

Abstract
In this selective review, we consider a number of unsolved questions regarding the glycogen storage diseases (GSD). Thus, the pathogenesis of Pompe disease (GSD II) is not simply explained by excessive intralysosomal glycogen storage and may relate to a more general dysfunction of autophagy. It is not clear why debrancher deficiency (GSD III) causes fixed myopathy rather than exercise intolerance, unless this is due to the frequent accompanying neuropathy. The infantile neuromuscular presentation of branching enzyme deficiency (GSD IV) is underdiagnosed and is finally getting the attention it deserves. On the other hand, the late-onset variant of GSD IV (adult polyglucosan body disease APBD) is one of several polyglucosan disorders (including Lafora disease) due to different etiologies. We still do not understand the clinical heterogeneity of McArdle disease (GSD V) or the molecular basis of the rare fatal infantile form. Similarly, the multisystemic infantile presentation of phosphofructokinase deficiency (GSD VII) is a conundrum. We observed an interesting association between phosphoglycerate kinase deficiency (GSD IX) and juvenile Parkinsonism, which is probably causal rather than casual. Also unexplained is the frequent and apparently specific association of phosphoglycerate mutase deficiency (GSD X) and tubular aggregates. By paying more attention to problems than to progress, we aimed to look to the future rather than to the past.
Key words: glycogen storage diseases, GSD, polyglucosan disorders
Why writing of glycogen storage diseases (GSD), which are "old hats" among the metabolic disorders affecting skeletal muscle? Why writing of GSD to honor Valerie Askanas and King Engel? Why writing with Ronen Spiegel? The answer to the first question is deeply personal: the very first paper of the senior (chronologically, not academically) author (SDM) described a little girl with Pompe disease (1) and his first project as a postdoctoral fellow with Dr. Lewis P. (Bud) Rowland was to unravel why glycogen accumulation is not limitless in muscle (2). The answer to the second question is an exciting, if ancient, collaboration showing that both morphological and biochemical features of Pompe disease were reproduced in muscle culture (3). The answer to the third question is a much more recent collaboration on a patient who had phosphoglycerate kinase (PGK) deficiency (GSD IX) and a pure myopathy, or so we thought initially (see below) (4).
The truth is that GSD are still very much an open chapter, where new entities are discovered (5, 6), apparently paradoxical myopathies due to lack, rather than excess, of glycogen (aglycogenosis or GSD 0) are being reported (7, 8), old disorders, such as Lafora disease, are now recognized as GSD (9), and therapy based on enzyme replacement is reasonably successful in GSD II (10, 11).
This is not meant to be a comprehensive review of the muscle glycogenoses. Rather, we will consider puzzling aspects of some GSD, following the Roman numerical order shown in Figure 1.
Figure 1.
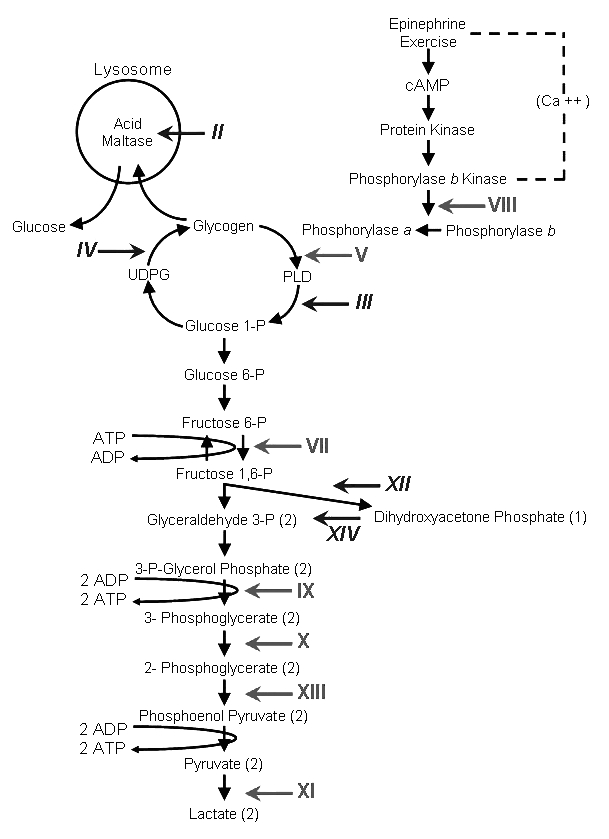
Scheme of glycogen metabolism and glycolysis. Roman numerals denote muscle glycogenoses due to defects in the following enzymes: II, acid α-glucosidase (AAG); III, debrancher; IV, brancher; V, myophosphorylase; VI, liver phosphorylase; VII, muscle phosphofructokinase (PFK); VIII, phosphorylase b kinase (PHK); IX, phosphoglycerate kinase (PGK); X, phosphoglycerate mutase (PGAM); XI, lactate dehydrogenase (LDH); XII, aldolase; XIII, β-enolase; IV, triosephosphate isomerase (TPI). Symbols in regular typeface denote glycogenoses characterized by exercise intolerance, cramps, and myoglobinuria. Symbols in italics denote glycogenoses characterized by fixed weakness.
GSD II (acid maltase deficiency, Pompe disease)
The first and oldest conundrum about this disorder was its clinical heterogeneity, with a severe generalized infantile form and a later-onset form largely confined to skeletal muscle and presenting in children (juvenile onset) or in adults (late-onset). As acid maltase (acid α-glucosidase, GAA) is a single ubiquitous protein, it is not surprising that initial findings of different residual GAA activities in muscle (12) have been confirmed and related to the severity of GAA mutations, with nonsense mutations prevailing in the infantile form and missense and splicing (i.e. "leaky") mutations prevailing in later onset cases (13, 14).
A second riddle regarded the pathogenesis of weakness: was it simply due to the mechanical disarray of the contractile material caused by the glycogen-laden lysosomes or to an energy defect? The former mechanistic hypothesis is questionable both because it does not explain the weakness of later onset GSD II and because equally disruptive overloads of lipid droplets in muscle were compatible with normal strength in a preclinical case of neutral lipid storage disease with myopathy (NLSDM) (15). The latter energetic hypothesis is supported by the notion that GAA activity normally "returns" to the cytoplasm the end product of lysosomal glycogen digestion, glucose. It is also telling that in muscle from patients with the infantile form of GSD II most glycogen is free in the cytoplasm, probably released by "burst lysosomes" (13).
A more compelling scenario for the pathogenesis of GSD II, as well as other lysosomal storage disorders (16), involves a disruption of the vital autophagic process, with accumulation of autophagosomes resulting from defective autophagosome-lysosome fusion (16, 17). In fact, Nishino and colleagues went as far as stating that "Pompe disease can no longer be viewed simply as a glycogen storage disease," but rather as a problem in handling excessive numbers of autophagosomes (13).
A unique feature of GSD II is the availability of a generally effective – and now widely utilized – enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA). There is already a vast literature on the subject (10), and a few problems have emerged, such as the immune reaction to rhGAA in infants with null mutations and no GAA protein (i.e. no cross-reactive immunological material, CRIM) (18). Assumption of rhGAA into lysosomes is probably hindered by the more general autophagic dysfunction mentioned above and several stratagems have been proposed to improve uptake, including conjugation of rhGAA with a synthetic oligosaccharide harboring mannose-6-phosphate (19), combining ERT with chaperones (20), and inhibition of glycogen synthesis (21, 22).
One final clinical note: besides skeletal muscle, smooth muscle must also be affected in late-onset GSD II because there have been a few reports of cerebral arteriopathies, often affecting the basilar artery (23, 24).
GSD III (debrancher enzyme deficiency; Cori-Forbes disease)
The debrancher is a "double duty" enzyme, with two catalytic functions, oligo-1,4-1,4-glucantransferase and amylo-1,6-glucosidase. Once phosphorylase has shortened the peripheral chains of glycogen to about four glucosyl units (this partially digested glycogen is called phosphorylase-limit dextrin, PLD), the debrancher enzyme removes the residual stumps in two steps. First, a maltotriosyl unit is transferred from a donor to an acceptor chain (transferase activity), leaving behind a single glucosyl unit, which is then hydrolyzed by the amylo-1- ,6-glucosidase, at which point the branch is off. A singlecopy gene, AGL, encodes the debrancher enzyme.
A comprehensive summary of clinical features and therapeutic options has been published recently (25).
Considering the predominantly myopathic presentation of GSD III, a clinician would likely question why the defect of an enzyme that acts hand-in-hand with myophosphorylase should cause weakness rather than cramps and myoglobinuria, the clinical hallmarks of McArdle disease. One reason for this discrepancy may be that in McArdle disease glycogen cannot be metabolized at all, whereas in GSD III the peripheral chains of normal glycogen can be utilized. However, this explanation postulates that the intact glycogenosynthetic pathway allows some turnover between normal glycogen and PLD, which is not unreasonable.
Another explanation for the fixed and mostly distal weakness of patients with GSD III (26) is the simultaneous involvement of muscle and nerve, as documented both electrophysiologically and by nerve biopsy (27, 28).
GSD IV (branching enzyme deficiency, Andersen disease)
The glycogen branching enzyme (GBE) is a single polypeptide encoded by one gene (GBE1). GBE deficiency results in the deposit of an amylopectin-like polysaccharide that has fewer branching points and longer outer chains than normal glycogen and is known as polyglucosan. Polyglucosan is periodate/Schiff (PAS)-positive and only partially digested by diastase, which makes it easily recognizable in various tissues and offers an important clue to the correct diagnosis.
It is gratifying to see that in the just published 22nd edition of Rudolph's Pediatrics, the neuromuscular presentation of GSD IV is given as much space as the hepatic form (29), which dominated previous textbook descriptions.
In fact, the neuromuscular presentation has been underdiagnosed, judging from the flurry of recent papers. As recognized in a seminal paper of 2004 (30), there are two main infantile presentations. The first is a perinatal disorder known as "fetal akinesia deformation sequence" or FADS, characterized by multiple congenital contractures (arthrogryposis multiplex congenita), hydrops fetalis, pulmonary hypoplasia, craniofacial abnormalities, intrauterine retarded growth (IURG), abnormal amniotic fluid volume, and perinatal death. The second, labeled "congenital," should probably be called "fatal infantile," as it presents at or soon after birth with hypotonia, muscle wasting, neuronal involvement, inconsistent cardiomyopathy, and early death.
Detailed neuropathology in a girl who died at 3 months showed PAS-positive polyglucosan inclusions in neurons of basal ganglia and thalamus, oculomotor and pontine nuclei, and in periaqueductal neurons (31). In the medulla, polyglucosan deposits were noted in the hypoglossal nucleus, the dorsal motor nucleus of the vagus, and the nucleus ambiguus. Similar findings were reported in two more infants (32, 33). The motor neurons of the spinal cord are also severely affected (32), explaining how one of the patients we studied was initially diagnosed as spinal muscular atrophy type I (SMA I) until mutations in the SMN1 gene were ruled out (34).
At the other end of the clinical spectrum, there is adult polyglucosan body disease (APBD), a neurological variant of GSD IV presenting late in life with progressive upper and lower motor neuron dysfunction (simulating amyotrophic lateral sclerosis), sensory neuropathy, neurogenic bladder (APBD patients often see a urologist before they see a neurologist), and – in about 50% of patients – dementia. The disease predominates among people of Askenazi Jewish descent (probably due to a founder effect) and is most commonly due to the Y329S mutation in GBE1. Not too surprisingly, this is a "mild" mutation, which probably explains the late onset of symptoms. Thanks to the energy and compassion of one patient, Gregory Weiss, a research foundation (APBDRF; www.apbdrf.org) has been created to develop therapeutic strategies.
GSD V (myophosphorylase deficiency, McArdle disease)
The clinical picture and the block in muscle glycogen breakdown were elegantly described by Brian McArdle in 1951 (35), the enzyme defects was discovered 8 years later (36-38), and it took 12 more years before the first mutations in PYGM were identified (39).
Despite its long history, McArdle disease still presents several riddles. First, although it was long considered a clinically homogeneous disease, its expression can vary from relatively mild exercise intolerance to a crippling condition with frequent cramps and recurrent episodes of myoglobinuria. Explanations have ranged from rare cases of "double trouble" (i.e. the coexistence in the same individuals of one mutation in PYGM and another in the gene encoding adenylate deaminase) to the association with insertion/deletion polymorphisms in the angiotensinconverting enzyme (ACE) (40). Probably more important is the protecting effect of even small amounts of myophosphorylase residual activity, which is determined by the type of mutations: for example, splice mutations are associated to milder clinical phenotypes (41).
What remains unexplained is the fatal infantile form of McArdle disease, which has been reported in a handful of cases (42-45). In these unfortunate infants muscle morphology, biochemistry, and molecular genetics [showing the "common" R50X null mutation (39, 45)] are no different from typical McArdle patients, despite the dismal outcome.
GSD VII (phosphofructokinase [PFK] deficiency, Tarui disease)
PFK is a tetrameric enzyme under the control of three autosomal genes: PFKM encodes the muscle subunit, PFKL encodes the liver subunit, and PFKP encodes the platelet subunit. Mature human muscle expresses only the M subunit and contains exclusively the M4 homotetramer, whereas erythrocytes, which express both the M and the L subunit, contain five isozymes, the two M4 and L4 homotetramers and three hybrid forms. In patients with typical PFK deficiency, mutations in PFKM cause total lack of activity in muscle but only partial deficiency in red blood cells.
Clinically, PFK deficiency, first described in 1965 in a Japanese family (46), is indistinguishable from McArdle disease, except for the absence of a typical second wind phenomenon and for the occasional presence of gouty arthritis due to "myogenic hyperuricemia" (47).
Two peculiar aspects of GSD VII are worth discussing: the presence of polyglucosan in muscle and the severe infantile presentation.
The presence – in addition to normal-looking glycogen – also of abnormal glycogen with the histochemical (diastase-resistance) and ultrastructural (fine granules and filaments instead of β-particles) features of polyglucosan was first noted in the muscle biopsy of two patients (48) and confirmed in a woman who had developed late-onset fixed weakness (49). We reasoned that this surprising finding could best be explained by the excessive accumulation of glucose-6-phosphate (G6P) upstream of the glycolytic block (49). As G6P is a functional activator of glycogen synthetase (GS), the finely balanced activity ratio of GS and GBE would be tilted in favor of GS and result in a polysaccharide with abnormally long and poorly branched chains, i.e. polyglucosan.
This pathogenic concept was confirmed by two experiments, one in the laboratory, the other an experiment of nature. First, when Nina Raben upregulated the expression of GS in the muscle of GAA-deficient mice, she unexpectedly obtained polyglucosan accumulation (50). Second, after a long search for the molecular basis of polyglucosan myopathy in horses, Stephanie Valberg and co-workers identified a gain-of-function mutation in GS, again altering the GS/GBE activity ratio in favor of GS (5).
The second riddle concerns the fatal infantile variant of GSD VII, reported in a dozen patients between 1987 and 2008. All infants were severely hypotonic at birth and a few developed joint contractures either in utero (51-53) or postnatally (54, 55). Decreased fetal movements were noted in two pregnancies (52, 53) and polyhydramnios in one (53). In all but two cases (53, 55), death occurred in infancy or early childhood due to pulmonary failure.
Most children showed evidence of multisystem involvent, including seizures, cortical blindness, developmental delay, dysmorphic features, and corneal ulcers. The encephalopathy was documented by neuroradiology or neuropathology, which showed dilated ventricles and cortical or cerebellar atrophy (51, 54-57).
Because of the early onset, multisystem involvement, and lack of any molecular evidence of mutations in the PFKM gene, the infantile variant of phosphofructokinase deficiency appears to be a separate entity from GSD VII, and its genetic basis (or bases) remain to be clarified, despite evidence that a transgenic PFKM-null mouse mimics the infantile more than the typical muscular form of the human disease (58).
GSD VIII (Phosphorylase b kinase [PHK] deficiency)
PHK is a multimeric enzyme composed of four different subunits, α, β, γ, and δ and the enzyme composition is (αβγδ)4. The γ subunit is catalytic and is regulated by the degree of phosphorylation of the α and β subunits. Calcium sensitivity is conferred by the δ subunit, which is tightly bound to calmodulin.
PHK deficiency has been associated with five main syndromes distinguished by inheritance and by tissue involvement: (i) a benign X-linked recessive hepatopathy of infancy or childhood (59); (ii) an autosomal recessive liver and muscle disease (60); (iii) a pure myopathy predominant in men (61); (iv) an autosomal recessive severe liver disease with cirrhosis (62); and (v) a fetal infantile cardiopathy, reported in a handful of patients (63-68).
The pure myopathy has thus far been described in detail only in men and is due to mutations in the X-linked gene (PHKA1) encoding the muscle-specific α subunit (69-73). Not surprisingly, patients with PHK deficiency have a clinical picture resembling McArdle disease, except much milder, a sort of "McArdle light." For example, patients usually have normal venous lactate rise after forearm ischemic exercise, no evidence of second wind, and modest accumulation of glycogen in the muscle biopsy. Formal cycle ergometry studies confirmed the mild impairment of glycogenolysis: there was no change in lactate during dynamic, submaximal exercise and IV glucose administration improved exercise tolerance, but less than in McArdle patients (72).
The molecular basis underlying the fatal infantile cardiomyopathy has been a puzzle for many years because there is no heart-specific PHK isozyme. The riddle was solved when Burwinkel et al. definitely excluded mutations in any of the PHK genes (74) but detected a single dominant mutation in the gene (PRKAG2) encoding the γ2 subunit of the AMP-activated protein kinase (AMPK). Later, we identified a second mutation in another infant (75). AMPK is an αβγ heterotrimer functioning as a "cellular fuel gauge," which is switched on by increases in the AMP:ATP ratio, an indicator of cellular energy deficit (76).
What remains a mystery is why mutations in AMPK should inhibit PHK and cause a "pseudo-PHK deficiency." We suspect that a similar mechanism may operate in the fatal infantile PFK deficiency that we discussed above.
GSD IX (phosphoglycerate kinase [PGK] deficiency)
PGK is a single polypeptide encoded by a gene (PGK1) on Xq13 and present in all tissues except spermatogenic cells. Although this enzyme is virtually ubiquitous, clinical presentations depend on the isolated or combined involvement of three tissues: erythrocytes (hemolytic anemia), skeletal muscle (exercise intolerance, cramps, myoglobinuria), and the central nervous system ([CNS] seizures, mental retardation, stroke).
In a recent review (4), we found that the most common association, seen in 11 of 33 patients (34%) was hemolytic anemia and CNS involvement. Isolated myopathy was a close second (9 of 33 patients, 27%). Isolated blood dyscrasia was reported in 6 patients (18%), the association of myopathy and CNS dysfunction in 4 patients (12%), the association of anemia and myopathy only in one patient (3%), and the involvement of all three tissues in 2 patients (6%).
However, the plot thickened when we found yet another patient with the association of myopathy and a peculiar CNS dysfunction, namely, severe juvenile Parkinsonism (77). What we found strange and a little disconcerting was that this young man harbored the same previously unreported mutation (p.T378P) that we had identified in our latest patient with pure myopathy (4). However, we recovered some confidence in genotype:phenotype correlation when Dr. Spiegel's patient also developed severe Parkinsonian symptoms and signs. Although this is an n of 2 series, our findings raise two interesting questions. First, is there, in fact, a causal relationship between the T378P mutation and Parkinsonism? Second, PGK deficiency was suspected in both patients because they presented initially with exercise intolerance, cramps, and myoglobinuria: Parkinsonism was a surprising clinical development. One cannot help wondering whether in some patients with PGK deficiency juvenile Parkinson disease may precede and overshadow the myopathy, thus escaping diagnosis. Certainly, this association has to be kept in mind.
GSD X (phosphoglycerate mutase [PGAM] deficiency)
PGAM is a dimeric enzyme composed of a musclespecific (M) subunit and a brain-specific (B) subunit. Normal adult human muscle contains predominantly the MM homodimer, which accounts for about 95% of the total activity.
Fourteen patients with PGAM deficiency in muscle have been reported, of whom nine were African American (78, 79). Although the first reported patient could not be studied at the molecular level (80), all other African American patients harbored the W78X mutation, at least in heterozygosity, suggesting a founder effect.
The most striking peculiarity of GSD X is its common association with tubular aggregates (TAs), which were seen in the muscle biopsies of 5 patients (36%) whereas they have never been reported in other glycogenoses. TAs are ordered stacks of tubules originating from the sarcoplasmic reticulum. Although they are a nonspecific pathological change seen in diverse conditions, including exposure to drugs, toxins, and hypoxia, their association with PGAM deficiency does not appear to be casual although the specific trigger remains unknown.
Conclusions
As stated at the outset, we did not intend to review all the muscle glycogenoses, but only to consider some conundrums still presented by "old" GSD. We have not considered Lafora disease because muscle involvement is overshadowed by the devastating encephalopathy. Likewise, we have not discussed some recently described glycogenoses, such as aldolase deficiency (81), β-enolase deficiency (82), and the two forms of glycogenosis type 0 (aglycogenosis?) (7, 8) because they have been described in single patients.
Thus, although we discussed more the problems than the progress promised in the title, we hope our considerations are an adequate homage to Valerie Askanas and W. King Engel.
References
- 1.Favini Sacerdoti F, DiMauro S, Angelini Dobrilla C. Un caso di glicogenosi tipo II (m. di Pompe). Studio clinico, morfologico e biochimico. Acta Paediatr Latina. 1967;20:651–677. [Google Scholar]
- 2.DiMauro S, Rowland LP, DiMauro PM. Control of glycogen metabolism in human muscle. Arch Neurol. 1970;23:534–540. doi: 10.1001/archneur.1970.00480300056007. [DOI] [PubMed] [Google Scholar]
- 3.Askanas V, Engel WK, DiMauro S, et al. Adult-onset acid maltase deficiency. Morphological and biochemical abnormalities reproduced in cultured muscle. New Engl J Med. 1976;294:573–578. doi: 10.1056/NEJM197603112941102. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel R, Area Gomez E, Akman HO, et al. Myopathic form of phosphoglycerate kinase (PGK) deficiency: A new case and pathogenic considerations. Neuromusc Disord. 2009;19:207–211. doi: 10.1016/j.nmd.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 5.McCue ME, Valberg SJ, Miller MB, et al. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91:458–466. doi: 10.1016/j.ygeno.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stojkovic T, Vissing J, Petit F, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. New Engl J Med. 2009;361:425–427. doi: 10.1056/NEJMc0901158. [DOI] [PubMed] [Google Scholar]
- 7.Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. New Engl J Med. 2007;357:1507–1514. doi: 10.1056/NEJMoa066691. [DOI] [PubMed] [Google Scholar]
- 8.Moslemi A-R, Lindberg C, Nilsson J, et al. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. New Engl J Med. 2010;362:1203–1210. doi: 10.1056/NEJMoa0900661. [DOI] [PubMed] [Google Scholar]
- 9.Turnbull J, Wang P, Girard J-M, et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann Neurol. 2010;68:925–933. doi: 10.1002/ana.22156. [DOI] [PubMed] [Google Scholar]
- 10.Ploeg AT. Where do we stand in enzyme replacement therapy in Pompe's disease? Neuromusc Dis. 2010;20:733–734. doi: 10.1016/j.nmd.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe's disease. New Engl J Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 12.Mehler M, DiMauro S. Residual acid maltase activity in late-onset acid maltase deficiency. Neurology. 1977;27:178–184. doi: 10.1212/wnl.27.2.178. [DOI] [PubMed] [Google Scholar]
- 13.Malicdan MC, Noguchi S, Nonaka I, et al. Lysosomal myopathies: An excessive build-up in autophagosomes is too much to handle. Neuromusc Disord. 2008;18:521–529. doi: 10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Nascimbeni AC, Fanin M, Tasca E, et al. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology. 2008;70:617–626. doi: 10.1212/01.wnl.0000299892.81127.8e. [DOI] [PubMed] [Google Scholar]
- 15.Akman HO, Davidzon G, Tanji K, et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromusc Dis. 2010;20:397–402. doi: 10.1016/j.nmd.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda T, Ewan L, Bauer M, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 18.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reacting immunological material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Jiang J-L, Gumlaw NK, et al. Glycoengineered acid alphaglucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther. 2009;17:954–963. doi: 10.1038/mt.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Douillard-Guilloux G, Raben N, Takikita S, et al. Restauration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum Mol Genet. 2010;19:684–696. doi: 10.1093/hmg/ddp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashe KM, Taylor KM, Chu Q, et al. Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Mol Genet Metab. 2010;100:309–315. doi: 10.1016/j.ymgme.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Laforet P, Petiot P, Nicolino M, et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology. 2008;70:2063–2066. doi: 10.1212/01.wnl.0000313367.09469.13. [DOI] [PubMed] [Google Scholar]
- 24.Sacconi S, Bocquet JD, Chanalet S, et al. Abnormalities of cerebral arteries are frequent in patients with late-onset Pompe disease. J. Neurol. 2010;257:1730–1733. doi: 10.1007/s00415-010-5618-0. [DOI] [PubMed] [Google Scholar]
- 25.Kishnani P, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–463. doi: 10.1097/GIM.0b013e3181e655b6. [DOI] [PubMed] [Google Scholar]
- 26.DiMauro S, Hartwig GB, Hays AP, et al. Debrancher deficiency: neuromuscular disorder in five adults. Ann Neurol. 1979;5:422–436. doi: 10.1002/ana.410050504. [DOI] [PubMed] [Google Scholar]
- 27.Powell HC, Haas R, Hall CL, et al. Peripheral nerve in type III glycogenosis: selective involvement of unmyelinated fiber Schwann cells. Muscle Nerve. 1985;8:667–671. doi: 10.1002/mus.880080808. [DOI] [PubMed] [Google Scholar]
- 28.Ugawa Y, Inoue K, Takemura T, et al. Accumulation of glycogen in sural nerve axons in adult-onset type III glycogenosis. Ann Neurol. 1986;19:294–297. doi: 10.1002/ana.410190313. [DOI] [PubMed] [Google Scholar]
- 29.Kishnani P, Chen Y-T. Disorders of glycogen metabolism. In: Rudolph CD, Rudolph AM, Lister GE, editors. Rudolph's Pediatrics. 22nd ed. New York: McGraw Hill; 2011. pp. 599–607. [Google Scholar]
- 30.Bruno C, Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV) Neurology. 2004;63:1053–1058. doi: 10.1212/01.wnl.0000138429.11433.0d. [DOI] [PubMed] [Google Scholar]
- 31.Taratuto AL, Akman HO, Saccoliti M, et al. Branching enzyme deficiency/ glycogenosis storage disease type IV presenting as a severe congenital hypotonia: Muscle biopsy and autopsy findings, biochemical and molecular studies. Neuromusc Dis. 2010;20:783–790. doi: 10.1016/j.nmd.2010.07.275. [DOI] [PubMed] [Google Scholar]
- 32.Herrick MK, Twiss JL, Vladutiu GD, et al. Concomitant branching enzyme and phosphorylase deficiencies. An unusual glycogenosis with extensive neuronal polyglucosan storage. J. Neuropath Exp Neurol. 1994;53:239–246. doi: 10.1097/00005072-199405000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinidou AE, Anninos H, Gyftodimou Y, et al. Neonatal neuromuscular variant of glycogen storage disease type IV: histopathological findings leading to the diagnosis. Histopathology. 2006;48:878–880. doi: 10.1111/j.1365-2559.2006.02425.x. [DOI] [PubMed] [Google Scholar]
- 34.Tay SKH, Akman HO, Chung WK, et al. Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromusc Disord. 2004;14:253–260. doi: 10.1016/j.nmd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 35.McArdle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci. 1951;10:13–33. [PubMed] [Google Scholar]
- 36.Schmidt R, Mahler R. Chronic progressive myopathy with myoglobinuria: demonstration of a glycogenolytic defect in the muscle. J Clin Invest. 1959;38:2044–2058. doi: 10.1172/JCI103983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mommaerts W, Illingworth B, Pearson CM, et al. A functional disorder of muscle associated with the absence of phosphorylase. Proc Nat Acad Sci U S A. 1959;45:791–797. doi: 10.1073/pnas.45.6.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larner J, Villar-Palasi C. Enzymes in a glycogen-storage myopathy. Proc Nat Acad Sci U S A. 1959;45:1234–1235. doi: 10.1073/pnas.45.8.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsujino S, Shanske S, DiMauro S. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle's disease) New Engl J Med. 1993;329:241–245. doi: 10.1056/NEJM199307223290404. [DOI] [PubMed] [Google Scholar]
- 40.Martinuzzi A, Sartori E, Fanin M, et al. Phenotype modulators in myophosphorylase deficiency. Ann Neurol. 2003;53:497–502. doi: 10.1002/ana.10499. [DOI] [PubMed] [Google Scholar]
- 41.Vissing J, Duno M, Schwartz M, et al. Splice mutations preserve myophosphorylase activity that ameliorates the phenotype in McArdle disease. Brain. 2009;132:1545–1552. doi: 10.1093/brain/awp065. [DOI] [PubMed] [Google Scholar]
- 42.DiMauro S, Hartlage P. Fatal infantile form of muscle phosphorylase deficiency. Neurology. 1978;28:1124–1129. doi: 10.1212/wnl.28.11.1124. [DOI] [PubMed] [Google Scholar]
- 43.Miranda AF, Nette G, Hartlage P, et al. Phosphorylase isoenzymes in normal and myophosphorylase deficient human heart. Neurology. 1979;29:1538–1541. doi: 10.1212/wnl.29.11.1538. [DOI] [PubMed] [Google Scholar]
- 44.Milstein JM, Herron TM, Haas JE. Fatal infantile muscle phosphorylase deficiency. J Child Neurol. 1989;4:186–188. doi: 10.1177/088307388900400305. [DOI] [PubMed] [Google Scholar]
- 45.El-Schahawi M, Bruno C, Tsujino S, et al. Sudden infant death syndrome (SIDS) in a family with myophosphorylase deficiency. Neuromusc Disord. 1997;7:81–83. doi: 10.1016/s0960-8966(97)00424-0. [DOI] [PubMed] [Google Scholar]
- 46.Tarui S, Okuno G, Ikua Y, et al. Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis. Biochem Biophys Res Comm. 1965;19:517–523. doi: 10.1016/0006-291x(65)90156-7. [DOI] [PubMed] [Google Scholar]
- 47.Mineo I, Kono N, Hara N, et al. Myogenic hyperuricemia. A common pathophysiologic feature of glycogenosis types III, V, and VII. New Engl J Med. 1987;317:75–80. doi: 10.1056/NEJM198707093170203. [DOI] [PubMed] [Google Scholar]
- 48.Agamanolis DP, Askari AD, DiMauro S, et al. Muscle phosphofructokinase deficiency: Two cases with unusual polysaccharide accumulation and immunologically active enzyme protein. Muscle Nerve. 1980;3:456–467. doi: 10.1002/mus.880030602. [DOI] [PubMed] [Google Scholar]
- 49.Hays AP, Hallett M, Delfs J, et al. Muscle phosphofructokinase deficiency: abnormal polysaccharide in a case of late-onset myopathy. Neurology. 1981;31:1077–1086. doi: 10.1212/wnl.31.9.1077. [DOI] [PubMed] [Google Scholar]
- 50.Raben N, Danon MJ, Lu N, et al. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739–1745. doi: 10.1212/wnl.56.12.1739. [DOI] [PubMed] [Google Scholar]
- 51.Moerman P, Lammens M, Fryns JP, et al. Fetal akinesia sequence caused by glycogenosis type VII. Genet. Couns. 1995;6:15–20. [PubMed] [Google Scholar]
- 52.Swoboda KJ, Specht L, Jones HR, et al. Infantile phosphofructokinase deficiency with arthrogryposis: Clinical benefit of a ketogenic diet. J. Pediatr. 1997;131:932–934. doi: 10.1016/s0022-3476(97)70048-9. [DOI] [PubMed] [Google Scholar]
- 53.Spriggs EL, Marles SL, Lacson A, et al. Long-term survival and normal cognitive development in infantile phosphofructokinase-1 deficiency. Clin Genet. 1999;56:235–237. doi: 10.1034/j.1399-0004.1999.560310.x. [DOI] [PubMed] [Google Scholar]
- 54.Danon MJ, Carpenter S, Manaligod JR, et al. Fatal infantile glycogen storage disease: Deficiency of phosphofructokinase and phosphorylase b kinase. Neurology. 1981;31:1303–1307. doi: 10.1212/wnl.31.10.1303. [DOI] [PubMed] [Google Scholar]
- 55.Al-Hassnan ZN, Al Budhaim M, Al-Owain M, et al. Muscle phosphofructokinase deficiency with neonatal seizures and nonprogressive course. J. Child Neurol. 2007;22:106–108. doi: 10.1177/0883073807299968. [DOI] [PubMed] [Google Scholar]
- 56.Servidei S, Bonilla E, Diedrich RG, et al. Fatal infantile form of muscle phosphofructokinase deficiency. Neurology. 1986;36:1465–1470. doi: 10.1212/wnl.36.11.1465. [DOI] [PubMed] [Google Scholar]
- 57.Pastoris O, Dossena M, Vercesi L, et al. Muscle phosphofructokinase deficiency in a myopathic child with severe mental retardation and aplasia of cerebellar vermis. Child Nerv Syst. 1992;8:237–241. doi: 10.1007/BF00262858. [DOI] [PubMed] [Google Scholar]
- 58.Garcia M, Pujol A, Ruzo A, et al. Phosphofructo-1-kinase deficiency leads to severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet. 2009;5:e1000615–e1000615. doi: 10.1371/journal.pgen.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hendrickx J, Lee P, Keating J, et al. Complete genomic structure and mutational spectrum of PHKA2 in patients with X-linked liver glycogenosis type I and II. Am J Hum Genet. 1999;64:1541–1549. doi: 10.1086/302399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burwinkel B, Maichele AJ, Aagenaes O, et al. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB) Hum Mol Genet. 1997;6:1109–1115. doi: 10.1093/hmg/6.7.1109. [DOI] [PubMed] [Google Scholar]
- 61.Wilkinson DA, Tonin P, Shanske S, et al. Clinical and biochemical features of 10 adult patients with muscle phosphorylase kinase deficiency. Neurology. 1994;44:461–466. doi: 10.1212/wnl.44.3_part_1.461. [DOI] [PubMed] [Google Scholar]
- 62.Kilimann MW. Glycogen storage disease due to phosphorylase kinase deficiency. In: Swallow DM, Edwards YH, editors. Protein dysfunction in human genetic disease. Oxford: Bios Scientific Publishers; 1997. pp. 57–75. [Google Scholar]
- 63.Mizuta K, Hashimoto E, Tsutou A, et al. A new type of glycogen storage disease caused by deficiency of cardiac phosphorylase kinase. Biochem Biophys Res Commun. 1984;119:582–587. doi: 10.1016/s0006-291x(84)80288-0. [DOI] [PubMed] [Google Scholar]
- 64.Servidei S, Metlay LA, Chodosh J, et al. Fatal infantile cardiopathy caused by phosphorylase b kinase deficiency. J Pediat. 1988;113:82–85. doi: 10.1016/s0022-3476(88)80535-3. [DOI] [PubMed] [Google Scholar]
- 65.Elleder M, Shin YS, Zuntova A, et al. Fatal infantile hypertrophic cardiomyopathy secondary to deficiency of hert specific phosphorylase b kinase. Virchows Arch A. 1993;423:303–307. doi: 10.1007/BF01606895. [DOI] [PubMed] [Google Scholar]
- 66.Regalado JJ, Rodriguez MM, Ferrer PL. Infantile hypertrophic cardiomyopathy of glycogenosis type IX: isolated cardiac phosphorylase kinase deficiency. Pediatr Cardiol. 1999;20:304–307. doi: 10.1007/s002469900471. [DOI] [PubMed] [Google Scholar]
- 67.Buhrer C, Landeghem FKH, Felderhoff-Mueser U, et al. Fetal bradycardia at 28 weeks of gestation associated with cardiac glycogen phosphorylase b kinase deficiency. Acta Paediatr. 2003;92:1352–1353. doi: 10.1080/08035250310006458. [DOI] [PubMed] [Google Scholar]
- 68.Eishi Y, Takemura T, Sone R, et al. Glycogen storage disease confined to the heart with deficient activity of cardiac phosphorylase kinase: a new type of glycogen storage disease. Hum Pathol. 1985;16:193–197. doi: 10.1016/s0046-8177(85)80071-x. [DOI] [PubMed] [Google Scholar]
- 69.Bruno C, Manfredi G, Andreu AL, et al. A splice junction mutation in the alpha-M gene of phosphorylase kinase in a patient with myopathy. Biochem Biophys Res Comm. 1998;249:648–651. doi: 10.1006/bbrc.1998.9211. [DOI] [PubMed] [Google Scholar]
- 70.Wehner M, Clemens PR, Engel AG, et al. Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet. 1994;3:1983–1987. doi: 10.1093/hmg/3.11.1983. [DOI] [PubMed] [Google Scholar]
- 71.Wuyts W, Reyniers E, Ceuterick C, et al. Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. Am J Med Genet. 2005;133A:82–84. doi: 10.1002/ajmg.a.30517. [DOI] [PubMed] [Google Scholar]
- 72.Orngreen MC, Schehaas HJ, Jeppesen TD, et al. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology. 2008;70:1876–1882. doi: 10.1212/01.wnl.0000289190.66955.67. [DOI] [PubMed] [Google Scholar]
- 73.Echaniz-Laguna A, Akman HO, Mohr M, et al. Muscle phosphorylase b kinase deficiency revisited. Neuromusc Disord. 2010;20:125–127. doi: 10.1016/j.nmd.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 74.Burwinkel B, Hu B, Schroers A, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003;11:516–526. doi: 10.1038/sj.ejhg.5200996. [DOI] [PubMed] [Google Scholar]
- 75.Akman HO, Sampayo JN, Ross FA, et al. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res. 2007;62:499–504. doi: 10.1203/PDR.0b013e3181462b86. [DOI] [PubMed] [Google Scholar]
- 76.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase – development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sotiriou E, Greene P, Krishna S, et al. Myopathy and Parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve. 2010;41:707–710. doi: 10.1002/mus.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tonin P, Bruno C, Cassandrini D, et al. Unusual presentation of phosphoglycerate mutase deficiency due to two different mutations in PGAM-M gene. Neuromusc Disord. 2009;19:776–778. doi: 10.1016/j.nmd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 79.Naini A, Toscano A, Musumeci O, et al. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol. 2009;66:394–398. doi: 10.1001/archneurol.2008.584. [DOI] [PubMed] [Google Scholar]
- 80.DiMauro S, Miranda AF, Khan S, et al. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science. 1981;212:1277–1279. doi: 10.1126/science.6262916. [DOI] [PubMed] [Google Scholar]
- 81.Kreuder J, Borkhardt A, Repp R, et al. Inherited metabolic myopathy and hemolysis due to a mutation in aldolase A. New Engl J Med. 1996;334:1100–1104. doi: 10.1056/NEJM199604253341705. [DOI] [PubMed] [Google Scholar]
- 82.Comi GP, Fortunato F, Lucchiari S, et al. B-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol. 2001;50:202–207. doi: 10.1002/ana.1095. [DOI] [PubMed] [Google Scholar]