

Abstract
Store-operated Ca2+ entry (SOCE) is a ubiquitous signaling process in eukaryotic cells in which the endoplasmic reticulum (ER)-localized Ca2+ sensor, STIM1, activates the plasma membrane-localized Ca2+ release-activated Ca2+ (CRAC) channel, Orai1, in response to emptying of ER Ca2+ stores. In efforts to understand this activation mechanism, we recently identified an acidic coiled-coil region in the C-terminus of Orai1 that contributes to physical association between these two proteins, as measured by fluorescence resonance energy transfer, and is necessary for Ca2+ influx, as measured by an intracellular Ca2+ indicator. Here, we present evidence that a positively charged sequence of STIM1 in its CRAC channel activating domain, human residues 384-386, is necessary for activation of SOCE, most likely because this sequence interacts directly with the acidic coiled-coil of Orai1 to gate Ca2+ influx. We find that mutation to remove positive charges in these residues in STIM1 prevents its stimulated association with wild type Orai1. However, association does occur between this mutant STIM1 and Orai1 that is mutated to remove negative charges in its C-terminal coiled-coil, indicating that other structural features are sufficient for this interaction. Despite this physical association, we find that thapsigargin fails to activate SOCE following co-expression of mutant STIM1 with either wt or the mutant Orai1, implicating STIM1 384-386 in transmission of the Ca2+ gating signal to Orai1 following store depletion.
Ca2+ release-activated Ca2+ (CRAC) channels play a key role in Ca2+ influx and mobilization in numerous cellular responses. These channels are activated by coupling of the ER-store Ca2+ sensor STIM1 (1,2) to the plasma membrane channel protein Orai1, also known as CRACM1 (3,4). Recently we showed that six acidic residues in a putative coiled-coil at the C-terminus of Orai1 contribute to STIM1-Orai1 interactions in response to Ca2+ store depletion (5). The mutant lacking these acidic residues, Orai1ΔDE, exhibits a clustered distribution at the plasma membrane in the absence of and following stimulation with thapsigargin. Similar clustering of wtOrai1 in unstimulated cells is caused by sphingosine derivatives that flip to the inner leaflet of the plasma membrane (5), suggesting that electrostatic repulsion prevents homo-oligomerization of unliganded Orai1. Stimulated oligomerization of Orai1 that is induced by clustered STIM1 is known to play a role in channel activation (6,7).
Based on these results, we hypothesized that electrostatic attraction and resulting neutralization of the acidic coiled-coil in Orai1 by basic residues in STIM1 facilitates oligomerization of Orai1 necessary for activation of the complex in response to store depletion. We therefore sought to identify a sequence on STIM1 that could mediate this charge neutralization. We first considered that the C-terminal polybasic sequence of STIM1 could be responsible for this coupling. However, our findings and those of others (8,9,10) demonstrated that this region is not necessary for robust SOCE or for formation of the STIM1-Orai1 complex. Furthermore, STIM1 residues 342-448 (human numbering), named the CRAC activating domain (CAD), was recently identified as a minimal region of STIM1 sufficient to activate Ca2+ mobilization (9, see also 8,10). Motivated by this information, we identified a short, highly conserved, basic sequence within CAD, STIM1 (382-387), as a potential region for interaction with the Orai1 acidic coiled-coil. We used fluorescence resonance energy transfer (FRET) and Ca2+ mobilization measurements of mutants to show that these residues are key for functional coupling of STIM1 and Orai1.
Materials and Methods
Constructs and cloning
STIM1 K(384-6)Q-mRFP cDNA was generated using the Stratagene Quickchange site directed mutagenesis kit on our previously constructed STIM1-mRFP vector (5). The primers used are: 5′-GCCAAGGAGGGGGCTGAGAAGATACAACAGCAGAGAAACACACTCTTTGGCACCTTCCAC-3′ and its reverse complement. Preparation of the constructs AcGFP-Orai1 and AcGFP-Orai1 ΔDE was previously described (5).
Cell culture
RBL-2H3 mast cells were cultured in minimal essential medium supplemented with 1 μg/ml gentamicin and 20% (v/v) fetal bovine serum. In preparation for transfection and imaging, cells were plated at 25% confluence into 35 mm MatTek wells. After approximately 20 h, cells were transfected with either mutant or wild-type versions of STIM1-mRFP and AcGFP-Orai1. These constructs were transfected using either Geneporter (Genlantis) or Fugene HD (Roche) per manufacturers’ instructions, with modifications to enhance transfection efficiency in the RBL cells previously described (11). Cells were imaged 24 h after transfection.
COS7 cells were cultured as monolayers in Dulbecco’s modified Eagle’s medium supplemented with 1 μg/ml gentamicin and 10% (v/v) fetal bovine serum. In preparation for imaging, cells were harvested and transfected with wt or mutant mRFP-STIM1 and Orai1 using Fugene HD according to manufacturer’s instructions. Cells were transfected with non-fluorescent derivatives of Orai1 and mRFP labeled derivatives of STIM1 as a marker for positive transfectants. Cells were imaged 24 h after transfection.
Confocal Microscopy
Immediately prior to imaging, RBL-2H3 cells were washed and incubated for 5 min at 37°C in 2.5 mL buffered salt solution (BSS: 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5.6 mM glucose, 1 mg/ml BSA, 20 mM HEPES, pH 7.4). Cells were then imaged on a Leica TCS SP2 confocal microscope with a Leica APO 63x dipping objective. Cells were excited at 488 nm and 543 nm, with laser intensity and phototube sensitivity adjusted to maximize signal/noise. Fluorescence emission was monitored at 495-540 nm and 555-675 nm. All live cell imaging was carried out at 37°C. After observing the resting state of the cells, they were stimulated for the time specified by the addition of 0.5 mL of BSS containing thapsigargin (150 nM final concentration). Leica Confocal Software was used during the experiment to acquire images, and ImageJ was used post acquisition to prepare composite micrographs by uniform contrast adjustment.
Ca2+ Measurements
Immediately prior to imaging, COS7 cells were incubated with 0.9 μM fluo-4-AM (Molecular Probes) for 10 minutes in BSS containing 0.5 mM sulfinpyrazone. Cells were then washed and resuspended in BSS/sulfinpyrazone. Fields of cells were imaged before and during thapsigargin stimulation under the same conditions and settings as described for multicolor imaging. For single-cell time courses, images were collected every 15 seconds for 2.5 min prior to stimulation and for 10 min after thapsigargin stimulation. For quantitation of pre- and post-stimulation Ca2+ levels, several fields of cells were imaged before stimulation and 10 minutes after thapsigargin stimulation. Cytoplasmic Fluo-4 fluorescence was quantified using ImageJ software. For each dish of cells, the average prestimulation fluorescence in untransfected cells was normalized to 1.0 to account for small differences in dye loading between multiple samples.
FRET Measurement
RBL-2H3 cells were imaged for FRET as previously described (5). Briefly, cells expressing a combination of mutant or wild-type STIM1-mRFP and AcGFP-Orai1 were imaged at 10 second intervals with excitation at 476 nm to minimize spectral bleedthrough. An automated mask-drawing algorithm in Matlab was used to select the pixels of interest at the plasma membrane for every time point using fluorescence from AcGFP as the template. The integrated red and green intensities under the mask were adjusted by background subtraction and correction for spectral bleedthrough. We report the ratio of the corrected red fluorescence to corrected green fluorescence as FRET (5).
Results and Discussion
The majority of the STIM1 CAD sequence shows homology to coiled-coil regions from a wide array of proteins (based on homology search using the Position-Specific Iterated Basic Local Alignment Search Tool, PSI-BLAST, provided by the NIH, http://blast.ncbi.nlm.nih.gov/Blast.cgi), but residues 382-395 have lower sequence homology to other proteins, suggesting a specialized function of this segment. We therefore mutated the three central lysines in this sequence (384-386) to glutamines to eliminate their positive charge while maintaining the hydrophilic and steric character of the endogenous sequence (STIM1 K(384-6)Q). Our modeling predicts that the flanking basic residues (K382 and R387) in this sequence are involved in intramolecular salt bridges with neighboring acidic amino acids and therefore might be of structural importance. The sequence we selected is similar to a slightly longer sequence of homologous, positively charged amino acids in the CAD region of the Bombyx mori (silkmoth) homologue of STIM1 that was recently shown to be important for translocation of STIM1 to plasma membrane punctae (12). It is very close to residue L373 that was identified by Fischauf et al. as being critical both for the interaction of STIM1 and Orai1 and for Ca2+ mobilization (13). We speculate that the mutation STIM1 L373S may disrupt the coiled-coil structure in the segment of STIM1 containing the basic residues we identified, thereby preventing proper engagement with Orai1.
We previously developed a microscopy-based FRET assay to monitor the physical association of wtSTIM1-mRFP with AcGFP-wtOrai1 and AcGFP-Orai1ΔDE (5). Using this method, we measured FRET between STIM1 K(384-6)Q-mRFP and AcGFP-wtOrai1 transfected into RBL mast cells. We found that these mutations in STIM1 almost completely prevent its association with wtOrai1 that is normally observed following stimulation by thapsigargin (Figure 1). In unstimulated cells, STIM1 K(384-6)Q-mRFP is expressed similarly to wtSTIM1-mRFP throughout the ER (Figure 2A,C) (5). However, unlike wt-STIM1-mRFP, this mutant fails to undergo thapsigargin-stimulated co-clustering with AcGFP-wtOrai1 in plasma membrane punctae (Figure 2D), consistent with the FRET results. Also shown in Figured 2A is the punctate distribution of AcGFP-Orai1ΔDE at the plasma membrane of this unstimulated RBL cell as previously described (5).
Figure 1.
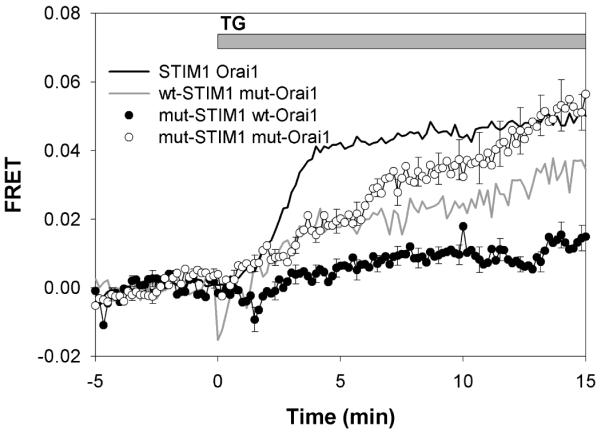
Thapsigargin-mediated FRET. Thapsigargin (150 nM)-stimulated FRET between STIM1-mRFP and AcGFP-Orai1 constructs in RBL mast cells: wtSTIM1-mRFP and AcGFP-wtOrai1 (black line), wtSTIM1-mRFP and AcGFP-Orai1ΔDE (mut-Orai1; grey line), STIM1 K(384-6)Q-mRFP (mut-STIM1) and AcGFP-Orai1ΔDE (○), and STIM1 K(384-6)Q-mRFP and AcGFP-wtOrai1 (●). Error bars show SEM for 12 to 18 cells from 4-5 different experiments, and every fifth error bar is shown for clarity.
Figure 2.

Cellular distributions of Orai1 and STIM1. Ventral surface confocal microscopy images of transfected RBL cells showing distributions of (A-B) AcGFP-Orai1ΔDE (green) and wtSTIM1-mRFP (red), (C-D) AcGFP-wtOrai1 (green) and STIM1 K(384-6)Q-mRFP (red), or (E-F) AcGFP-Orai1ΔDE and STIM1 K(384-6)Q-mRFP in the absence (A,C,E) or presence (B,D,F) of 150 nM thapsigargin for 30 min at 37°C. Scale bars represent 10 m.
To evaluate the functional capacity of this STIM1 mutant, we monitored Ca2+ mobilization in COS7 cells using Fluo-4 as previously described (5). COS7 cells exhibit little or no endogenous SOCE, but they exhibit a robust response to thapsigargin when wtSTIM1-mRFP and wtOrai1 are co-expressed (Figure 3A, blue circles). In contrast, when STIM1 K(384-6)Q-mRFP is co-expressed with wtOrai1 at similar levels, no thapsigargin-stimulated SOCE is observed (Figure 3A, green triangles). Results in Figures 1 and 3 indicate that STIM1 lysine residues 384-386 are necessary for coupling to Orai1 and consequent activation of SOCE.
Figure 3.

Stimulated Ca2+ responses. A) Ca2+ responses to 150 nM thapsigargin in COS7 cells expressing wtSTIM1-mRFP and wtOrai1 ( ), STIM1 K(384-6)Q-mRFP and wtOrai1 (
), STIM1 K(384-6)Q-mRFP and wtOrai1 ( ), wtSTIM1-mRFP and Orai1ΔDE (
), wtSTIM1-mRFP and Orai1ΔDE ( ), STIM1 K(384-6)Q-mRFP and Orai1ΔDE (
), STIM1 K(384-6)Q-mRFP and Orai1ΔDE ( ), and untransfected cells (
), and untransfected cells ( ). Fluo-4 fluorescence was normalized relative to the average prestimulation basal Ca2+ level in untransfected cells in the same field. Each time course represents the average of 10-18 cells, and error bars show SEM. Cells with basal Ca2+ levels >3x larger than the average for untransfected cells were excluded. B) Mean Ca2+ levels before and 10 min after addition of 150 nM thapsigargin for averaged individual COS7 cells expressing a combination of wtSTIM1-mRFP or mutant STIM1 K(384-6)Q-mRFP with wtOrai1 or mutant Orai1ΔDE. Each bar represents the mean for 20-60 cells from 3 or more different experiments, and error bars show SEM.
). Fluo-4 fluorescence was normalized relative to the average prestimulation basal Ca2+ level in untransfected cells in the same field. Each time course represents the average of 10-18 cells, and error bars show SEM. Cells with basal Ca2+ levels >3x larger than the average for untransfected cells were excluded. B) Mean Ca2+ levels before and 10 min after addition of 150 nM thapsigargin for averaged individual COS7 cells expressing a combination of wtSTIM1-mRFP or mutant STIM1 K(384-6)Q-mRFP with wtOrai1 or mutant Orai1ΔDE. Each bar represents the mean for 20-60 cells from 3 or more different experiments, and error bars show SEM.
These observations, together with our previous results, led us to hypothesize that basic residues 384-386 of STIM1 directly engage the acidic coiled-coil at the C-terminus of Orai1, involving electrostatic attraction between the oppositely charged residues. If this is the case, wtOrai1 might not couple with STIM1 K(384-6)Q without this electrostatic attraction. Alternatively, electrostatics may contribute but may not be the dominant energetic contribution to the interaction between STIM1 and Orai1. In this latter case, the stimulated association of the wild type proteins might be restored if the charged sequences on both of these proteins are mutated to neutral amino acids. To distinguish these possibilities, we measured FRET between STIM1 K(384-6)Q-mRFP and AcGFP-Orai1ΔDE, and we found that thapsigargin stimulates their physical association to a similar extent as for the wild type proteins, but with substantially slower kinetics (Figure 1). This finding is similar to the slow FRET-detected association between wtSTIM1-mRFP and AcGFP-Orai1ΔDE that we previously reported (5; Figure 1). Consistent with the FRET observed, STIM1 K(384-6)Q-mRFP co-patches with AcGFP-Orai1ΔDE in plasma membrane punctae in response to thapsigargin, similar to that seen with wtSTIM1-mRFP and AcGFP-Orai1ΔDE (Figure 2B,F). As with FRET-detected association, this visible co-patching of STIM1 K(384-6)Q-mRFP with AcGFP-Orai1ΔDE requires longer stimulation times with thapsigarin than for the corresponding wt constructs.
This finding that STIM1 K(384-6)Q undergoes stimulated association with Orai1ΔDE, but does not associate with its wild-type partner, suggests a model for the interaction between these two proteins and the role of the charged sequences in this interaction (Figure 4). Although the charged sequence on STIM1 and its resulting electrostatic attraction to the acidic sequence on Orai1 appears to contribute little to the overall binding energy of the active complex, mutation to eliminate the charged residues on STIM1 clearly reduces the interaction energy with its wild type partner. Thus, the simplest model to account for these observations is one in which this acidic sequence on Orai1 normally interacts with the basic sequence on STIM1 in an attractive electrostatic interaction.
Figure 4.

Cartoon depicting basic features of proposed model for wild type and mutant STIM1 and Orai1 interactions. A) In unstimulated cells, the negatively charged acidic amino acids on wtOrai1 and the positively charged lysine sequence (K384-386) in the STIM1 CAD domain (dark grey) defined by our mutations are shown associated with solvated counter ions. Upon stimulation, these ions are displaced from the binding cleft between the two proteins, allowing these charged regions to interact with each other and causing the Ca2+-selective channel to open in an allosteric manner. B) When K384-386 in STIM1 is mutated to glutamines, STIM1 no longer binds and activates wtOrai1 due to the loss of electrostatic attraction and resulting repulsion at this interaction subsite. C) When these charged sequences on Orai1 and STIM1 are both mutated, STIM1 CAD regains the capacity to bind Orai1 via interactions at other sites because the electrostatic repulsion has been eliminated. However, this association does not cause allosteric activation of the Ca2+ gate. The oligomerization state of these proteins is omitted for clarity: Orai1 is oligomerized in the active wtSTIM-wtOrai1 complex and in all cases involving Orai1ΔDE.
To determine whether stimulated association between STIM1 K(384-6)Q and Orai1ΔDE results in SOCE, we monitored Ca2+ mobilization in COS7 cells expressing these mutants. As shown in Figure 3A, we did not detect a significant Ca2+ response to thapsigargin (red squares), compared to untransfected COS7 cells (pink circles). The absence of SOCE with these constructs, which clearly undergo thapsigargin-stimulated association detected by FRET (Figure 1) and co-patching (Figure 2F), implicates these sequences in the gating of Ca2+ influx. Our proposed model accounts for these results: Ca2+ gating is controlled by the C-terminal acidic coiled-coil of Orai1, and engagement of this sequence with basic residues 383-387 in the CAD region of STIM1 induces the formation of the active CRAC channel (Figure 4A).
Our previous results provided evidence that the acidic coiled-coil of Orai1 controls its oligomerization state, possibly based on mutual electrostatic repulsion (5). In our current model, STIM1 facilitates Ca2+ mobilization in part by inducing the oligomerization of Orai1. Modulation of electrostatics at the faces of coiled-coils has been implicated previously in the control of the oligomerization state of different proteins (14,15), consistent with our view that, by neutralizing the acidic coiled-coil on Orai1, STIM1 383-387 reduces the charge-charge repulsion between individual Orai1 monomers and allows them to oligomerize into an active state. Our model is also consistent with our observations that neutralization of the acidic residues on the C-terminal coiled-coil of Orai1 induces plasma membrane patching of this protein in the absence of store depletion or STIM1 translocation (Figure 2A) (5).
We investigated possible Ca2+ influx through spontaneous oligomerization of Orai1ΔDE. Figure 3B summarizes average Ca2+ levels before and 10 min after addition of 150 nM thapsigargin for a large number of individual cells. For unstimulated cells, there is a small but significant increase in the average Ca2+ levels of cells expressing Orai1ΔDE compared to untransfected cells or cells transfected with wtOrai1. We find that a small fraction (~10-20%) of these cells have a basal state of cytoplasmic Ca2+ that is >3-fold above that of untransfected cells, suggesting they have spontaneously activated CRAC channels (supplemental Figure S1A). However, cells expressing Orai1ΔDE with both high and low resting Ca2+ levels are similarly unresponsive to thapsigargin, consistent with the incapacity of Orai1ΔDE to be activated by STIM1 (Figure S1B). This variation in resting Ca2+ levels we observe for cells expressing clustered Orai1ΔDE indicates that there must be additional factor(s) controlling the gating of Ca2+ entry through Orai1 that varies among cells; one possibility is population differences in polyphosphoinositide levels (16).
To account for the activation of SOCE, we propose that, in addition to inducing Orai1 oligomerization, binding of the STIM1 K(384-386) sequence to the acidic residues in the coiled-coil of Orai1 causes an allosteric change that directly opens the Ca2+ selective pore in the STIM1-Orai1 complex. Park et al. showed that STIM1 CAD interacts with both the N and C termini of Orai1 (9). A possible scenario is that the C-terminal acidic coiled-coil of Orai1 interacts with its own N-terminus in the resting state, and STIM1 (346-348) disengages this interaction in activated cells, thereby opening the channel in an allosteric manner (Figure 4A). The N-terminus of Orai1 contains a poly-arginine sequence (Orai1 28-33) that could serve as a potential binding partner for its acidic coiled-coil in unstimulated cells. Yuan, et al. found that the N-terminus of Orai1 is important for activation of CRAC channels with full length STIM1, but not with a minimal interaction domain that is similar to CAD (10), suggesting additional changes in Orai1 structure may be involved in this activation process.
In summary, we find that the basic sequence in the C-terminus of STIM1, K384-386, is important for functional coupling of STIM1 with Orai1 that is driven by thapsigargin-mediated Ca2+ store depletion (Figure 4A,B). Futhermore, we demonstrate that mutation to neutralize acidic residues in the C-terminal coiled-coil of Orai1 restores its stimulated association with STIM1 K(384-6)Q, but does not restore SOCE (Figure 4C). Our results support a model in which both oligomerization of Orai1 and Ca2+ gating are dependent on electrostatic interaction between these sequences in Orai1 and STIM1. In addition, our results reveal that these two processes can be uncoupled from each other in this exquisitely regulated mechanism of SOCE activation.
Supplementary Material
Footnotes
Supporting Information Available: Figure S1. Population distribution of Ca2+ levels in cells expressing mutant and wild type STIM1 and Orai1. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by Chemistry/Biology Interface Training Grant (to N. Calloway), the American Chemical Society, Division of Medicinal Chemistry Predoctoral Fellowship (to N. Calloway), and by National Institutes of Health grant AI022449.
References
- 1.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 4.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calloway N, Vig M, Kinet JP, Holowka D, Baird B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol. Biol. Cell. 2009;20:389–399. doi: 10.1091/mbc.E07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Nat. Acad. Sci. 2007;104:9301–06. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:814–816. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gosse JA, Wagenknecht-Wiesner A, Holowka D, Baird B. Transmembrane sequences are determinants of immunoreceptor signaling. J. Immunol. 2005;175:2123–2131. doi: 10.4049/jimmunol.175.4.2123. [DOI] [PubMed] [Google Scholar]
- 12.Hull JJ, Lee JM, Kajigaya R, Matsumoto S. Bombyx mori homologs of STIM1 and Orai1 are essential components of the signal transduction cascade that regulates sex pheromone production. J. Biol. Chem. 2009;284:31200–31213. doi: 10.1074/jbc.M109.044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K, Romanin C. Molecular determinants of the coupling between STIM1 and Orai channels: Differential activation of Orai1,2,3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 2009;284:21696–21706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woolfson DN. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 15.Fairman R, Chao HG, Lavoie TB, Villafranca JJ, Matsueda GR, Novotny J. Design of heterotetrameric coiled coils: evidence for increased stabilization by Glu(−)-Lys(+) ion pair interactions. Biochemistry. 1996;35:2824–2829. doi: 10.1021/bi952784c. [DOI] [PubMed] [Google Scholar]
- 16.Vasudevan L, Jeromin A, Volpicelli-Daley L, De Camilli P, Holowka D, Baird B. The beta- and gamma-isoforms of type I PIP5K regulate distinct stages of Ca2+ signaling in mast cells. J. Cell Sci. 2009;122:2567–2574. doi: 10.1242/jcs.048124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.