

Abstract
Mussel-inspired adhesive hydrogels represent innovative candidate medical sealants or glues. In the present work, we describe an enzyme-degradable mussel-inspired adhesive hydrogel formulation, achieved by incorporating minimal elastase substrate peptide Ala-Ala into the branched poly(ethylene glycol) (PEG) macromonomer structure. The system takes advantage of neutrophil elastase expression upregulation and secretion from neutrophils upon recruitment to wounded or inflamed tissue. By integrating adhesive degradation behaviors that respond to cellular cues, we expand the functional range of our mussel-inspired adhesive hydrogel platforms. Rapid (<1 min) and simultaneous gelation and adhesion of the proteolytically active, catechol-terminated precursor macromonomer was achieved by addition of sodium periodate oxidant. Rheological analysis and equilibrium swelling studies demonstrated that the hydrogel is appropriate for soft tissue-contacting applications. Notably, hydrogel storage modulus (G′) achieved values on the order of 10 kPa, and strain at failure exceeded 200% strain. Lap shear testing confirmed the material’s adhesive behavior (shear strength: 30.4 ± 3.39 kPa). Although adhesive hydrogel degradation was not observed during short-term (27 h) in vitro treatment with neutrophil elastase, in vivo degradation proceeded over several months following dorsal subcutaneous implantation in mice. This work represents the first example of an enzymatically degradable mussel-inspired adhesive and expands the potential biomedical applications of this family of materials.
Introduction
Present work in our laboratory seeks to address the challenge of developing biocompatible adhesive hydrogel materials for soft-tissue adhesion and repair. One approach takes inspiration from mussel species such as Mytilus edulis: these bivalve mollusks produce an adhesive holdfast called the byssus that mediates attachment to heterogeneous organic and inorganic substrates in the turbulent aquatic environment.1,2 The byssus is composed of fibrous threads which connect the internal byssal retractor muscle to terminal adhesive plaques anchored to the external substrate surface. Each byssal thread and adhesive plaque is formed in an elegant injection molding process that begins with secretion of liquid protein precursors into a cavity defined by the mussel foot, followed rapidly by solidification of the thread and plaque over several minutes. The process concludes with disengagement of the mussel foot from the newly formed thread and plaque, after which the entire process is repeated numerous times to generate a full complement of byssal threads. It has been demonstrated that byssal plaque proteins mediate adhesive hardening and interfacial adhesion and are enriched in 3,4-dihydroxyphenylalanine (DOPA), a post-translationally modified amino acid.3−5 The DOPA amino acid side chain is catecholic, and under oxidizing conditions this structure covalently cross-links both to itself and to biologically relevant nucleophiles such as primary amines and thiols.6−9
Incorporation of DOPA and DOPA-mimetic catechols into hydrogel platforms has allowed us10−12 and others13−15 to create candidate adhesive biomaterials with desirable physical properties. Recently, we confirmed mussel-inspired adhesive performance and tissue compatibility in both ex vivo and in vivo model systems.16,17 In an ex vivo analysis of adhesive sealing of punctured human fetal membrane, our material demonstrated minimal cellular toxicity, with excellent tissue adhesion and integrity following applied radial strain.(16) We also utilized mussel-inspired adhesive hydrogel for experimental islet transplantation, employing the material as a sealant to immobilize islets on tissue surfaces: the preservation of islet viability and function yielded diabetes reversal in mice.(17) This “islet sealant” application represented the first in vivo analysis of a mussel-inspired adhesive and established the biocompatibility and persistence of the material. In these studies, the adhesive hydrogel material was composed of a branched poly(ethylene glycol) (PEG) polymer functionalized with catechols through a nondegradable linker. We now seek to expand the functional applications of the mussel-inspired adhesive platform by incorporating cell-directed enzymatic degradation to its cadre of desirable performance characteristics.
Modular design of catechol, linker, and polymer backbone offers multiple opportunities to fine-tune adhesive hydrogel material properties through architectural and compositional changes to achieve specific goals such as mechanism and rate of degradation. For example, peptidic substrate sequences may be incorporated into the precursor backbone, thereby promoting enzyme-directed hydrogel degradation behavior for cell- and tissue-contacting applications. Indeed, proteolytically responsive PEG hydrogels are well-documented in the literature for tissue engineering,(18) regenerative medicine,(19) and molecular delivery(20) applications as well as for the development of stem cell niches.(21) In the context of adhesive hydrogel use for tissue repair and reconstruction, it is appropriate to introduce susceptibility to extracellular matrix proteases involved in matrix restructuring and wound healing processes. Neutrophil elastase is a serine protease secreted from activated neutrophils in the context of their recruitment to a wound or site of local inflammation. Addition of an elastase substrate peptide into the polymer precursor structure thereby represents a means for tailoring hydrogel degradation behaviors at these sites. As such, incorporation of elastase-responsive behavior into PEG-based hydrogels has been utilized for biomolecule delivery22,23 and for structural degradation in extracellular matrix-mimetic materials.(24)
Here we describe the design, in vitro characterization, and in vivo performance of an elastase degradable mussel-inspired adhesive hydrogel for soft tissue-contacting applications at sites of injury or inflammation. A branched PEG reagent was terminally modified with a DOPA-mimetic catechol linked to the polymer backbone through an Ala-Ala dipeptide substrate of elastase to generate an adhesive precursor macromonomer (cAAPEG). Under oxidizing conditions, intermolecular cross-linking of catechol groups drives rapid macromonomer gelation, while catechol cross-linking to tissue surfaces simultaneously imparts adhesive behavior. Rheological analysis, equilibrium swelling studies, and lap shear testing confirmed cAAPEG’s gelation and adhesive characteristics. Subcutaneous implantation in mice revealed favorable tissue compatibility and in vivo degradation of the material by cell-mediated elastase activity.
Experimental Section
Materials and Animal Subjects
Amine-terminated four-arm poly(ethylene glycol) (P4AM-10, MW 10 kDa) was purchased from SunBio (Anyang City, South Korea). Fmoc-Ala-Ala-OH was purchased from Bachem (Torrance, CA). Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) was purchased from Novabiochem (EMD, Gibbstown, NJ). 3,4-Dihydroxyhydrocinnamic acid (DOHA), N,N-diisopropylethylamine (DIPEA), triethylamine, and piperidine were purchased from Sigma (St. Louis, MO). FITC-conjugated human sputum elastase was purchased from Elastin Products Co. (Owensville, MO). Decellularized porcine dermis extracellular matrix was generously supplied by Kensey Nash Corp. (Exton, PA). Human neutrophil elastase (HNE) was purchased from Innovative Research (Novi, MI). Mouse-reactive anti-rabbit neutrophil elastase was purchased from Abcam, (Cambridge, MA), and the EnVision+ system containing anti-rabbit secondary antibody conjugated to horseradish peroxidase-modified dextran polymer was supplied by Dako (Carpinteria, CA).
Healthy, weight-matched male CD1 mice were purchased from Charles River (Wilmington, MA). All animal studies were performed under approval of the Northwestern Animal Care and Use Committee.
Synthesis and Characterization of Degradable cAAPEG Macromonomer
5.0 g of P4AM-10 was dissolved in 20 mL of chloroform. 2.2945 g (6 mmol) of Fmoc-Ala-Ala-OH, 3.122 g (6 mmol) of PyBOP, and 2.090 mL (12 mmol) of DIPEA were added to 20 mL of dimethylformamide (DMF). These reagent solutions were mixed and allowed to stir at room temperature for 3 h. The reaction solution was precipitated overnight in cold anhydrous diethyl ether. The resulting white precipitate was collected and dried overnight under vacuum. The crude dried product (Fmoc-Ala-Ala-modified four-arm PEG) was resuspended in 35 mL of methanol, and the resulting yellow solution was precipitated overnight in cold anhydrous diethyl ether. The secondary precipitate was collected and dried overnight under vacuum. Dry product was resuspended in HCl-acidified water (pH 5.2) and subjected to extensive dialysis (3500 MWCO) in HCl-acidified water (pH 5.0–5.5). The aqueous dialyzed product was flash frozen and lyophilized to yield the Fmoc-Ala-Ala-modified four-arm PEG intermediate, and a negative Kaiser test indicated successful modification.
Fmoc-Ala-Ala-modified four-arm PEG was reacted in 20% piperidine in DMF for 2 h to remove the Fmoc protecting group, after which the crude product was recovered by overnight precipitation in cold anhydrous diethyl ether. The flocculent precipitate was collected by centrifugation at 4 °C, dried, reconstituted in 35 mL of methanol, and precipitated overnight in cold anhydrous diethyl ether. The product (H2N-Ala-Ala-modified four-arm PEG) was isolated by centrifugation at 4 °C and dried overnight under vacuum. A positive Kaiser test indicated successful Fmoc deprotection.
4.0 g of H2N-Ala-Ala-modified four-arm PEG was dissolved in 20 mL of chloroform and combined with 0.7949 g (4.36 mmol) of DOHA, 2.2704 g (4.36 mmol) of PyBOP, and 1.216 mL (8.72 mmol) of triethylamine in 20 mL of DMF. The mixture was allowed to stir at room temperature 2 h before overnight precipitation in cold anhydrous diethyl ether. The resulting white precipitate was collected, dried overnight under vacuum, resuspended in 12 mM HCl, and subjected to extensive dialysis (3500 MWCO) in HCl-acidified water (pH 3.5–4.0). The aqueous dialyzed product was filtered, flash frozen, and lyophilized to yield the purified product (DOHA-Ala-Ala-modified four-arm PEG; cAAPEG). A negative Kaiser test indicated complete DOHA conjugation to terminal free amines of the H2N-Ala-Ala-modified four-arm PEG intermediate.
Matrix-assisted laser desorption ionization (MALDI) mass spectrometry was performed on the four-arm amine-terminated PEG reagent P4AM-10, synthetic intermediates, and the final product cAAPEG macromonomer. Samples were analyzed in positive mode using linear detection on an Autoflex III Smartbeam MALDI mass spectrometer (Bruker Daltonics, Billerica, MA), using the flexControl and flexAnalysis operational and processing software package. cAAPEG macromonomer was dissolved in 12 mM HCl to a concentration of 0.3 mg/mL, prior to UV–vis analysis (model U-2010; Hitachi, San Jose, CA). Additionally, the 1H NMR spectrum of cAAPEG macromonomer was obtained on an Inova 500 MHz spectrometer (Varian, Palo Alto, CA) in deuterated chloroform at room temperature.
All cAAPEG macromonomer utilized in enzymatic or tissue-contacting studies was ethylene oxide gas sterilized on a 12 h cycle prior to use (AN74i Anprolene sterilizer; Andersen Sterilizers, Haw River, NC). Endotoxin analysis (BioTest Laboratories, Brooklyn Park, MN) showed that undiluted cAAPEG macromonomer (14.5 mg in 40 mL of pyrogen-free water) contained less than 0.4 endotoxin units (EU) in total sample, as measured by the Limulus Amebocyte Lysate test.
Preparation of cAAPEG Hydrogels
cAAPEG macromonomer was dissolved in 2× phosphate-buffered saline (PBS) pH 7.4 to a concentration of 230 mg/mL (19.2 mM). Hydrogel formation was induced by addition of an equal volume of 76.7 mM sodium periodate in water. In all experiments excepting the gelation assay, homogeneous mixing of the macromonomer solution and the sodium periodate oxidizing solution was achieved by gel delivery from a dual-barrel mixing tip device (Nordson Micromedics, St. Paul, MN). In the gelation assay, solutions were briefly vortexed in a microcentrifuge tube and periodically inverted to observe the time at which gelation occurred. For equilibrium swelling, in vitro hydrogel degradation, and subcutaneous implantation studies, hydrogels were cast into PDMS masks (diameter D = 20 mm, depth d = 2 mm; Sigma-Aldrich, St. Louis, MO), and disk samples were generated from biopsy punch (D = 5 mm) of formed gel.
Rheological Characterization
Rheological analysis was performed on a Paar Physica modular compact rheometer (MCR) 300, with Peltier hood accessory for temperature and humidity control, using US200/32 control software. Analysis of in situ gel formation was carried out on mixtures of cAAPEG macromonomer and sodium periodate delivered directly onto the rheometer plate fixture from the dual-barrel mixing tip device described above. Changes in storage and loss modulus corresponding to gel formation were monitored in oscillatory mode using a 15 mm parallel plate with 1.0 mm gap, at 5% strain and 10 rad/s with constant temperature (25 °C). Frequency sweep was performed at 5% strain, 0.1 to 100 rad/s. Strain sweep was performed at angular frequency 10 rad/s, 0.01% to 1000% strain, to gel failure.
Equilibrium Swelling
cAAPEG hydrogel discs were weighed immediately following formation (Wi) and swollen in sterile-filtered deionized water at room temperature for 4 days. Water was changed once per day. Samples were removed from water, blotted, and weighed to obtain swollen mass (Ws). Gel swelling ratio was defined as follows:Swollen samples were flash frozen and lyophilized to obtain gel polymer dry mass (Wd).
Lap Shear Testing
The adhesive performance of cAAPEG hydrogels was analyzed in a lap shear test adapted from ASTM standard F2255-05 (reapproved 2010). Dry, decellularized porcine dermis substrates were cut to 2.5 cm × 3 cm and rehydrated 1 h in PBS. Dermal tissue substrates were immobilized on aluminum test fixtures using cyanoacrylate glue, loosely covered with PBS-soaked gauze, and allowed to cure for 1 h prior to adhesive hydrogel deposition. cAAPEG adhesive hydrogel (100 μL) was applied to an immobilized tissue substrate, and a second immobilized tissue substrate was placed over the first, generating a single test sample. Final dimensions of the area of substrate overlap and adhesive interface were approximately 1.2 cm × 2.5 cm. PBS-soaked gauze was placed over test samples (n = 4), and the cAAPEG adhesive was allowed to cure at ambient conditions in air for 2 h. Immediately prior to tensile testing, the area of substrate overlap was measured using digital calipers. Samples were tested to failure in lap shear on an Instron 5544 testing platform with Bluehill operating and analytical software, at ambient conditions with 5 mm min–1 strain rate. Shear strength was calculated from the measured maximum load and the known area of adhesive overlap.
In Vitro Hydrogel Degradation
cAAPEG hydrogel disks were swollen in sterile-filtered PBS + 0.3 mg/mL sodium azide at room temperature for 4 days. The disks were incubated at 37 °C in PBS containing 0.3 mg/mL sodium azide with (n = 4) or without (n = 4) 6.6 μM human neutrophil elastase (HNE). At predetermined time points, samples were removed for weighing and immediately returned to corresponding treatment solutions. At 0, 5, and 10 h of incubation, and following termination of the experiment, HNE activity in treatment solutions was assayed by monitoring evolution of absorbance signal at 405 nm generated by neutrophil elastase-mediated cleavage of commercially available chromogenic substrate MeOSuc-Ala-Ala-Pro-Val-pNA (EMD Chemicals, Gibbstown, NJ) on SpectraMax M5 plate reader with SoftPro Max analysis software.
Hydrogel Degradation Following Subcutaneous Implantation in Mice
All cAAPEG hydrogel casting, disk preparation, and sample handling were performed under sterile conditions immediately prior to animal surgery. Substrate-free cPEG hydrogels were prepared as previously described;(17) implantable cAAPEG and cPEG disk samples were otherwise handled identically. In advance of subcutaneous implantation, mice were anesthetized with nebulized isoflurane (Isothesia, Butler Schein, Dublin, OH). Anesthesia was maintained over the course of surgery by continuous isoflurane delivery through nose cone. The dorsal surgical site was shaved, and the skin was sterilized by alternating applications of betadine solution and 70% ethanol and allowed to dry. A single, 1 cm incision was made at the dorsal implantation site, and a subcutaneous pocket was created by no-cut blunt dissection of the fascia between the dermis and the underlying muscle tissue. A single cAAPEG or cPEG hydrogel disk sample was implanted into the subcutaneous pocket prior to closure with stainless steel wound clip. Mice were monitored until regaining consciousness and recovery of normal grooming and feeding activities. At 1, 3, 6, and 16 weeks postimplant, the hydrogel samples and associated dermal, muscle, and fascia tissue were surgically removed from anesthetized mice, prior to euthanasia. Samples were fixed overnight in 10% neutral-buffered formalin at 4 °C and mounted in paraffin. 4 μm tissue sections were subjected to hematoxylin and eosin (HE) staining. Immunohistochemical identification of elastase was achieved utilizing mouse-reactive antirabbit neutrophil elastase primary antibody detected with the EnVision+ staining system utilizing anti-rabbit secondary antibody conjugated to horseradish peroxidase-modified dextran polymer.
Statistics
Data are presented as mean ± SEM, except where indicated.
Results and Discussion
Synthesis and Characterization of Degradable cAAPEG Macromonomer
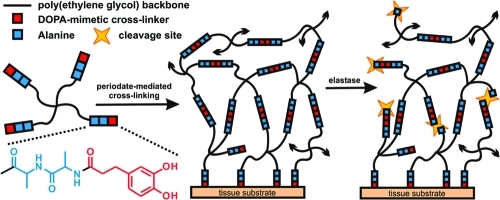
To generate a rapidly forming and mussel-inspired adhesive hydrogel material susceptible to neutrophil elastase-mediated degradation, the cAAPEG macromonomer (Figure 1) was first prepared in three-step solution-phase modification of a four-arm PEG reagent to yield a branched precursor terminated with the cross-linkable, elastase substrate-presenting DOHA-Ala-Ala moiety. Given that periodate-mediated cross-linking of oxidized DOHA catechol is responsible for hydrogel network formation, selection of the putative elastase substrate was not trivial, as nucleophile-presenting peptide sequences must be assiduously avoided. Otherwise, the competitive chemical cross-linking of highly reactive, oxidized catechol moieties such o-quinone to nucleophilic amino acid(s) would render the substrate nonresponsive to elastase activity. Another important element of elastase substrate design involved minimizing substrate length. Although longer peptides are generally better substrates and are predicted to promote more rapid degradation, minimal substrates enjoy many benefits such as simplified synthesis, lower cost, and reduced immunogenicity. Taking these parameters into consideration, as well as the preference of elastase for small, hydrophobic amino acids at the site of enzymatic cleavage,(25) the dipeptide Ala-Ala was selected as the elastase substrate in this degradable hydrogel formulation. Notably, elastase-mediated cleavage at an Ala-Ala sequence has been previously demonstrated in peptide–lipid conjugates(26) and utilized for molecular payload delivery from hydrogel(27) and mesoporous silica particles.(28)
Figure 1.

Oxidative cross-linking of enzymatically degradable cAAPEG macromonomer yields rapid and simultaneous gelation and tissue adhesion. In the presence of neutrophil elastase, the Ala-Ala dipeptide linker (blue) is cleaved to provide cell-mediated structural degradation. Black arrowheads indicate continuation of the cross-linked hydrogel matrix.
The cAAPEG macromonomer was synthesized without complication on the multigram scale with relatively high yield (∼80%), facilitating extensive in vitro and in vivo analysis of the material. The purified, lyophilized, off-white cAAPEG powder demonstrated excellent shelf life and dissolved easily in aqueous and organic solvents to yield a solution with faint yellow tint. Solutions of the macromonomer in water and neutral pH buffer darkened over several days, suggesting oxidative mechanisms. For this reason, all solutions containing macromonomer were freshly prepared, immediately prior to use.
The cAAPEG macromonomer was characterized by MALDI mass spectrometry, UV–vis spectrophotometry, and 1H NMR. MALDI mass spectrometry data of branched P4AM-10 reagent, synthetic intermediates, and cAAPEG macromonomer confirmed successful preparation of cAAPEG macromonomer (Figure 2a). In this figure, spectrum 2 (Fmoc-Ala-Ala four-arm PEG) demonstrated a clear increase in molecular weight from the PEG reagent (spectrum 1), and the subsequent Fmoc deprotection step generated an obvious shift to lower molecular weight (H2N-Ala-Ala four-arm PEG, spectrum 3). Finally, terminal DOHA addition increased cAAPEG product peak molecular weight to the predicted value of ∼12 kDa (spectrum 4). UV–vis spectrophotometric analysis of cAAPEG macromonomer showed an absorbance maximum of the dihydroxybenzyl (catechol) moiety at 280 nm, with minimal contribution from undesirable oxidized components at 320 nm and higher wavelengths(29) (Figure 2b), demonstrating that terminal catechol groups were unoxidized at completion of cAAPEG macromonomer synthesis. Interpolation of macromonomer absorbance at 280 nm against a standard curve of known catechol concentration confirmed quantitative cAAPEG terminal modification by DOHA (0.762 μmol of catechol per mg of polymer, Supporting Information Figure S1). NMR analysis of cAAPEG macromonomer identified the protons contributed by the PEG polymer backbone, the Ala-Ala dipeptide linker, and the terminal catechol of DOHA. As shown in Figure 2c, signals d, d′ and e, e′ associated with the Ala-Ala dipeptide are observed at 4.4 and 0.6 ppm, respectively, while DOHA-associated methylenic signals b and c and aromatic hydrogens (a) are clearly present. A strong signal from ether hydrogens of the PEG backbone (f) is centered near 3.6 ppm.
Figure 2.

Characterization of the cAAPEG macromonomer. Matrix-assisted laser deionization (MALDI) mass spectrometry (a) confirmed the changes in molecular mass expected during stepwise synthesis of cAAPEG macromonomer (∼12 kDa). UV–vis spectrophotometry of cAAPEG macromonomer (b) revealed a peak at 280 nm corresponding to catechol, with little evidence of oxidized species (>300 nm). Structural confirmation of cAAPEG macromonomer by 1H NMR (c) was performed in deuterated chloroform; peak assignments corresponding to PEG backbone, Ala-Ala linker, and catechol protons are indicated in italics.
Physical Characterization of cAAPEG Adhesive Hydrogels
Neutrophil elastase-degradable cAAPEG was designed as an enzyme-reactive hydrogel to expand the functional range and potential applications of mussel-inspired adhesive materials. Catechol end groups of the cAAPEG macromonomer allow for rapid and oxidation-mediated cross-linking upon mixing with periodate.(29) Immediately upon mixing cAAPEG macromonomer and sodium periodate solutions, a transition from colorless to medium brown was observed, followed rapidly by gel formation within 20–30 s. As cAAPEG is a candidate soft-tissue adhesive or sealant material, this color change represents a desirable performance characteristic: upon in situ delivery, the location of adhesive deposition would be visible. This evolution is related to periodate-induced catechol oxidation to highly reactive groups such as o-quinone, with corresponding absorbance shifts into visible wavelengths. Chemical cross-linking of these reactive groups is responsible for macromonomer intermolecular cross-linking and hydrogel formation.(29) It is notable that introduction of the Ala-Ala dipeptide substrate between terminal catechol groups and the PEG core did not significantly impact cAAPEG gelation behavior when compared to peptide linker-free catechol-modified cPEG adhesive hydrogel.(17)
In situ rheological analysis of cAAPEG hydrogels confirmed rapid formation of an elastic gel upon mixing with periodate. Because of rapid gelation kinetics (<1 min), it was not possible to observe G′/G″ crossover (Figure 3a), a commonly employed indicator of gelation.(30) Nevertheless, storage modulus G′ rapidly increased 1–2 min after mixing cAAPEG macromonomer and periodate solutions, before achieving plateau values on the order of 10 kPa. This value conforms with rheological analyses of other enzymatically degradable branched PEG hydrogels(31) and underlines the material’s potential utility in soft tissue-contacting applications. Comparatively low loss modulus G″ indicated solidlike behavior of the cAAPEG adhesive hydrogel. Gel stability was demonstrated in frequency sweep analysis, which verified that storage and loss moduli were independent of applied angular frequency (Figure 3b). Strain sweep analysis showed strain at failure (secondary G′/G″ crossover) above 200% strain, illustrating the mechanical robustness of the hydrogel (Figure 3c).
Figure 3.

Rheological characterization of cAAPEG adhesive hydrogel formation (a), frequency sweep (b), and strain sweep (c) analyses.
In a complementary set of studies, equilibrium swelling and macromolecular infiltration into the cAAPEG hydrogel network were assessed. Equilibrium swelling analysis indicated a cAAPEG swelling ratio of 34.3% (>90% water), suggesting a well-cross-linked network with robust mechanical strength. Confocal microscopy of hydrogels immersed in buffer containing fluorescently tagged elastase (∼30 kDa) revealed homogeneous fluorescence signal, confirming the ability of elastase enzyme to uniformly infiltrate the cross-linked network within 12 h (Supporting Information, Figure S2). Cross-linked PEG-diacrylate hydrogels have demonstrated similar HNE infiltration behavior.(22)
In addition to characterization of cross-linked cAAPEG as a hydrogel material, lap shear testing established its potential for tissue adhesion. cAAPEG hydrogel was applied to porcine dermal tissue substrates attached to aluminum tags and allowed to cure for a short period (2 h) prior to tensile testing. These experimental conditions best approximate early postapplication performance, in contrast to the 12–24 h cure times utilized in other studies. The mean shear strength of the adhesive was 30.4 ± 3.39 kPa. Although it is difficult to directly compare this result with other studies due to differences in methodology and tissue types, this value is similar to those obtained from PEG-based adhesives developed in our group(12) and elsewhere(32) and is several times greater than fibrin glue shear strength measurements obtained in those studies (<10 kPa). In all tests the bonded samples underwent cohesive failure (Figure 4), indicating excellent adhesion of the cAAPEG hydrogel to the tissue. Similar performance is anticipated for in situ cAAPEG adhesion to living biological tissues.
Figure 4.

Photograph of decellularized porcine dermis bonded with cAAPEG adhesive hydrogel and then fractured under lap shear after 2 h. Cohesive failure is indicated by the presence of cAAPEG adhesive hydrogel (dark) on both fracture surfaces.
cAAPEG Adhesive Hydrogel Degradation Analysis
Cross-linked cAAPEG hydrogels presenting Ala-Ala dipeptide were designed to degrade in the presence of the proteolytic enzyme neutrophil elastase. The in vitro response of cAAPEG hydrogels to elastase proteolytic activity was assessed over a 27 h period. Samples were exposed to neutrophil elastase-containing buffer solutions at the outset of the experiment, and no repeat dosing was performed. Enzyme activity assays confirmed that human neutrophil elastase (HNE) activity decreased with time: the protease lost ∼90% of its initial activity over the course of the experiment performed at 37 °C (Supporting Information, Figure S3a). This study was designed to assess very short-term response to a single enzyme treatment. Under these stringent treatment conditions, HNE-mediated cAAPEG hydrogel degradation was not detected by mass loss measurements (Supporting Information, Figure S3b). In contrast, it has previously been shown that thiol–ene photopolymerized PEG hydrogels with elastase-sensitive 12-mer peptide cross-links degraded in a matter of hours in an enzyme concentration-dependent fashion.(23) It is important to consider that the minimal Ala-Ala dipeptide utilized in the present study was predicted to be a relatively inefficient elastase substrate, selected to permit material degradation over longer periods. This explains the lack of mass loss observed in stringent, short-term in vitro analysis.
Based on the finding that neutrophil elastase-mediated degradation of cAAPEG hydrogels likely occurs over a time period poorly suited to in vitro assessment, cAAPEG hydrogel implantation was performed to characterize material degradation over an extended period in vivo. This approach furthermore represented a more realistic assessment of its performance, as an in vitro platform cannot fully reconstitute the complex chemical environment present in an intact, living tissue. cAAPEG hydrogel samples were implanted subcutaneously in mice as preformed 5 mm disks and subsequently removed at specified time points for histological and immunohistochemical analysis. Hydrogel disks exhibited good integration with surrounding tissues at the time of explant surgery. At 1 week postimplantation, there was evidence of cellular infiltration into the hydrogel (Figure 5a,b), and immunohistochemical staining for elastase was positive at the cAAPEG–tissue interface, suggesting the presence of elastase secreted by activated neutrophils (Figure 5b and inset). At subsequent explant time points, the degree of cellular infiltration and hydrogel surface erosion demonstrably increased with time. In hydrogel samples removed 3 weeks following implantation, significant cellular infiltration and gel restructuring corresponded with dramatic elastase staining at the interface of the infiltrating cell population and the cAAPEG surface (Figure 5c,d). At 6 weeks following implantation, distinctive hydrogel surface irregularities were associated with cellular infiltration several hundreds of micrometers into the bulk hydrogel network (Figure 5e,f). Surface irregularities were also present at the 16 week time point, and histological processing of these explanted samples suggested that hydrogel restructuring by infiltrating cells is followed by fibrous matrix deposition at extended time points, similar to the outcome of wound healing (Figure 5g). Notably, positive elastase staining and cellular infiltration were observed within the bulk cAAPEG hydrogel, made more evident with slight sample separation that occurred as an artifact of processing (Figure 5h and inset).
Figure 5.

Histological (a, c, e, g) and immunohistochemical (b, d, f, h) analysis of subcutaneously implanted cAAPEG confirmed in vivo neutrophil elastase-mediated degradation of this material. Scale bars: 200 μm, all images. White arrows: site of cellular infiltration into the cAAPEG adhesive hydrogel. Black arrows: positive neutrophil elastase stain.
The impact of incorporating elastase-responsive Ala-Ala dipeptide substrate into the macromolecular cAAPEG hydrogel network was underlined by contrasting its degradation behavior to that of substrate-free cPEG hydrogel. Facile addition of the minimal elastase substrate dramatically altered this behavior, converting an inert implantable adhesive (cPEG; Figure 6a) into a material undergoing extensive cell-mediated degradation at 16 weeks, across the millimeter scale (cAAPEG; Figure 6b). In these sagittal sections, implanted cPEG maintained distinct linear surfaces, whereas ongoing cAAPEG degradation led to significant cellular infiltration, surface roughness, and disruption of original sample geometry. Following implantation at the dorsal subcutaneous site, cAAPEG degrades via enzyme-mediated surface erosion, preferentially from the underside and edges of the sample. This finding was consistent within cAAPEG groups and at each time point assessed. In contrast, cPEG demonstrated no cellular infiltration and no degradation, in agreement with our previous in vivo studies.(17)
Figure 6.

Histological analysis of subcutaneously implanted substrate-free cPEG (a) and elastase-responsive cAAPEG (b) mussel-inspired adhesive hydrogels highlighted the role of the Ala-Ala dipeptide substrate in cAAPEG degradation at 16 weeks following implantation. Scale bars: 500 μm, both images.
cAAPEG adhesive hydrogel was designed to degrade in a wounded or inflamed tissue environment through the enzymatic activity of elastase secreted from activated neutrophils. Histological and immunohistochemical analysis confirmed that cAAPEG degradation processes remained active over a period of several months in vivo. In explanted hydrogel samples, evidence of elastase expression at the cAAPEG–tissue interface suggested that this material experienced structural degradation mediated, at least in part, by this enzyme. In this set of experiments, tissue infiltration by activated neutrophils represented a nonspecific inflammatory response to dermal incision and subcutaneous implantation of cAAPEG hydrogel samples. cAAPEG was not designed to initiate a chronic inflammatory response; therefore, elastase expression by activated neutrophils appeared to decrease over time. However, this outcome should not prohibit further cAAPEG degradation at extended time points: given that the Ala-Ala dipeptide is a minimal substrate, it is likely that other ECM-resident proteases such as matrix metalloproteinases (MMPs) participated in cAAPEG hydrogel degradation in vivo, as suggested for other PEG-based hydrogels incorporating minimal substrates for degradation by plasmin.(33) In the present work it is expected that longer neutrophil elastase substrate peptide sequences would shorten the timeline of in vivo hydrogel degradation, while increasing the risk of antigen-specific immunogenicity of the material.
Conclusions
We have developed an adhesive hydrogel inspired by marine mussel holdfasts and engineered to degrade by the proteolytic activity of neutrophil elastase. Under oxidative cross-linking conditions, cAAPEG gelation and adhesion to tissue occurred in less than 1 min. Rheological analysis, equilibrium swelling studies, and lap shear testing elucidated the material’s hydrogel and adhesive properties. Adhesive hydrogels presenting minimal Ala-Ala dipeptide linker for neutrophil elastase-mediated degradation were not found to degrade during short-term in vitro analysis; however, in vivo subcutaneous implantation studies in mice confirmed that this material slowly degraded and elicited a minimal inflammatory tissue response. cAAPEG is the first example of an enzyme-degradable mussel-inspired adhesive, and this material demonstrated attractive properties for use immediately following soft-tissue injury, for applications in nonstatic tissues including the gut and the vasculature, and for adhesion in a chronically inflamed environment such as hernia. Other recently introduced candidate medical adhesives have similarly focused on tissue-specific applications, including wound care for diabetic ulcers,(34) suture adjunct for corneal tissue,(35) and surgical repair of peripheral nerves.(36) As such, future experiments will interrogate the performance and degradation of in situ-formed adhesive in a wound healing or chronic inflammation model.
Acknowledgments
We thank Nicholas Talarico and Bella Shmaltsuyev of the Pathology Core Facility, and Donna Emge of the Mouse Histology and Phenotyping Laboratory, Robert H. Lurie Comprehensive Cancer Center of Northwestern University, for assistance with histology and immunohistochemistry. The authors thank Kensey Nash Corporation for their generous donation of decellularized porcine dermis. This work was supported by National Institutes of Health Grants R01 DE021215, R01 DE021104, and R37 DE014193.
Supporting Information Available
Standard curve of the catechol end group concentration assay; confocal fluorescence microscopy of cAAPEG adhesive hydrogel infiltration by FITC-elastase; elastase enzymatic activity assays; raw sample mass data from in vitro elastase degradation of adhesive hydrogel. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Waite J. H. Integr. Comp. Biol. 2002, 42, 1172–1180. [DOI] [PubMed] [Google Scholar]
- Lee B. P.; Messersmith P. B.; Israelachvili J. N.; Waite J. H. Annu. Rev. Mater. Res. 2011, 41, 99–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite J. H.; Tanzer M. L. Science 1981, 212, 1038–1040. [DOI] [PubMed] [Google Scholar]
- Papov V. V.; Diamond T. V.; Biemann K.; Waite J. H. J. Biol. Chem. 1995, 270, 20183–20192. [DOI] [PubMed] [Google Scholar]
- Waite J. H.; Qin X. Biochemistry 2001, 40, 2887–2893. [DOI] [PubMed] [Google Scholar]
- Yu M. E.; Hwang J. Y.; Deming T. J. J. Am. Chem. Soc. 1999, 121, 5825–5826. [Google Scholar]
- Burzio L. A.; Waite J. H. Biochemistry 2000, 39, 11147–11153. [DOI] [PubMed] [Google Scholar]
- Lee H.; Scherer N. F.; Messersmith P. B. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12999–13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Waite J. H. J. Biol. Chem. 2006, 281, 26150–26158. [DOI] [PubMed] [Google Scholar]
- Huang K.; Lee B. P.; Ingram D. R.; Messersmith P. B. Biomacromolecules 2002, 3, 397–406. [DOI] [PubMed] [Google Scholar]
- Catron N. D.; Lee H.; Messersmith P. B. Biointerphases 2006, 1, 134–141. [DOI] [PubMed] [Google Scholar]
- Burke S. A.; Ritter-Jones M.; Lee B. P.; Messersmith P. B. Biomed. Mater. 2007, 2, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood G.; Horton T. N.; Wilker J. J. Macromolecules 2007, 40, 3960–3964. [Google Scholar]
- Shazly T. M.; Baker A. B.; Naber J. R.; Bon A.; Van Vliet K. J.; Edelman E. R. J. Biomed. Mater. Res. A 2010, 95, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Chung H. J.; Yeo S.; Ahn C. H.; Lee H.; Messersmith P. B.; Park T. G. Soft Matter 2010, 6, 977–983. [Google Scholar]
- Bilic G.; Brubaker C.; Messersmith P. B.; Mallik A. S.; Quinn T. M.; Haller C.; Done E.; Gucciardo L.; Zeisberger S. M.; Zimmermann R.; Deprest J.; Zisch A. H. Am. J. Obstet. Gynecol. 2010, 202 (85), e81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker C. E.; Kissler H.; Wang L. J.; Kaufman D. B.; Messersmith P. B. Biomaterials 2010, 31, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. Biomaterials 2010, 31, 4639–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. C.; Anseth K. S. Pharm. Res. 2009, 26, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. J.; Chung E. H.; Rodriguez R. T.; Firpo M. T.; Healy K. E. J. Biomed. Mater. Res., Part A 2006, 79A, 1–5. [DOI] [PubMed] [Google Scholar]
- Aimetti A. A.; Tibbitt M. W.; Anseth K. S. Biomacromolecules 2009, 10, 1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimetti A. A.; Machen A. J.; Anseth K. S. Biomaterials 2009, 30, 6048–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B. K.; Gobin A. S.; Tsai A. T.; Schmedlen R. H.; West J. L. Biomaterials 2001, 22, 3045–3051. [DOI] [PubMed] [Google Scholar]
- Sweeney P. J.; Walker J. M. In Methods in Molecular Biology; Burrell M. M., Ed.; Humana Press: Totowa, NJ, 1993; Vol. 16, pp 277–303. [DOI] [PubMed] [Google Scholar]
- Pak C. C.; Ali S.; Janoff A. S.; Meers P. Biochim. Biophys. Acta 1998, 1372, 13–27. [DOI] [PubMed] [Google Scholar]
- Thornton P. D.; Mart R. J.; Webb S. J.; Ulijn R. V. Soft Matter 2008, 4, 821–827. [DOI] [PubMed] [Google Scholar]
- Thornton P. D.; Heise A. J. Am. Chem. Soc. 2010, 132, 2024–2028. [DOI] [PubMed] [Google Scholar]
- Lee B. P.; Dalsin J. L.; Messersmith P. B. Biomacromolecules 2002, 3, 1038–1047. [DOI] [PubMed] [Google Scholar]
- Ross-Murphy S. B. Polym. Gels Networks 1994, 2, 229–237. [Google Scholar]
- van Dijk M.; van Nostrum C. F.; Rijkers D. T. S.; Liskamp R. M. J.; Hennink W. E. Biomacromolecules 2010, 11, 1608–1614. [DOI] [PubMed] [Google Scholar]
- Strehin I.; Nahas Z.; Arora K.; Nguyen T.; Elisseeff J. Biomaterials 2010, 31, 2788–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y. S.; Rizzi S. C.; Ehrbar M.; Weber F. E.; Hubbell J. A.; Lutolf M. P. J. Biomed. Mater. Res., Part A 2010, 93, 870–877. [DOI] [PubMed] [Google Scholar]
- Choi J. S.; Yoo H. S. J. Biomed. Mater. Res., Part A 2010, 95A, 564–573. [DOI] [PubMed] [Google Scholar]
- Oelker A. M.; Berlin J. A.; Wathier M.; Grinstaff M. W. Biomacromolecules 2011, 12, 1658–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickett T. A.; Amoozgar Z.; Tuchek C. A.; Park J.; Yeo Y.; Shi R. Y. Biomacromolecules 2011, 12, 57–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.