

Abstract
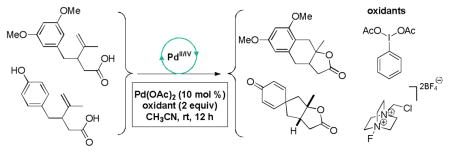
An intramolecular cyclization cascade reaction has been developed utilizing a high valent palladium intermediate that generates a carbon–carbon and carbon–oxygen bond in a single transformation. This method provides rapid access to highly functionalized tricyclic scaffolds, including spirocyclic cyclohexadienones. Good yields and mild conditions are reported with high tolerance towards oxygen and water.
High valent palladium mediated olefin difunctionalizations are versatile transformations in organic chemistry. Recent examples of palladium mediated amino acetoxylations,1 dihydroxylations,2 and diaminations3 have greatly expanded the scope of synthetic methods available to organic chemists.4 Seminal contributions by Semmelhack and Hegedus exploited Wacker intermediates generated from PdII-mediated cyclization reactions to make the key carbon–carbon bonds (Figure 1).5 Methods inspired by their synthetic strategies have been successful in generating a high degree of molecular diversity6 and found application in natural product synthesis.7 Although carbon–carbon bond forming olefin difunctionalizations have been well developed in the context of Heck8 and π-allyl palladium9 chemistry, high valent palladium mediated carbon–carbon bond forming alkene difunctionalization has not been studied to the same extent.
Figure 1.

Pd-Mediated Oxidative C–C Bond Forming Cyclizations
Recently, the Michael group reported an intermolecular carboamination reaction using Pd(OAc)2 and N-fluorobenzenesulfonimide (NFSI) to synthesize substituted pyrrolidines from tethered amino alkenes in moderate to high yields.10 Another report by Waser and coworkers described an intermolecular oxyalkynylation of olefins employing an alkynyl λ3-iodane oxidant, which enabled them to generate substituted tetrahydrofuran and benzofuran derivatives.4f Furthermore, the groups of Kita12b,d–f and Swenton12a,c have synthesized spirocyclohexadieneones through chemical and electrochemical dearomatization of phenols, respectively. Hypervalent iodine or anodic oxidations are most effective in forging carbon heteroatom bonds, although there are a few reported examples of carbon–carbon bond forming dearomatizations that produce ipso substituted dienones. With this in mind, we envisioned using the increased reactivity of high valent palladium obtained by oxidizing Wacker intermediates to allow for the formation of multiple bonds in a single operation, without the need to prefunctionalize our substrates.
Our research in transition metal catalyzed olefin difunctionalization was initiated during efforts directed toward the synthesis of the terpenoid antibiotic platensimycin.11 The key transformation in our proposal was an oxidative dearomatization of a para-substituted phenol to form a spirocyclic cyclohexadienone as a precursor for a radical conjugate addition to efficiently provide the bicyclic fragment of platensimycin (Figure 2).
Figure 2.

Natural Product Synthesis as Inspiration for Methods Development
Unfortunately, hypervalent iodine, halocyclization or conventional Wacker cyclization conditions proved to be ineffective toward oxidative dearomatization, often causing extensive substrate oligomerization or decomposition.12,13 Consequently, we decided to address these shortcomings using a PdII/IV catalytic cycle.
As illustrated in Table 1, our initial results provided only trace quantities of the desired dearomatized cyclohexadienes (Table 1, entries 1–3). After extensive optimization, the electrophilic fluorination reagent Selectfluor was found to provide the best results, affording spirocycle 2 in 43% yield as a single diastereoisomer. Treating substrate 1 with oxidants in the absence of a PdII source (Table 1, entries 5 and 6) afforded only the oxygen trapping product 2a. We proceeded to screen for the optimal nucleopalladation coupling partners other than pendant alcohols (Figure 3). Unfortunately, carboxylic acid and cyanohydrin derivatives (see 3 and 4, respectively, for representative examples) gave comparable yields without providing deeper insight into reaction optimization alternatives. In spite of low isolated yields, this transformation remains a useful method of generating complex polycyclic compounds and represents a unique example of a high valent palladium–mediated carbon–carbon bond forming oxidative dearomatization reaction.14
Table 1.
Optimization for Oxidative Dearomatization
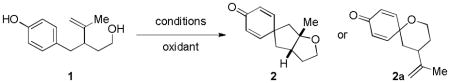 | ||
|---|---|---|
| entry | conditions | yield of 2a |
| 1 | Pd(OAc)2 (10 mol %), PhI(OAc)2 (2.0 equiv) PhMe, rt, 12 h | 0% |
| 2 | Pd(OAc)2 (10 mol %), PhI(OAc)2 (2.0 equiv) CH3CN, rt, 12 h | 5–10% |
| 3 | Pd(OAc)2 (10 mol %), PhI=O, (3.0 equiv) CH3CN, rt, 12 h | N. R. |
| 4 | Pd(OAc)2 (10 mol %), Selectfluor (2.0 equiv) CH3CN, rt, 12 h | 43% |
| 5 | PhI(OAc)2 (1.1 equiv), CH3CN | 0%b |
| 6 | PhI(O2CCF3)2 (1.1 equiv), (F3C)2CHOH | 0%b |
Isolated yield (%) after purification by chromatography on SiO2;
2a was the only isolable product.
Figure 3.
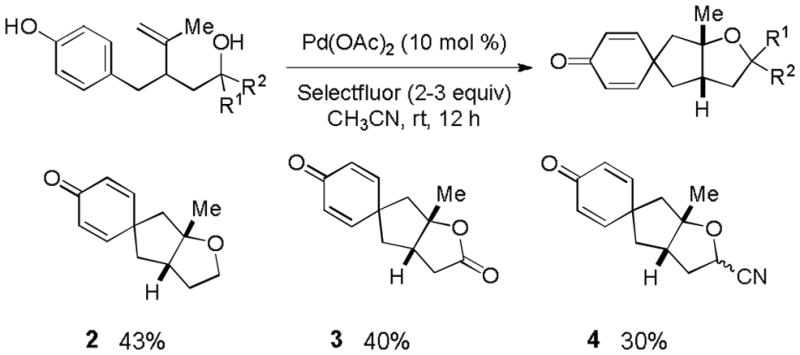
Examples of Wacker Cyclization/Dearomatization
We subsequently explored the effects of different substituents on the arene moiety. In the event, we decided to investigate substrates with pendant carboxylic acids due to their known ability to participate in nucleopalladation processes. Thus, upon exposure of 7 to our reaction conditions, we isolated the desired adduct 8a in only 20% yield. Further analysis of the reaction mixture revealed C–H insertion product 8 to be the major product (isolated in 68% yield). Intrigued by the crossover between oxidative dearomatization and C–H functionalization, we sought to explore the possibility of utilizing the ω-unsaturated carboxylic acids for oxidative C–H functionalization reactions. We sought to favor this transformation by varying the substitution pattern and electronic properties of the arene and to suppress the formation of dearomatization products.15,16
Upon screening oxidants and palladium complexes to promote C–H functionalization (not shown), it was found that PhI(OAc)2 (2 equiv) and Pd(OAc)2 (10 mol %) in CH3CN provided optimal conditions for this transformation. These reactions were unsucessful when performed under traditional Pd0/II wacker oxidation conditions (stoichiometric and catalytic). Applying these conditions to substrate 5 afforded tricyclic compound 6 in excellent yield (92%) with a 1.75:1 diastereomeric ratio (Table 2, Entry 1). High conversions were observed for electron rich and electron neutral substrates providing the tricyclic products in good to moderate yields (Table 1). Substrates that formed smaller fused ring systems (entries 3–9) showed a consistent decline in yield compared with entries 1 and 2. Further analysis of the reaction mixture obtained upon conversion of 11 to 12 (Table 2, entry 4) indicated that the attenuated yield was due to the competitive formation of pyranone (24) and olefin acetoxylation (23) side-products. Prevention of these side-products proved challenging, however they provided some mechanistic insight for this reaction (vide infra).
Table 2.
Substrate Scope for C–H Functionalization
| entry | substrate | product | yieldb |
|---|---|---|---|
| 1 |
 5 |
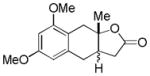 6 |
92%c |
| 2 |
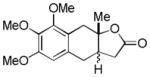 7 |
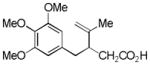 8 |
68%d,e |
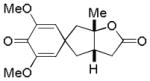 8a |
20% | ||
| 3 |
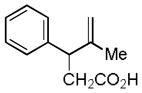 9 |
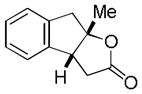 10 |
60% |
| 4 |
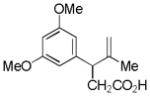 11 |
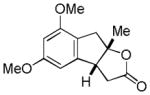 12 |
61% |
| 5 |
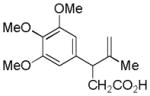 13 |
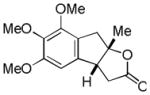 14 |
53% |
| 6 |
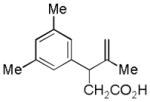 15 |
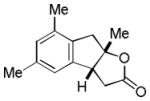 16 |
57% |
| 7 |
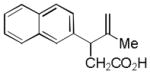 17 |
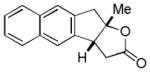 18 |
56% |
| 8 |
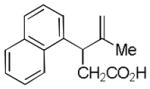 19 |
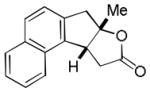 20 |
51% |
| 9 |
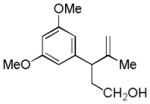 21 |
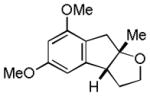 22 |
46% |
Reaction Conditions: Pd(OAc)2 (10 mol %), PhI(OAc)2 (2.0 equiv), CH3CN, rt, 12 h;
Isolated yield (%) after purification by chromatography on SiO2;
dr 1.75:1;
dr 1.3:1;
see ref 21
A plausible catalytic cycle is outlined in Figure 4. Beginning with the C–H functionalization pathway (Section I, Path A), PdII coordinates to the olefin of the substrate and undergoes an oxypalladation to form a Wacker intermediate. A PdII C–H metallation, followed by oxidation by PhI(OAc)2 provides a palladacycle that undergoes reductive elimination, producing the C–H insertion product.17 For phenols, the catalytic cycle proceeds through a dearomatization pathway (Path B). In turn, the Wacker intermediate undergoes oxidation by Selectfluor, generating the highly electrophilic alkylpalladiumIV intermediate. This undergoes a direct reductive nucleophilic substitution by the phenol to form the spirocyclohexadienone product.
Figure 4.

Proposed Mechanisms for Difunctionalization Cascade and Degradation Pathways
With the proposed mechanistic hypothesis established, we began to formulate an explanation for the unavoidable formation of the olefin acetoxylation (23) and pyranone (24) side-products (Figure 4, Section II) Using substrate 11 as a representative example, we hypothesized that 23 and 24 were generated as a consequence of the relative configuration of the Wacker intermediate formed during the initial nucleopalladation step, which can either result in a trans or cis substituted lactone (intermediates A and B, Figure 3, Section II). Since the subsequent reductive elimination of B would yield a highly strained trans-fused [3.3.0] bicyclic system (compound C),18 intermediate B can undergo a Meerwein-Wagner shift19,20 to form an oxocarbenium intermediate with concomitant trapping by acetate (i.e. 22). As an alternative pathway, intermediate B can also undergo acetoxylation to form acetate 23.
To further support our mechanistic hypothesis, we, exposed acid 11 to our reaction conditions, using the deficient oxidant PhI(O2CCF3)2 as the stoichiometric oxidant. As a result, the trifluoroacetate derivative of pyranone 24 was isolated as the major product in 30% yield along with trace amounts of the trifluoroacetate derivative of 23 was also observed. We speculate that the poor level of substrate–directed diastereoselectivity in the nucleopalladation step is the most likely explanation for the low overall yield of the reaction (Table 2, entries 3–9), which is consistent with the observed side-product.
In conclusion, we have developed a mild, carbon– carbon bond forming olefin difunctionalization methodology employing a PdII/IVcatalytic cycle. In addition, phenolic substrates underwent oxidative dearomatization in moderate yields, an unprecedented transformation in high valent palladium catalysis. Further investigations on the mechanism, scope and applications of this reaction, especially in context of the oxidative dearomatization, are currently being explored.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. David Freeman (Boston University) for helpful suggestions and comments on this manuscript. Financial support for this research from the NIH-NIGMS (R01-GM096129), Alfred P. Sloan Foundation, Amgen, Boehringer Ingelheim, and Boston University is gratefully acknowledged. GJK thanks the Boston University Undergraduate Research Program for research support. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the NSF.
Footnotes
Supporting Information Available Experimental procedures, 1H and 13C NMR spectra for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (b) Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (c) Desai LV, Sanford MS. Angew Chem Int Ed. 2007;46:5737. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]
- 2.(a) Li Y, Song D, Dong VM. J Am Chem Soc. 2008;130:2962. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]; (b) Wang A, Jiang H, Chen H. J Am Chem Soc. 2009;131:3846. doi: 10.1021/ja900213d. [DOI] [PubMed] [Google Scholar]
- 3.(a) Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (b) Muñiz K. J Am Chem Soc. 2007;129:14542. doi: 10.1021/ja075655f. [DOI] [PubMed] [Google Scholar]; (c) Iglesias Á, Pérez EG, Muñiz K. Angew Chem Int Ed. 2010;49:8109. doi: 10.1002/anie.201003653. [DOI] [PubMed] [Google Scholar]
- 4.(a) Podhajsky SM, Sigman MS. Organometallics. 2007;26:5680. doi: 10.1021/om700675z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Sigman MS. J Am Chem Soc. 2007;129:3076. doi: 10.1021/ja070263u. [DOI] [PubMed] [Google Scholar]; (c) Tong X, Beller M, Tse MK. J Am Chem Soc. 2007;129:4906. doi: 10.1021/ja070919j. [DOI] [PubMed] [Google Scholar]; (d) Welbes LL, Lyons TW, Cychosz KA, Sanford MS. J Am Chem Soc. 2007;129:5836. doi: 10.1021/ja071204j. [DOI] [PubMed] [Google Scholar]; (e) Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870. doi: 10.1021/ja103472a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nicolai S, Erard S, Fernández-Gonzáles D, Waser J. Org Lett. 2010;12:384. doi: 10.1021/ol9027286. [DOI] [PubMed] [Google Scholar]; (g) Yeung CS, Zhao X, Borduas N, Dong VM. Chem Sci. 2010;1:331. [Google Scholar]; (h) Jaegli S, Dufour J, Wei HL, Piou T, Duan XH, Vors JP, Neuville L, Zhu J. Org Lett. 2010;12:4498. doi: 10.1021/ol101778c. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hegedus LS, Allen GF, Waterman EL. J Am Chem Soc. 1976;98:2674. [Google Scholar]; (b) Hayashi T, Hegedus LS. J Am Chem Soc. 1977;99:7093. [Google Scholar]; (c) Hegedus LS, Allen GF, Bozell JJ, Waterman EL. J Am Chem Soc. 1978;100:5800. [Google Scholar]; (d) Semmelhack MF, Bodurow C. J Am Chem Soc. 1984;106:1496. [Google Scholar]; (e) Semmelhack MF, Zhang N. J Org Chem. 1989;54:4483. [Google Scholar]; (f) Semmelhack MF, Epa WR. Tetrahedron Lett. 1993;34:7205. [Google Scholar]; (g) Wolfe JP, Rossi MA. J Am Chem Soc. 2004;126:1620. doi: 10.1021/ja0394838. [DOI] [PubMed] [Google Scholar]; (h) Lira R, Wolfe JP. J Am Chem Soc. 2004;126:13906. doi: 10.1021/ja0460920. [DOI] [PubMed] [Google Scholar]; (i) Leathen ML, Rosen BR, Wolfe JP. J Org Chem. 2009;74:5107. doi: 10.1021/jo9007223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Rosen BR, Ney JE, Wolfe JP. J Org Chem. 2010;75:2756. doi: 10.1021/jo100344k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Bloome KS, Alexanian EJ. J Am Chem Soc. 2010;132:12823. doi: 10.1021/ja1053913. [DOI] [PubMed] [Google Scholar]
- 6.For recent reviews on high valent palladium catalysis see: Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083. doi: 10.1039/b813246a.Muñiz K. Angew Chem Int Ed. 2009;48:9412. doi: 10.1002/anie.200903671.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Sehnal P, Taylor RJK, Fairlamb IJS. Chem Rev. 2010;110:824. doi: 10.1021/cr9003242.Xu LM, Li BJ, Yang Z, Shi ZJ. Chem Soc Rev. 2010;39:712. doi: 10.1039/b809912j.
- 7.Nicolai S, Piemontesi C, Waser J. Angew Chem Int Ed. 2011;50:4680. doi: 10.1002/anie.201100718. [DOI] [PubMed] [Google Scholar]
- 8.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. and references therein. [DOI] [PubMed] [Google Scholar]
- 9.Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. and references therein. [DOI] [PubMed] [Google Scholar]
- 10.(a) Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659. [DOI] [PubMed] [Google Scholar]; (b) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- 11.(a) Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Alloco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barret J, Schmatz D, Becker JW, Cully D, Singh SB. Nature. 2006;441:358. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]; (b) Saleem M, Hussain H, Ahmed I, van Ree T, Krohn K. Nat Prod Rep. 2011;28:1534. doi: 10.1039/c1np00010a. [DOI] [PubMed] [Google Scholar]
- 12.For oxidative cyclizations of phenols: Morrow GW, Swenton JS. Tetrahedron Lett. 1987;28:5445.Tamura Y, Yakura T, Haruta J, Kita Y. J Org Chem. 1987;52:3927.Callinan A, Chen Y, Morrow GW, Swenton JS. Tetrahedron Lett. 1990;31:4551.Kita Y, Tohma H, Kikuchi K, Inagaki M, Yakura T. J Org Chem. 1991;56:435.Dohi T, Maruyama A, Minamitsuji Y, Takenaga N, Kita Y. Chem Commun. 2007:1224. doi: 10.1039/b616510a.Dohi T, Maruyama A, Takenaga N, Senami K, Minamitsuji Y, Fujioka H, Caemmerer SB, Kita Y. Angew Chem Int Ed. 2008;47:3787. doi: 10.1002/anie.200800464.
- 13.For carbon-carbon bond forming dearomatization reactions, see: Moriarty RM, Prakash O, Duncan MP, Vaid RK, Musallam HA. J Org Chem. 1987;52:150.Kita Y, Tohma H, Inagaki M, Hatanaka K, Yakura T. J Am Chem Soc. 1992;114:2175.Ley SV, Schucht O, Thomas AW, Murray JP. J Chem Soc, Perkin Trans 1. 1999:1251.Zhang X, Larock RC. J Am Chem Soc. 2005;127:12230. doi: 10.1021/ja053079m.Lalic G, Corey EJ. Org Lett. 2007;9:4921. doi: 10.1021/ol702323s.Andrez JC, Giroux MA, Lucien J, Canesi S. Org Lett. 2010;12:4368. doi: 10.1021/ol101851z.Desjardins S, Andrez JS, Canesi S. Org Lett. 2011;13:3406. doi: 10.1021/ol201149u.
- 14.For palladium mediated arene dearomatization reactions, see: Nemoto T, Ishige Y, Yoshida M, Kohno Y, Kanematsu M, Hamada Y. Org Lett. 2010;12:5020. doi: 10.1021/ol102190s.Rousseaux S, Garciá-Fortanet J, Angel Del Aguila Sanchez M, Buchwald SL. J Am Chem Soc. 2011;133:9282. doi: 10.1021/ja203644q.
- 15.(a) Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (b) Furuya T, Kaiser HM, Ritter T. Angew Chem Int Ed. 2008;47:5993. doi: 10.1002/anie.200802164. [DOI] [PubMed] [Google Scholar]; (c) Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]
- 16.Engle KM, Mei TS, Wang X, Yu JQ. Angew Chem Int Ed. 2011;50:1478. doi: 10.1002/anie.201005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The oxidation of the alkylpalladium complex may occur prior to C–H metallation. Control experiments on substrate 5 revealed the necessity of a high valent palladium cycle. Stoichiometric Pd(OAc)2 or catalytic variants using CuII salts failed to produce any of 6. For an example of a PdIV mediated C-H metallation see: Racowski JM, Ball ND, Sanford MS. J Am Chem Soc. doi: 10.1021/ja2051099.
- 18.Anslyn EV, Dougherty DA. In: Modern Physical Organic Chemistry. Murdzek John., editor. University Science Books; California: 2006. p. 111. [Google Scholar]
- 19.(a) Boontanonda P, Grigg R. J Chem Soc, Chem Commun. 1977:583. [Google Scholar]; (b) Clark GR, Thiensathit S. Tetrahedron Lett. 1985;26:2503. [Google Scholar]
- 20.This byproduct may also be the result of β-carbon elimination/endo cyclization. For examples of β-carbon elimination, see: Nishimura T, Ohe K, Uemura S. J Am Chem Soc. 1999;121:2645.Nishimura T, Uemura S. J Am Chem Soc. 1999;121:11010.For a review on the subject see: Leemans K, D’hooge M, De Kimpe N. Chem Rev. 2011;111:3268. doi: 10.1021/cr100295j.
- 21.When Selectfluor was used as the stoichiometric oxidant, there was extensive decomposition of the starting material
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.