

Abstract
Impaired apoptosis is a cancer hallmark, and some types of lymphomas and other cancers harbor mutations that directly affect key cell death regulators, such as Bcl-2 family members. However, because the majority of tumors seem to lack such mutations, we are examining the hypothesis that tumorigenesis can be sustained at least initially by the normal expression of specific endogenous pro-survival Bcl-2 family members. We previously demonstrated that the lymphomagenesis in Εμ-myc transgenic mice, which constitutively overexpress the c-Myc oncoprotein in B-lymphoid cells and develop pre-B and B-cell lymphomas, does not require endogenous Bcl-2. In striking contrast, we report here that loss in these mice of its close relative Bcl-xL attenuated the pre-neoplastic expansion of pro-B and pre-B cells otherwise driven by c-Myc overexpression, sensitized these cells to apoptosis and ablated lymphoma formation. Remarkably, even loss of a single bcl-x allele delayed the lymphomagenesis. These findings identify Bcl-xL as a prerequisite for the emergence of c-Myc–driven pre-B/B lymphoma and suggest that BH3 mimetic drugs may provide a prophylactic strategy for c-Myc–driven tumors.
Introduction
Impaired apoptosis is considered a prerequisite for the development of most, if not all, cancers,1–3 yet the mechanisms that promote the survival of most nascent malignant cells during the process of neoplastic transformation remain unknown. Apoptosis is regulated by opposing factions of the Bcl-2 family, which include both proteins essential for cell survival and those that drive cell death.4,5 Despite very similar biochemical functions, the pro-survival family members (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1) have proven to have essential functions in specific cell types.5 The critical initiators of apoptosis are the “BH3-only” proteins (eg, Bim, Puma, Bid), so-called because they share only a single Bcl-2 homology (BH) domain with other family members. They are activated by developmental cues and diverse stress stimuli, including cytokine deprivation, DNA damage and activation of oncogenes, such as c-Myc.6–10 A second proapoptotic Bcl-2 sub-family, principally represented by the multi-BH domain proteins Bax and Bak, mediates the pivotal downstream step of mitochondrial outer membrane permeabilization (MOMP), which evokes activation of the caspase cascade that demolishes the cell.4,11 The BH3-only proteins may provoke activation of Bax and Bak by their direct engagement, by sequestering pro-survival relatives or both ways.4,5,12–14
Studies using transgenic mice have established that overexpression of Bcl-215–18 or its antiapoptotic relatives, including Bcl-xL19 and Mcl-1,20,21 or loss of proapoptotic Bcl-2 family members, such as Bim10 or Puma,22–24 contribute to tumor development, particularly in conjunction with mutations that deregulate cell cycle control, such as enforced c-Myc expression. Importantly, lymphomas evoked by combined overexpression of Bcl-2 and c-Myc require sustained Bcl-2 overexpression,25 most likely to counter the apoptosis promoted by c-Myc overexpression under stress conditions, such as limiting supply of growth factors.26–29 However, because only a proportion of cancers contain cytogenetic alterations (chromosomal translocations or somatically acquired copy number alterations) that directly promote the overexpression of Bcl-2 or one of its homologs,2,3,30 we surmise that most cells undergoing neoplastic transformation are sustained (at least initially) by the normal expression of endogenous Bcl-2–like antiapoptotic proteins. Identification of the pro-survival Bcl-2 proteins critical for development of a tumor is expected to guide the design of prophylactic and early intervention strategies.
We previously investigated the contribution of endogenous Bcl-2 to lymphoma development31 by interbreeding Eμ-myc transgenic mice32 with Bcl-2–deficient mice33,34 and using fetal liver-derived stem cells from their offspring to reconstitute lethally irradiated wt mice with an Eμ-myc/bcl-2−/− (or control genotype) hematopoietic system. In the transgenic pre-malignant B lymphoid cells, Bcl-2 proved critical for the survival of mature (sIg+) B cells but largely dispensable for that of pro-B and pre-B cells. Despite the marked deficit in mature pre-leukemic B lymphoid cells, however, the onset and incidence of Eμ-myc pre-B/B lymphoma were unaffected.31 This suggested that the tumors arise from B cell precursors, whose survival during neoplastic transformation might be sustained by a Bcl-2 relative. An appealing candidate is Bcl-xL, because it is highly expressed in early B lineage cells35 and required for their maintenance.36 Indeed, the studies reported here demonstrate that Bcl-xL is essential for the survival of Myc-driven pre-leukemic cells and for lymphoma development.
Methods
Mice
Experiments with mice were conducted with approval from The Walter and Eliza Hall Institute Animal Ethics Committee. Eμ-myc transgenic32 and bcl-x+/− 36 mice have been described. These strains had been backcrossed to the C57BL/6-Ly5.2 genetic background for > 30 or > 10 generations, respectively. The rag-2−/− mice37 (Taconic) were backcrossed to the C57BL/6-Ly5.1 genetic background for > 15 generations.
Hematopoietic stem cell reconstitution
Eμ-myc/bcl-x−/− E12.5 (Ly5.2) embryos were generated as follows: Eμ-myc transgenic males were first crossed with bcl-x+/− females and their Eμ-myc/bcl-x+/− male offspring were then mated with bcl-x+/− females. The day when the vaginal plug was detected was deemed embryonic day 0.5. Embryos from timed matings were harvested at E12.5 and genotyped by PCR on tail-derived DNA. Immediately before injection, single cell suspensions were prepared from fetal livers and ∼ 2 × 106 cells mixed with ∼ 1 × 105 rag-2−/− (C57BL/6-Ly5.1) bone marrow leukocytes and injected into the tail vein of lethally irradiated (2 × 5.5 Gy, at 3-hour intervals) C57BL/6-Ly5.1 mice. Mice were maintained on neomycin sulfate-supplemented drinking water for 14 days after irradiation to prevent infection.
Lymphoma monitoring and statistical analysis
Mice were monitored daily for signs of lymphoma development. Tumor-free survival was defined as the time from lethal irradiation and hematopoietic reconstitution with fetal liver cells to the time the animal was deemed ill by an experienced animal technician. To verify that mice used for pre-leukemic analysis lacked malignant cells, 1 × 106 of their bone marrow and/or spleen cells were transplanted into non-irradiated histocompatible recipient mice, which were then monitored for 90 days for the development of any tumor. Kaplan-Meier survival curves were constructed using GraphPad Prism Version 5.0, and statistical analysis performed using a 1-way ANOVA test (for comparison of 3 cohorts), or log-rank (Mantel-Cox) test (for comparison of 2 cohorts).
Analysis of the hematopoietic system of reconstituted mice
Bone marrow (both femora), spleen and lymph nodes (combined mesenteric, inguinal and axillary) were harvested and single cell suspensions prepared. Total cell numbers were determined by trypan blue staining and counting in a hemocytometer. Absolute numbers in each cellular compartment were calculated by multiplying the percentage of a cell type (as determined by FACS analysis) by the total organ cellularity. Donor-derived cell numbers were calculated by multiplying the percentage donor-derived (Ly5.2+) cells by the total organ cellularity. Peripheral blood was harvested via retro-orbital bleed or at sacrifice by cardiac puncture and collected into heparinized vessels. Total white blood cells (WBCs), red blood cells (RBCs), the percentage of hematocrit and hemoglobin levels were determined using an Advia 120 blood analyzer equipped with a mouse analysis software module (Bayer).
Immunofluorescent staining, flow cytometric analysis, and cell sorting
Before FACS-based immuno-phenotyping, peripheral blood was depleted of red blood cells by incubation (2 × 5 minutes) with red cell lysis buffer (56mM NH4Cl, 0.1mM EDTA, 12mM NaHCO3; pH 7.3) at 4°C. To prevent non-specific antibody binding, cells were incubated in the presence of 2.4G2 (anti-FcgRII) antibody plus 2% normal rat serum. Host (Ly5.1+) and donor-derived (Ly5.2+) cells were discriminated by staining with anti-Ly5.1 (A201.1) and anti-Ly5.2 (5.450.15.2) monoclonal antibodies. Pre-leukemic pro-B (B220+c-Kit+sIgM−sIgD−), pre-B (B220+c-Kit−sIgM−sIgD−) and sIg+ (B220+c-Kit−sIg+) B lymphoid populations were purified by FACS sorting after staining with monoclonal antibodies to: B220 (RA3-6B2), c-Kit (ACK2 or ACK4), IgM (5.1 or 333.12) and IgD (11-26C). Antibodies were produced in our laboratory and conjugated to biotin, FITC (both from Molecular Probes), cyanine 5 (Cy5, Amersham), R-phycoerythin (R-PE) or allophycocyanin (APC, both from Prozyme) according to the manufacturers' instructions. Biotinylated antibodies were detected by secondary staining with FITC-, PE- or Tricolor-coupled streptavidin (Caltag). Dead cells were excluded by staining with propidium iodide (2 μg/mL). Purification of B lymphoid sub-populations was performed by multi-parameter FACS sorting using a MoFlo (Cytomation) or DiVa (BD Biosciences) high-speed flow cytometer.
Cell cycle analysis
Bone marrow was harvested from the femora of pre-leukemic mice at 8-12 weeks after reconstitution. Fetal liver donor-derived pro-B (Ly5.2+B220+c-Kit+sIg−) and pre-B cells (Ly5.2+B220+c-Kit−sIg−) were purified by multi-parameter FACS sorting, fixed in 70% ice-cold ethanol and placed at −20°C overnight. Cells were washed twice in MT-PBS and incubated with 200 μL PI solution (38mM Na3C6H5O7, pH 7.4, 690nM propidium iodide, 5 μg/mL RNaseA) at 37°C for 20 minutes. DNA content was measured by acquiring an equal number of events (> 104 cells/sample) using a FACScan (BD Biosciences) with linear amplification settings.
Cell culture and cell survival analysis
FACS-sorted B lymphoid sub-populations were cultured in a humidified incubator (10% CO2) at 37°C in flat bottom 96-well microtitre plates at 2-5 × 104 cells/100 μL (per time point) in the high glucose version of DMEM supplemented with 250μM L-asparagine, 50μM 2-mercaptoethanol and 10% heat-inactivated FCS (JRH Biosciences). Cells were cultured in simple medium (no added growth factors) to assess the effects of cytokine deprivation. Cells were harvested at the indicated time points and viability determined by staining with PI and FITC-conjugated annexin V followed by flow cytometric analysis using a FACScan analyzer (BD Biosciences).
Results
Loss of a single bcl-x allele delays Εμ-myc–driven lymphomagenesis
Because endogenous Bcl-2 proved dispensable for Myc-induced lymphomagenesis31 and Bcl-xL is expressed at relatively high levels in pro-B and pre-B cells,35 the stages from which Εμ-myc lymphomas are thought to arise,32,38,39 we sought to examine the role of Bcl-xL in this process. To do so, we first crossed Εμ-myc transgenic32 mice with bcl-x+/− mice36 and compared lymphoma development in their progeny (Figure 1). Remarkably, loss of a single bcl-x allele significantly delayed the lymphomagenesis: the median survival rose from 116 days for Εμ-myc mice to 174 days for Εμ-myc/bcl-x+/− animals (P = .015). In contrast, and consistent with our previous study,31 loss of 1 allele of bcl-2 did not significantly affect Εμ-myc–induced lymphomagenesis (Figure 1; P = .303).
Figure 1.

Loss of one bcl-x allele significantly delays Εμ-myc lymphoma development. Kaplan-Meier analysis of tumor development comparing control Εμ-myc transgenic mice with Εμ-myc/bcl-2+/− and Εμ-myc/bcl-x+/− mice. Median tumor onset was 116 days for Εμ-myc, 154 days for Εμ-myc/bcl-2+/− (P = .303) and 174 days for Εμ-myc/bcl-x+/− mice (P = .023).
Bcl-xL loss reduces accumulation of Εμ-myc pre-leukemic B lymphoid cells
To explore the impact of complete loss of endogenous Bcl-xL in Myc-driven lymphomagenesis, we circumvented the embryonic lethality caused by germ line bcl-x deficiency36 by generating hematopoietic chimeric mice. To this end, lethally irradiated C57BL/6-Ly5.1+ (wt) mice were transplanted with hematopoietic stem/progenitor cells from the fetal liver of E12.5 Eμ-myc/bcl-x−/− (Ly5.2+) embryos or, as controls, Eμ-myc/bcl-x+/+ (hereafter “Eμ-myc”), bcl-x−/− or wt embryos (Figure 2A). The erythroid deficit caused by Bcl-xL deficiency36 was overcome by coinjecting hematopoietic stem/progenitor cells from rag-2−/− (C57BL/6-Ly5.1) mice (Figure 2B), which contribute to erythroid (and myeloid) but not lymphoid development.37 Analysis of the peripheral blood also revealed that the Eμ-myc/bcl-x−/− fetal liver cells could readily engraft and yield cells of the B lymphoid and other hematopoietic lineages (Figure 2C).
Figure 2.

Eμ-myc/bcl-x−/− fetal liver cells engraft and restore hematopoiesis in lethally irradiated recipient mice. (A) Generation of mice reconstituted with an Eμ-myc/bcl-x−/− (Ly5.2+) hematopoietic system. Fetal liver cell (FLC) suspensions were prepared from Eμ-myc/bcl-x−/− or, as controls, Eμ-myc, bcl-x−/− or non-transgenic wt E12.5 embryos (all C57BL/6-Ly5.2). The FLC (2 × 106) were combined with ∼ 1 × 105 bone marrow cells from rag-2−/− (C57BL/6-Ly5.1) mice and injected intravenously into lethally irradiated C57BL/6-Ly5.1 recipient mice. (B) Functional erythropoiesis in lethally irradiated mice coreconstituted with Eμ-myc/bcl-x−/− fetal liver cells plus rag-2−/− bone marrow leukocytes. Peripheral blood was harvested from reconstituted mice generated as shown in panel A and subjected to automated analysis (Advia 120; Bayer). Data shown demonstrate that (left panel) red blood cell numbers (× 109/mL), (middle panel) hematocrit (%) and (right panel) hemoglobin content (g/dL) were all within the normal range (normal ranges for these parameters for un-manipulated, adult C57BL/6 mice are indicated by brackets on the right side of the graphs). Data represent means ± SEM from 6-12 recipient mice transplanted with fetal liver cells of each genotype and were derived from at least 3 separate sets of analyses. (C) Peripheral blood leukocytes from reconstituted animals (generated as described in panel A) were stained with fluorochrome-conjugated monoclonal antibodies to Ly5.1, Ly5.2, B220, IgM and IgD and analyzed by flow cytometry. Representative FACS profiles demonstrate the relative frequencies of fetal liver donor-derived (gated on Ly5.2+) B lymphoid cells (B220+sIg+) in peripheral blood of mice reconstituted with fetal liver cells of the indicated genotypes. A summary of such data from analysis of bloods from multiple animals of each genotype is shown in Figure 3B.
To determine how loss of endogenous Bcl-xL affected development of Εμ-myc transgenic B cells, we isolated the blood, bone marrow, spleen and lymph nodes of pre-leukemic mice at 8-12 weeks after reconstitution and enumerated pro-B, pre-B and sIg+ B cells by flow cytometric analysis (Figure 3). Because deregulated c-Myc expression enhances proliferation of B lymphoid precursors but retards their subsequent differentiation, the bone marrow of Eμ-myc mice displayed a marked increase in pre-B cells (B220+c-Kit− sIg−) over non-transgenic mice (Figure 3A), but the bone marrow, blood, spleen and lymph nodes all had several-fold fewer sIg+ B cells (Figure 3A-D), consistent with previous observations.38 Notably, Bcl-xL loss significantly reduced the level of Εμ-myc pro-B (P < .05) and pre-B cells (P < .01) in the bone marrow (Figure 3A), as well as the sIg+ B cells (P < .01) in the bone marrow, blood, spleen and lymph nodes (Figure 3A-D).
Figure 3.

Loss of Bcl-xL causes a reduction in Εμ-myc transgene expressing pro-B, pre-B, and sIg+ B cells. Lethally irradiated C57BL/6-Ly5.1 mice were reconstituted (as shown in Figure 2A) with fetal liver cells from wt, bcl-x−/−, Eμ-myc or Eμ-myc/bcl-x−/− embryos. Bone marrow (A), peripheral blood (B), spleen (C) and lymph nodes (axillary, brachial, inguinal plus mesenteric; D) were harvested from pre-leukemic mice 8-12 weeks after reconstitution. Flow cytometric analysis was used to determine the absolute numbers of fetal liver donor–derived (Ly5.2+) total B lymphoid cells, pro-B cells (Ly5.2+B220+c-Kit+sIg−), pre-B cells (Ly5.2+B220+c-Kit−sIg−) and sIg+ B cells (Ly5.2+B220+c-Kit−sIg+) within these tissues. Data represent mean ± SEM from 4-6 recipient mice transplanted with fetal liver cells of each genotype. *P < .05 and **P < .01 denote statistically significant differences between Eμ-myc/bcl-x−/− and Eμ-myc reconstituted mice.
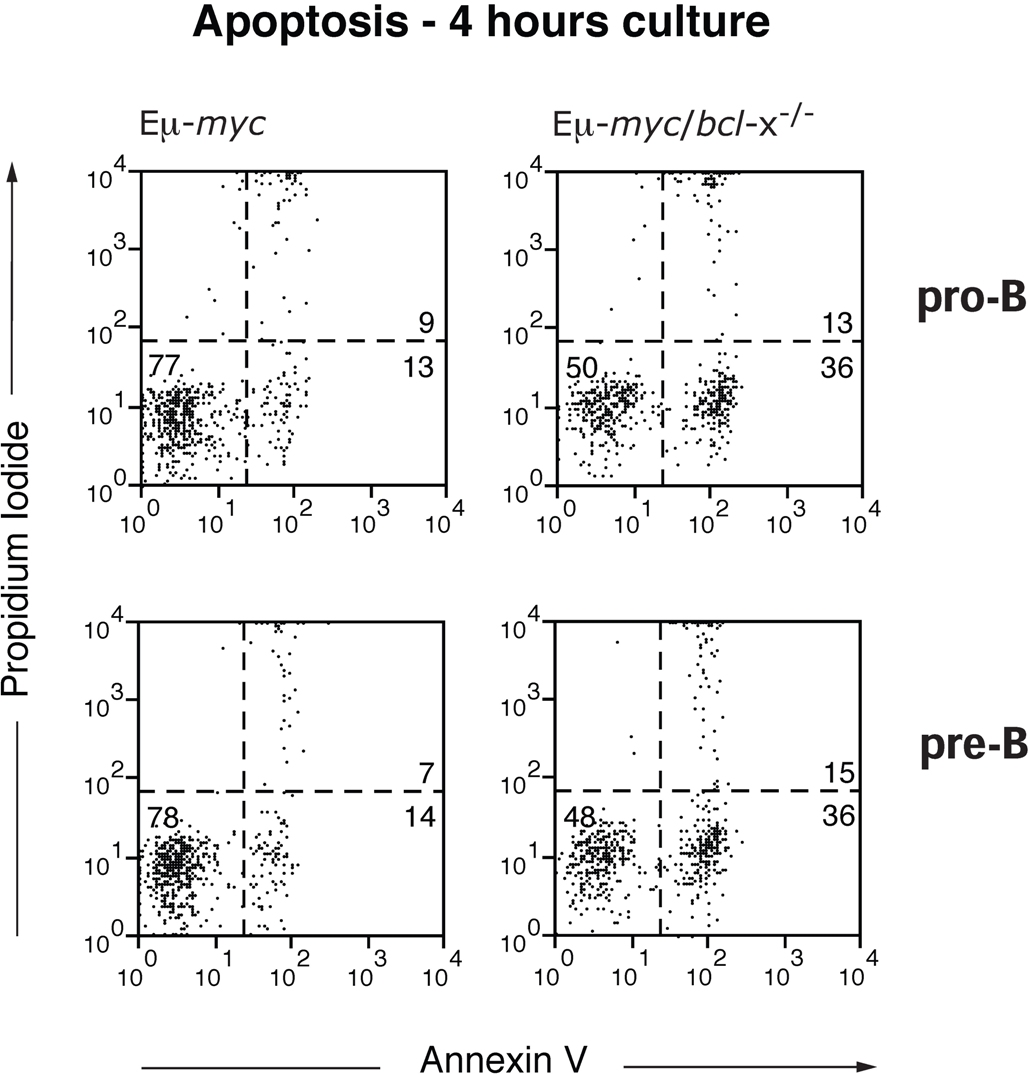
Overexpression of c-Myc enhances the apoptosis of diverse cell types (including lymphocytes) subjected to many stress stimuli, including growth factor deprivation.26,29 To determine whether the reduction in early B lymphoid cells in Eμ-myc/bcl-x−/− mice reflected enhanced c-Myc-induced apoptosis, we analyzed their survival when cultured without cytokine support (Figure 4A). As reported,29,31 Eμ-myc pro-B and pre-B cells died faster than their non-transgenic counterparts (not shown). Notably, Bcl-xL deficiency provoked significant (P < .05) further acceleration of cell death: by 16 hours of culture, the viability of Eμ-myc/bcl-x−/− pro-B or pre-B cells was only ∼ 25 to ∼ 30% that of their Eμ-myc counterparts (Figure 4A). Flow cytometric analysis confirmed that the exacerbated cell death in culture of the Myc-driven cells lacking Bcl-xL reflected apoptosis (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). As expected, loss of Bcl-xL did not affect the enhanced cycling of the B lymphoid cells evoked by Εμ-myc transgene expression (Figure 4B). Thus, whereas endogenous Bcl-2 is critical primarily for the survival of pre-leukemic sIg+ Εμ-myc B lymphoid cells,31 endogenous Bcl-xL is also essential to maintain their precursors.
Figure 4.

Loss of Bcl-xL accelerates the death of Εμ-myc pro-B and pre-B cells in culture but has no impact on their cell cycling. (A) Accelerated apoptosis of Eμ-myc/bcl-x−/− pro-B and pre-B cells in culture. In vitro cell survival assays were performed with FACS-purified donor-derived pro-B cells (Ly5.2+B220+c-Kit+sIg−) or pre-B cells (Ly5.2+B220+c-Kit−sIg−) from the bone marrow of pre-leukemic mice 8-12 weeks after hematopoietic reconstitution (generated as shown in Figure 2A). The cells were cultured in the absence of cytokines (simple medium) for the indicated periods and cell viability measured by staining with PI plus annexin V-FITC, followed by FACS analysis. Viable cells were defined as PI−/annexin V−. Data represent mean ± SEM from at least 3 independent experiments with cells of each genotype derived from 3-5 reconstituted mice. Because of the rapid death of Eμ-myc/bcl-x−/− pro-B cells in culture, times after 24 hours were not examined. *Significantly faster death (P < .05) of Eμ-myc/bcl-x−/− than Eμ-myc B lymphoid cells. (B) Bcl-xL loss does not affect the increased cycling of pre-leukemic Εμ-myc B lymphoid cells Donor fetal liver–derived pro-B cells (Ly5.2+B220+c-Kit+sIg−) and pre-B cells (Ly5.2+B220+c-Kit−sIg−) were FACS sorted from the bone marrow of pre-leukemic reconstituted mice (generated as shown in Figure 2A). Sorted cells were fixed in 70% ethanol and stained with propidium iodide to quantify DNA content. The indicated proportions of cells in the G0/G1 and S/G2/M stages of the cycle were determined by FACS analysis and quantification using manual gating. Data shown are representative of 3 independent experiments.
Loss of Bcl-xL prevents Εμ-myc–induced pre-B/B lymphoma development
To determine how loss of endogenous Bcl-xL affects lymphomagenesis, we monitored cohorts of the reconstituted mice. Eμ-myc reconstituted mice developed donor-derived (Ly5.2+) lymphoma with a median onset of 22 weeks and 40% mortality by 65 weeks (Figure 5), yielding ∼ 75% pre-B (B220+sIg−) and ∼ 25% sIg+ B lymphomas. In striking contrast, none of 25 Eμ-myc/bcl-x−/− animals monitored for 70 weeks developed a tumor (Figure 5; P < .001), and autopsy after this time revealed no overt signs of lymphoma.
Figure 5.

Bcl-xL is required for the development of Eμ-myc–induced B cell lymphoma. Kaplan-Meier analysis of tumor development in mice reconstituted (as shown in Figure 2A) with fetal liver cells from Eμ-myc (n = 41) or Eμ-myc/bcl-x−/− (n = 25) embryos. Mice were killed when deemed sick by an animal technician blinded to their genotype, and the presence of fetal liver donor-derived Εμ-myc pre-B or B lymphoma was confirmed by histological analysis and immuno-phenotyping. Cohorts of control mice (lethally irradiated C57BL/6-Ly5.1 coreconstituted with bcl-x−/− (n = 21) or wt (n = 27) fetal liver cells plus rag-2−/− bone marrow cells) were also followed. As anticipated, none developed pre-B or B-cell lymphoma within 70 weeks.
Discussion
The discovery that the bcl-2 gene, which is commonly translocated in follicular lymphoma,40 promotes cell survival15 and lymphomagenesis in transgenic mice16–18 engendered the now widely accepted concept that evasion of apoptosis is a critical step in neoplastic transformation.1–3 Nevertheless, because most tumors do not contain genomic aberrations that directly cause overexpression of a Bcl-2 homolog, it seems likely that the development of such cancers relies on the normal expression of an endogenous Bcl-2–like protein. Although Bcl-2 overexpression greatly accelerates lymphomagenesis in Eμ-myc mice,16 we found that endogenous Bcl-2 is dispensable for lymphoma development in these cancer-prone animals.31 In contrast, we show here that endogenous Bcl-xL is essential for Eμ-myc–driven lymphomagenesis (Figure 5).
The finding that Bcl-xL loss but not Bcl-2 deficiency prevented Myc-induced lymphoma development initially appeared surprising because loss of either gene reduced the accumulation of Eμ-myc–expressing pre-leukemic cells. However, whereas loss of either endogenous Bcl-231 or Bcl-xL (Figure 3) provoked a ∼ 5- to 10-fold drop in pre-leukemic sIg+ Eμ-myc B cells, only Bcl-xL loss substantially reduced the numbers of pre-leukemic pro-B and pre-B cells, sensitized them to apoptosis (Figure 3A) and abrogated lymphomagenesis (Figure 5). We surmise that loss of Bcl-xL but not of Bcl-2 ablated tumorigenesis primarily because Bcl-xL35 is expressed more highly than Bcl-241 in pro-B and pre-B cells, the cell types that most readily acquire collaborating oncogenic mutations because of their high proliferation rate and the genomic instability associated with Ig gene rearrangement.42
Two of our findings help to explain how the absence of Bcl-xL leads to ablation of Myc-induced lymphomagenesis (see model in Figure 6). The first is the approximately 5-fold lower number of pro-B and pre-B cells in the bone marrow of Eμ-myc/bcl-x−/− than Eμ-myc mice (Figure 3A). This decrease in the likely target cells for transformation almost certainly reflects their greater sensitivity to in vivo stresses, including in particular the apoptotic impetus promoted by Myc under conditions of stress, such as cytokine deprivation (Figure 4A). However, reduction in target cell numbers alone should markedly reduce but not ablate malignant transformation. Hence, the increased apoptosis evident on culture of the transgenic pro-B and pre-B cells lacking Bcl-xL suggests that the demise of these cells continues during the early stages of transformation, with Myc-driven proliferation balanced by Myc-induced apoptosis (Figure 6A).
Figure 6.

A model describing how loss of Bcl-xL prevents Εμ-myc–induced lymphoma development. (A) The combination of Myc-induced apoptosis and the absence of Bcl-xL pro-survival activity leads to a much smaller pool of pro-B and pre-B cells, the likely target cells for transformation, and the increased apoptosis at early stages of transformation balances Myc-induced proliferation, precluding tumor development. (B) The molecular basis of Myc-driven apoptosis. Myc overexpression up-regulates the BH3-only proteins Puma and Bim, in a p53-dependent manner for Puma and a p53-independent route for Bim.10,22–24 In bcl-x+/+ Myc-driven pre-leukemic cells, Bcl-xL and its pro-survival relatives (eg, Bcl-2) can sequester the up-regulated Puma and Bim, limiting the apoptosis. In the absence of Bcl-xL, however, the activated Bim and Puma overwhelm the remaining pro-survival proteins and (directly or indirectly) provoke activation of Bax and Bak. By balancing Myc-induced proliferation, the resulting apoptosis stops tumor formation.
Myc-driven apoptosis is understood in outline (Figure 6B). Over-expression of c-Myc increases expression of Bim and Puma, and loss of either of these proapoptotic BH3-only proteins accelerates Εμ-myc–induced lymphomagenesis by reducing Myc-induced apoptosis10,22–24,43. Hence, we surmise that in the absence of Bcl-xL the elevated levels of Bim and Puma induced by c-Myc overwhelm the remaining antiapoptotic Bcl-2 relatives (eg, Bcl-2) in early B lymphoid cells and provoke the activation of Bax and Bak (Figure 6B). The resulting apoptosis may both reduce the target population for transformation and eliminate pre-neoplastic clones otherwise destined for malignancy (Figure 6A). Pertinently, ∼ 20% of human mantle cell lymphomas exhibit bi-allelic BIM deletion44 and the c-MYC translocations that drive Burkitt lymphomas are reportedly sometimes accompanied by point mutations in c-MYC that prevent it from increasing Bim expression.43
Given that c-Myc is abnormally highly expressed in the great majority of human cancers,45 our demonstration that Bcl-xL is essential for Εμ-myc–induced lymphoma development has important implications for both oncogenesis and anti-cancer therapy. Notably, in many diverse human malignancies the genes for Bcl-xL (or Mcl-1) are amplified, most often in concert with MYC amplification.30 In addition, Burkitt lymphomas frequently express Bcl-xL,46 and notably one such lymphoma was recently found to bear not only the hallmark MYC translocation but also a translocation of BCL-X that drove its high expression.47
The BH3 mimetic ABT-73748 and the closely related ABT-263 (navitoclax),49 which target Bcl-2, Bcl-xL, and Bcl-w but not Mcl-1, are showing promise as anti-cancer agents.14 Our results suggest that specific BH3 mimetic drugs could also prevent the development of certain types of tumors by unleashing the proapoptotic impetus of the oncogenic changes they have sustained (eg, deregulated c-Myc expression). Hence, such drugs could contribute to the management of individuals with hereditary predisposition to malignancies and to reduce the likelihood of relapses or secondary malignancies in patients with sporadic cancers. A potential hazard would be induction of immune deficiency, but a recent study showed that conditional deletion of Bcl-x in antigen-activated B cells of mice had no effect on development of germinal centers or memory B cells and little on plasma cells, whereas deletion of Mcl-1 essentially abolished all 3 populations.50 Hence, a BH3 mimetic specific for Bcl-xL could have prophylactic potential for individuals at high risk of developing lymphomas and perhaps other tumors driven by Myc.
Supplementary Material
Acknowledgments
The authors thank Drs S. Cory, D. Lo, D. Huang, S. Nutt, P. Bouillet, M. Bath, and L. Tai for mice, reagents, and advice; K. Vella, G. Siciliano, D. Cooper, N. Iannarella, and J. Coughlin for animal care; J. Corbin for automated blood analysis; B. Helbert and C. Young for genotyping; Dr F. Battye, V. Milovac, C. Tarlinton, and J. Garbe for cell sorting; and D. Quilici, T. Nikolaou, and G. Thomas for irradiation.
This work was supported by grants and fellowships from the Cancer Council of Victoria (to P.N.K.), the National Health and Medical Research Council (Program Grant #461221, NHMRC Australia Fellowship), the National Institutes of Health (CA43540), and the Leukemia & Lymphoma Society (SCOR grant #7413). This work was made possible through State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.N.K. planned research, conducted experiments, interpreted data, and wrote manuscript; S.G. and A.R.D.D. conducted experiments, interpreted data, and helped with writing of manuscript; and J.M.A. and A.S. conceived the study, planned experiments, interpreted data, and wrote manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for P.N.K. is Metabolism Branch, National Cancer Institute/National Institutes of Health, Bethesda, MD.
Correspondence: Andreas Strasser, PhD or Jerry Adams, PhD, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, 3052, Victoria, Australia; e-mail: strasser@wehi.edu.au or adams@wehi.edu.au.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 3.Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8(2):121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 4.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 6.Bouillet P, Metcalf D, Huang DCS, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 7.Huang DCS, Strasser A. BH3-only proteins – essential initiators of apoptotic cell death. Cell. 2000;103(6):839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 8.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302(5647):1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 9.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 10.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101(16):6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6(6):1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 13.Merino D, Giam M, Hughes PD, et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186(3):355–362. doi: 10.1083/jcb.200905153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 16.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348(6299):331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 17.McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14;18). Nature. 1991;349(6306):254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 18.Strasser A, Harris AW, Cory S. Εmu-bcl-2 transgene facilitates spontaneous transformation of early pre-B and immunoglobulin-secreting cells but not T cells. Oncogene. 1993;8(1):1–9. [PubMed] [Google Scholar]
- 19.Swanson PJ, Kuslak SL, Fang W, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172(11):6684–6691. doi: 10.4049/jimmunol.172.11.6684. [DOI] [PubMed] [Google Scholar]
- 20.Zhou P, Levy NB, Xie H, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood. 2001;97(12):3902–3909. doi: 10.1182/blood.v97.12.3902. [DOI] [PubMed] [Google Scholar]
- 21.Campbell KJ, Bath ML, Turner ML, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116(17):3197–3207. doi: 10.1182/blood-2010-04-281071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci U S A. 2004;101(25):9333–9338. doi: 10.1073/pnas.0403286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrison SP, Jeffers JR, Yang C, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28(17):5391–5402. doi: 10.1128/MCB.00907-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michalak EM, Jansen ES, Happo L, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16(5):684–696. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6(3):241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69(1):119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 27.Fanidi A, Harrington EA, Evan GI. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature. 1992;359(6395):554–556. doi: 10.1038/359554a0. [DOI] [PubMed] [Google Scholar]
- 28.Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359(6395):552–554. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- 29.Strasser A, Elefanty AG, Harris AW, Cory S. Progenitor tumours from Εmu-bcl-2-myc transgenic mice have lymphomyeloid differentiation potential and reveal developmental differences in cell survival. EMBO J. 1996;15(15):3823–3834. [PMC free article] [PubMed] [Google Scholar]
- 30.Beroukhim R, Mermel C, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly PN, Puthalakath H, Adams JM, Strasser A. Endogenous bcl-2 is not required for the development of Emu-myc-induced B-cell lymphoma. Blood. 2007;109(11):4907–4913. doi: 10.1182/blood-2006-10-051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318(6046):533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama K, Nakayama K, Negishi, et al. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261(5128):1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 34.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75(2):229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 35.Grillot DAM, Merino R, Pena JC, et al. Bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183(2):381–391. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motoyama N, Wang FP, Roth KA, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x deficient mice. Science. 1995;267(5203):1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 37.Shinkai Y, Rathbun G, Lam K-P, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangements. Cell. 1992;68(5):855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 38.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in Emu-myc transgenic mice. Cell. 1986;47(1):11–18. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 39.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Emu-myc transgenic mouse: a model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167(2):353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 41.Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nuñez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. EMBO J. 1994;13(3):683–691. doi: 10.1002/j.1460-2075.1994.tb06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang JH, Gostissa M, Yan CT, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460(7252):231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436(7052):807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tagawa H, Karnan S, Suzuki R, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24(8):1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 45.Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genet Dev. 2010;20(1):91–95. doi: 10.1016/j.gde.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pujals A, Renouf B, Robert A, Chelouah S, Hollville E, Wiels J. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis. 2011;2:e184. doi: 10.1038/cddis.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turkmen S, Riehn M, Klopocki E, Molkentin M, Reinhardt R, Burmeister T. A BACH2-BCL2L1 fusion gene resulting from a t(6;20)(q15;q11.2) chromosomal translocation in the lymphoma cell line BLUE-1. Genes Chromosomes Cancer. 2011;50(6):389–396. doi: 10.1002/gcc.20863. [DOI] [PubMed] [Google Scholar]
- 48.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 49.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 50.Vikstrom I, Carotta S, Luethje K, et al. Mcl-1 is essential for germinal center formation and B cell memory. Science. 2010;330(6007):1095–1099. doi: 10.1126/science.1191793. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}