GFP signals (490–560 nm) were acquired using 810 nm excitation (Verdi/Mira laser). SPNs were patched using video microscopy with a Hitachi CCD camera and an Olympus 60X/0.9 NA lens. Alexa 568 fluorescence was used for visualization of cell bodies, dendrites, and spines. Following patch rupture, the internal solution was allowed to equilibrate for 15–20 minutes before imaging. Maximum projection images of the soma and dendrites were acquired with 0.36 μm2 pixels with 10 μs pixel dwell time; ~80 images are taken with 1 μm focal steps.
Ca2+ imaging
Drugs were either bath applied by dissolving them in the external aCSF (for experiments involving NMDA and glycine) or superfused via the perfusion pump used to apply MNI-glutamate described below. In the latter instances drugs were dissolved in HEPES-based aCSF containing (in mM): 124 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1 NaH2PO4, 26 HEPES and 16.66 glucose. Dendritic changes in Ca2+ were measured using Fluo-4 as previously described 8. Ca2+ transient were expressed as ΔF/Fo. Green fluorescent line scan signals were acquired at 6 ms per line and 512 pixels per line with 0.08 μm pixels and 10 μs pixel dwell time. The laser-scanned images were acquired with 810 nm light pulsed at 90 MHz (~250 fs pulse duration). Power attenuation was achieved with two Pockels cells electro-optic modulators (models 350–80 and 350–50, Con Optics, Danbury, CT). The two cells were aligned in series to provide an enhanced modulation range for fine control of the excitation dose (0.1% steps over four decades). The line scan was started 200 ms before the stimulation protocol and continued 4 seconds after the stimulation to obtain the background fluorescence and to record the decay of the optical signal after stimulation. To reduce photo-damage and photo-bleaching, the laser was fully attenuated using the second Pockels cell at all times during the scan except for the period directly flanking the evoked state transition.
2PLU
2P uncaging of MNI-glutamate was performed simultaneously with Ca2+ imaging using a Chameleon-XR laser system (Coherent Laser Group, Santa Clara, CA). MNI-glutamate (Tocris Cookson, Ellisville, MO) was superfused (0.4 ml/hr) at 5 mM using a system of syringe pumps (WPI, Sarasota, FL) and a multi-barreled perfusion manifold fitted with a small-volume mixing tip that allowed rapid switching between solutions (Cell MicroControls, Norfolk, VA). MNI-glutamate was uncaged using 1ms pulses of 720 nm light independently controlled by a third Pockels cell modulator (model 302, Con Optics, Danbury, CT). Experiments involved uncaging on a single spine to evoke a somatic EPSP measuring 1–4 mV. The uncaging pulses were 1 ms in duration and typically about 15 mW in strength measured at the sample plane. Photolysis power was adjusted to closely mimic spontaneously occurring EPSPs and tuned to achieve the predetermined somatic EPSP amplitude. We opted to use this strategy of tuning laser power, as opposed to relying on photobleaching 48, because of our method of MNI glutamate delivery. As this study required same-cell pharmacological manipulations, MNI glutamate was delivered via a superfusion pump rather than a recirculating pump, introducing chemical penetration as an addition variable, making photobleaching an inappropriate measure of total glutamate uncaging. State transitions were evoked by stimulating approximately 10 neighboring coplanar spines in rapid succession (500 Hz), with each spine stimulated 1–4 times. The distance from soma was measured by a straight line drawn from the site of stimulation to the origin of the dendrite at the soma. Simultaneous photolysis and line-scan images were made from shafts/spines. As described above, the custom-written software package WinFluor automated and synchronized the Ca2+ imaging with the electrophysiological stimulation and the photolysis.
Neuron modeling
A SPN was modeled using NEURON version 6.0 49. The presented model is based upon anatomical reconstructions of SPNs and similar to one previously published by our group 50 containing 6 primary dendrites having primary, secondary and tertiary branch points. In addition, 1396 spines were added to 4 tertiary dendrites. The other dendrites were modeled in a way that mimicked the presence of spines, but computationally simplified the model 25. Soma dendrites were populated with ion channels known to contribute to SPN excitability 26, with the exception of Nav1 Na+ channels, which were shown experimentally in the present study not to be participants in up-state generation; this omission simplified the simulations by eliminating the complication of somatic spikes. Spines were endowed with L-type, Cav3.2 T-type and R-type voltage gated Ca2+ channels, KLeak channels and Kir2 potassium channels. Presynaptic glutamate releasing terminals were added to 10 neighboring spines on a single tertiary dendrite (mimicking a distal stimulation site) to simulate a corticostriatal synapse. These 10 corresponding spines were further endowed with AMPA and NMDA receptors. Tertiary dendrite and spine Ra was adjusted to 125 Ωcm. Cav3.2 conductances matching those in spines were added to tertiary dendrites. NMDA/AMPA/Cav3.2/R-type channel conductances were tuned to give somatic voltage responses similar to those observed experimentally. Kv1 and KCNQ potassium conductances were increased in the initial axon segment to favor somatic state transition kinetics similar to those observed experimentally.
Statistical analysis
Differences in state transition duration and Ca2+ transients were examined using the Mann-Whitney U nonparametric test of significance and the Wilcoxin signed rank test. Differences were considered statistically significant if p<0.05.
Supplementary Material
Supplementary Figure 1. Glutamate uncaging and electrical stimulation produce similar somatic responses. (A) Example somatic EPSPs generated by electrical intrastriatal stimulation with a theta glass electrode placed within 20 μm of a distal dendrite branch (black) vs. glutamate uncaging on a single spine (red). EPSPs are in response to 3 stimuli delivered at 20 Hz in a iSPN.
Supplementary Figure 2. The direction of synapse number (i.e. towards (in) or away from (out) soma) had no effect on up-state direction. (n=27, in; n=20, out).
Supplementary Figure 3. Distal and proximal uncaging events produce similar postsynaptic responses. (A) Alpha function fits of averaged single somatic EPSPs induced by glutamate uncaging on a single distal (black) or proximal (red) dendritic spine (left). Ca2+ transients (measured with Fluo 4) corresponding to EPSPs on the left (right). (B) EPSP amplitudes (left) and spine Ca2+ transients (middle) evoked by single EPSPs used to trigger state transitions were similar in proximal (red, 47.6 ± 0.97 μm from soma) and distal (black, 118.7 ± 2.1 μm from soma) dendritic spines (n=13–15, 6 cells). Rise times of large somatic EPSPs induced by proximal dendritic activation are similar to the rise times of state transitions evoked by distal dendritic activation (right; n=8).
Supplementary Figure 4. Spontaneous synaptic inputs to distal and proximal dendrites yield similar somatic depolarizations. (A) MIP image of a D1 SPN showing the locations at which high osmolarity ACSF (HEPES-buffered, adjusted to 600 mOsM with sucrose) was locally applied with a puffer pipette to increase spontaneous synaptic events73. Puffer sucrose solution contained 10 μM Alexa Fluor 568 for visualization. (B) Local administration of high osmolarity ACSF significantly increased the frequencies of somatically recorded spontaneous depolarizing events generated at both distal (n=6 cells) and proximal (n=5 cells) dendritic locations, but had no effect on the distribution of mini epsp amplitudes. Normalized mini epsp amplitude distributions are fit to alpha functions. All recordings were performed in 1 μM TTX. *p<0.05, paired t-test. (C) NEURON simulation showing somatic EPSPs triggered by stimulating AMPA/NMDA receptors on a single spine located 50, 100 or 200 μm from soma predict a negative correlation between somatic EPSPs amplitude/rise and distance from soma (left). Tapering the diameter of the dendritic shaft eliminates this distance dependence, and recapitulates the results observed experimentally (right).
Supplementary Figure 5. Stimulation of distal dendrites also produce up-states at physiological temperatures. (A) State transitions were evoked in dSPNs by uncaging at distal (black) dendritic spines; the same protocol was then used to excite a proximal dendrite (red), similarly to Fig. 1c–d. Recordings were performed at 33-35 °C. (B) Somatic voltage traces aligned to the last uncaging stimulus and normalized show up-states are readily induced by distal but not proximal activation at elevated temperature. Box plots indicate time between end of uncaging and 25% voltage fall (n=5, p=0.008, Mann-Whitney rank sum test). Though readily generated at elevated temperatures, cells were less stable than at room temperature, and often did not remain healthy enough for multiple comparisons after an up-state induction (1/3 of SPNs remained stable enough for multiple comparisons).
Supplementary Figure 6. Voltage gated sodium channels and L type voltage gated Ca2+ channels do not regulate state transition kinetics or duration. (A) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of 2 μM TTX. Traces shown to the right are aligned to the end of the last uncaging pulse and the maximum amplitude of the state transition. Box plots to the right represent the time from the last uncaging event to 25% fall in somatic plateau potential. TTX had no effect on up-state duration (n=4, Mann-Whitney rank sum test). (B) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence isradipine (5 μM). Isradipine had no effect on up-state duration (n=5, Mann-Whitney rank sum test). Shaded regions indicate timing of glutamate uncaging. (C) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of rCalciseptin (0.4 μM). rCalciseptin had no effect on up-state duration (n=5, Mann-Whitney rank sum test).
Supplementary Figure 7. Activation of NMDA receptors increases cortical pyramidal neuron firing. (A) Somatic voltage recording of a cortical pyramidal neuron in the absence and presence of 5 μM NMDA + 5 μM glycine shows this excitatory cocktail increases pyramidal neuron firing. (B) NMDA/glycine-induced firing is inhibited by 1 μM TTX puffed directly on the soma. Photomicrograph showing the pyramidal neuron cell body, patch electrode (left) and puffer electrode placement (right) is shown to the right.
Supplementary Figure 8. Somatic depolarization reduces the number of inputs required for an up-state. The NEURON model presented in figure 5 was (black) was modified to contain only 9 excitatory inputs (blue), which fails to evoke an up-state. Depolarizing the soma with 40 pA current before synaptic stimulation allows the 9 excitatory inputs to evoke an up-state.
Supplementary Figure 9. A single remote glutamatergic input modulates up state duration. The NEURON model presented in figure 5 was modified to contain a single extra spine (containing AMPA receptors) located on a distal (equivalent to the distance at which the plateau potential is evoked) tertiary dendrite not used to evoke a plateau potential (top). This added synapse was activated by a separate juxtaposed presynaptic terminal 60 ms after the initiation of an up state (bottom). The upstate is shown with or without activation of the added spine.
Acknowledgments
NIH NS 034696, MH074866, the Picower Foundation and CHDI foundation supported this work. We thank Dr. John Dempster, Karen Saporito, Nicholas Schwarz, Sasha Ulrich and Dr. David Wokosin for excellent technical assistance.
Footnotes
Author contributions J.L.P. conducted experiments and data analysis; M.D. conducted the experiments in cortical pyramidal neurons and provided technical assistance with uncaging; D.J.S. supervised the project; D.J.S. and J.L.P. designed experiments and prepared the manuscript.
References
- 1.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- 2.Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- 3.Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- 4.Surmeier DJ, et al. The role of dopamine in modulating the structure and function of striatal circuits. Prog Brain Res. 2010;183:149–167. doi: 10.1016/S0079-6123(10)83008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–2410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plenz D, Kitai ST. Up and down states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantia nigra organotypic cultures. J Neurosci. 1998;18:266–283. doi: 10.1523/JNEUROSCI.18-01-00266.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stern EA, Jaeger D, Wilson CJ. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature. 1998;394:475–478. doi: 10.1038/28848. [DOI] [PubMed] [Google Scholar]
- 8.Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Poirazi P, Mel BW. Impact of active dendrites and structural plasticity on the memory capacity of neural tissue. Neuron. 2001;29:779–796. doi: 10.1016/s0896-6273(01)00252-5. [DOI] [PubMed] [Google Scholar]
- 11.Bargas J, Galarraga E, Aceves J. Dendritic activity on neostriatal neurons as inferred from somatic intracellular recordings. Brain Res. 1991;539:159–163. doi: 10.1016/0006-8993(91)90700-6. [DOI] [PubMed] [Google Scholar]
- 12.Vergara R, et al. Spontaneous voltage oscillations in striatal projection neurons in a rat corticostriatal slice. J Physiol. 2003;553:169–182. doi: 10.1113/jphysiol.2003.050799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28:6483–6492. doi: 10.1523/JNEUROSCI.0435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Branco T, Clark BA, Hausser M. Dendritic Discrimination of Temporal Input Sequences in Cortical Neurons. Science. 2010 doi: 10.1126/science.1189664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larkum ME, Nevian T, Sandler M, Polsky A, Schiller J. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science. 2009;325:756–760. doi: 10.1126/science.1171958. [DOI] [PubMed] [Google Scholar]
- 18.Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- 19.Holthoff K, Kovalchuk Y, Konnerth A. Dendritic spikes and activity-dependent synaptic plasticity. Cell Tissue Res. 2006;326:369–377. doi: 10.1007/s00441-006-0263-8. [DOI] [PubMed] [Google Scholar]
- 20.Galarraga E, Hernandez-Lopez S, Reyes A, Barral J, Bargas J. Dopamine facilitates striatal EPSPs through an L-type Ca2+ conductance. Neuroreport. 1997;8:2183–2186. doi: 10.1097/00001756-199707070-00019. [DOI] [PubMed] [Google Scholar]
- 21.Day M, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- 22.McRory JE, et al. Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem. 2001;276:3999–4011. doi: 10.1074/jbc.M008215200. [DOI] [PubMed] [Google Scholar]
- 23.Bargas J, Howe A, Eberwine J, Cao Y, Surmeier DJ. Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J Neurosci. 1994;14:6667–6686. doi: 10.1523/JNEUROSCI.14-11-06667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson CJ. In: Single Neuron Computation. McKenna T, Davis J, Zornetzer SF, editors. Academic Press; 1992. pp. 141–171. [Google Scholar]
- 26.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci. 30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pomata PE, Belluscio MA, Riquelme LA, Murer MG. NMDA receptor gating of information flow through the striatum in vivo. J Neurosci. 2008;28:13384–13389. doi: 10.1523/JNEUROSCI.4343-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charpier S, Mahon S, Deniau JM. In vivo induction of striatal long-term potentiation by low-frequency stimulation of the cerebral cortex. Neuroscience. 1999;91:1209–1222. doi: 10.1016/s0306-4522(98)00719-2. [DOI] [PubMed] [Google Scholar]
- 35.Kasanetz F, Riquelme LA, O’Donnell P, Murer MG. Turning off cortical ensembles stops striatal Up states and elicits phase perturbations in cortical and striatal slow oscillations in rat in vivo. J Physiol. 2006;577:97–113. doi: 10.1113/jphysiol.2006.113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.West AR, Grace AA. Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci. 2002;22:294–304. doi: 10.1523/JNEUROSCI.22-01-00294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sjostrom PJ, Rancz EA, Roth A, Hausser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- 38.Pidoux M, Mahon S, Deniau JM, Charpier S. Integration and propagation of somatosensory responses in the corticostriatal pathway: an intracellular study in vivo. J Physiol. 2011;589:263–281. doi: 10.1113/jphysiol.2010.199646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahon S, Deniau JM, Charpier S. Various synaptic activities and firing patterns in cortico-striatal and striatal neurons in vivo. J Physiol Paris. 2003;97:557–566. doi: 10.1016/j.jphysparis.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Centonze D, Picconi B, Gubellini P, Bernardi G, Calabresi P. Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur J Neurosci. 2001;13:1071–1077. doi: 10.1046/j.0953-816x.2001.01485.x. [DOI] [PubMed] [Google Scholar]
- 41.Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 44.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 45.Tepper JM, Wilson CJ, Koos T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008;58:272–281. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Czubayko U, Plenz D. Fast synaptic transmission between striatal spiny projection neurons. Proc Natl Acad Sci U S A. 2002;99:15764–15769. doi: 10.1073/pnas.242428599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaczorowski CC, Disterhoft J, Spruston N. Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J Physiol. 2007;578:799–818. doi: 10.1113/jphysiol.2006.124586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 49.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- 50.Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Glutamate uncaging and electrical stimulation produce similar somatic responses. (A) Example somatic EPSPs generated by electrical intrastriatal stimulation with a theta glass electrode placed within 20 μm of a distal dendrite branch (black) vs. glutamate uncaging on a single spine (red). EPSPs are in response to 3 stimuli delivered at 20 Hz in a iSPN.
Supplementary Figure 2. The direction of synapse number (i.e. towards (in) or away from (out) soma) had no effect on up-state direction. (n=27, in; n=20, out).
Supplementary Figure 3. Distal and proximal uncaging events produce similar postsynaptic responses. (A) Alpha function fits of averaged single somatic EPSPs induced by glutamate uncaging on a single distal (black) or proximal (red) dendritic spine (left). Ca2+ transients (measured with Fluo 4) corresponding to EPSPs on the left (right). (B) EPSP amplitudes (left) and spine Ca2+ transients (middle) evoked by single EPSPs used to trigger state transitions were similar in proximal (red, 47.6 ± 0.97 μm from soma) and distal (black, 118.7 ± 2.1 μm from soma) dendritic spines (n=13–15, 6 cells). Rise times of large somatic EPSPs induced by proximal dendritic activation are similar to the rise times of state transitions evoked by distal dendritic activation (right; n=8).
Supplementary Figure 4. Spontaneous synaptic inputs to distal and proximal dendrites yield similar somatic depolarizations. (A) MIP image of a D1 SPN showing the locations at which high osmolarity ACSF (HEPES-buffered, adjusted to 600 mOsM with sucrose) was locally applied with a puffer pipette to increase spontaneous synaptic events73. Puffer sucrose solution contained 10 μM Alexa Fluor 568 for visualization. (B) Local administration of high osmolarity ACSF significantly increased the frequencies of somatically recorded spontaneous depolarizing events generated at both distal (n=6 cells) and proximal (n=5 cells) dendritic locations, but had no effect on the distribution of mini epsp amplitudes. Normalized mini epsp amplitude distributions are fit to alpha functions. All recordings were performed in 1 μM TTX. *p<0.05, paired t-test. (C) NEURON simulation showing somatic EPSPs triggered by stimulating AMPA/NMDA receptors on a single spine located 50, 100 or 200 μm from soma predict a negative correlation between somatic EPSPs amplitude/rise and distance from soma (left). Tapering the diameter of the dendritic shaft eliminates this distance dependence, and recapitulates the results observed experimentally (right).
Supplementary Figure 5. Stimulation of distal dendrites also produce up-states at physiological temperatures. (A) State transitions were evoked in dSPNs by uncaging at distal (black) dendritic spines; the same protocol was then used to excite a proximal dendrite (red), similarly to Fig. 1c–d. Recordings were performed at 33-35 °C. (B) Somatic voltage traces aligned to the last uncaging stimulus and normalized show up-states are readily induced by distal but not proximal activation at elevated temperature. Box plots indicate time between end of uncaging and 25% voltage fall (n=5, p=0.008, Mann-Whitney rank sum test). Though readily generated at elevated temperatures, cells were less stable than at room temperature, and often did not remain healthy enough for multiple comparisons after an up-state induction (1/3 of SPNs remained stable enough for multiple comparisons).
Supplementary Figure 6. Voltage gated sodium channels and L type voltage gated Ca2+ channels do not regulate state transition kinetics or duration. (A) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of 2 μM TTX. Traces shown to the right are aligned to the end of the last uncaging pulse and the maximum amplitude of the state transition. Box plots to the right represent the time from the last uncaging event to 25% fall in somatic plateau potential. TTX had no effect on up-state duration (n=4, Mann-Whitney rank sum test). (B) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence isradipine (5 μM). Isradipine had no effect on up-state duration (n=5, Mann-Whitney rank sum test). Shaded regions indicate timing of glutamate uncaging. (C) State transitions were generated in dSPNs by glutamate uncaging on distal dendritic spines, and then the response to the same uncaging protocol was measured in the presence of rCalciseptin (0.4 μM). rCalciseptin had no effect on up-state duration (n=5, Mann-Whitney rank sum test).
Supplementary Figure 7. Activation of NMDA receptors increases cortical pyramidal neuron firing. (A) Somatic voltage recording of a cortical pyramidal neuron in the absence and presence of 5 μM NMDA + 5 μM glycine shows this excitatory cocktail increases pyramidal neuron firing. (B) NMDA/glycine-induced firing is inhibited by 1 μM TTX puffed directly on the soma. Photomicrograph showing the pyramidal neuron cell body, patch electrode (left) and puffer electrode placement (right) is shown to the right.
Supplementary Figure 8. Somatic depolarization reduces the number of inputs required for an up-state. The NEURON model presented in figure 5 was (black) was modified to contain only 9 excitatory inputs (blue), which fails to evoke an up-state. Depolarizing the soma with 40 pA current before synaptic stimulation allows the 9 excitatory inputs to evoke an up-state.
Supplementary Figure 9. A single remote glutamatergic input modulates up state duration. The NEURON model presented in figure 5 was modified to contain a single extra spine (containing AMPA receptors) located on a distal (equivalent to the distance at which the plateau potential is evoked) tertiary dendrite not used to evoke a plateau potential (top). This added synapse was activated by a separate juxtaposed presynaptic terminal 60 ms after the initiation of an up state (bottom). The upstate is shown with or without activation of the added spine.