

Abstract
Recipient antigen-presenting cells (APCs) initiate GVHD by directly presenting host minor histocompatibility antigens (miHAs) to donor CD8 cells. However, later after transplantation, host APCs are replaced by donor APCs, and if pathogenic CD8 cells continue to require APC stimulation, then donor APCs must cross-present host miHAs. Consistent with this, CD8-mediated GVHD is reduced when donor APCs are MHC class I−. To study cross-presentation, we used hosts that express defined MHC class I Kb-restricted miHAs, crossed to Kb-deficient backgrounds, such that these antigens cannot be directly presented. Cross-priming was surprisingly efficient, whether antigen was restricted to the hematopoietic or nonhematopoietic compartments. Cross-primed CD8 cells were cytolytic and produced IFN-γ. CD8 cells were exclusively primed by donor CD11c+ cells, and optimal cross-priming required that they are stimulated by both type I IFNs and CD40L. In studying which donor APCs acquire host miHAs, we made the surprising discovery that there was a large-scale transfer of transmembrane proteins from irradiated hosts, including MHC class I–peptide complexes, to donor cells, including dendritic cells. Donor dendritic cells that acquired host MHC class I–peptide complexes were potent stimulators of peptide-specific T cells. These studies identify new therapeutic targets for GVHD treatment and a novel mechanism whereby donor APCs prime host-reactive T cells.
Introduction
Allogeneic hematopoietic stem cell transplantation (alloSCT) can be a life-saving therapy for hematologic malignancies and acquired or inherited nonmalignant disorders of blood cells. Mature donor T cells in allografts play important roles in alloSCT. They contribute to T-cell reconstitution in the recipient, promote donor hematopoietic engraftment, and mediate an anti–tumor effect called GVL. Unfortunately, donor T cells can broadly target host tissues causing GVHD.1 Because of GVHD, all patients receive some type of prophylactic immunosuppression, either by depleting T cells from the allograft, or more commonly, with pharmacologic agents and that inhibit T-cell function. However, even with pharmacologic immunosuppression, GVHD remains a major cause of morbidity and mortality. Novel approaches are clearly needed.
GVHD is initiated by antigen-presenting cells (APCs) that prime alloreactive donor T cells.1 In MHC-matched alloSCT, donor T cells target minor histocompatibility antigens (miHAs), which are the peptide products of polymorphic genes that distinguish the host from the donor. We previously found that direct presentation of MHC class I–restricted miHAs by recipient APCs is necessary for CD8-mediated GVHD in an MHC-matched, multiple miHA-mismatched model.2
However, although host APCs are key players, there was reason to believe that donor APCs should also play a significant role, as GVHD can begin and persist at times after transplantation when host hematopoietic cells, including APCs, are largely if not completely replaced by donor-derived cells. If, at these later times, alloreactive CD8 cells still need to be primed by professional APCs to be pathogenic, one would predict that donor APCs cross-presenting host miHAs would be important. On the other hand, it is possible that GVHD is maintained by a small number of persistent host APCs.3 In studies to address the role of donor APCs, we previously reported that CD8-mediated GVHD against miHAs is reduced when donor BM, and therefore donor APCs, lack MHC class I.4 One explanation for this result was that donor APCs cross-prime miHA-reactive donor CD8 cells. However, GVHD could have been reduced for a number of other reasons, including reduced survival and antigen responsiveness of donor CD8 cells in an MHC I− environment.5,6 Furthermore, even if cross-priming did occur, we could not determine whether naive donor CD8 cells were cross-primed or if donor APCs were only capable of stimulating CD8 cells that were previously primed by host APCs.
In this study, we directly address the hypothesis that cross-priming of MHC I–presented miHAs contributes significantly to GVHD. We took advantage of B6 mice that naturally or transgenically express H607 or transgenically express chicken ovalbumin (actOVA).8 Both H60 and OVA contain Kb-restricted immunodominant CD8 targets (LTFNYRNL and SIINFEKL, respectively).9 Importantly, MHC class I–tetramers and intracellular IFN-γ assays can readily detect and quantitate LTFNYRNL-10 and SIINFEKL-reactive CD8 cells. To prevent direct presentation of these peptides by donor APCs, we crossed these mice to B6 Kb−/− mice. We then used these mice as recipients in allogeneic bone marrow transplant (alloBMT) experiments.
Cross-priming was surprisingly efficient, whether antigen was restricted to the hematopoietic or nonhematopoietic compartments. CD8 cells were exclusively primed by donor CD11c+ cells, and optimal cross-priming required they be stimulated by both type I IFNs and CD40L. In studying which donor APCs acquired host miHAs, we made the surprising discovery that there was a large-scale transfer of transmembrane proteins from irradiated hosts, including MHC class I–peptide complexes, to donor cells, including dendritic cells (DCs). Donor DCs that acquired host MHC class I–peptide complexes were potent stimulators of peptide-specific T cells. These studies identify new therapeutic targets for GVHD treatment and a novel mechanism whereby donor APCs prime host-reactive T cells.
Methods
Mice
B6 mice were purchased from the NCI. C3H.SW mice (H-2b), CD40−/−, and OT-1 RAG−/− B6 mice were purchased from The Jackson Laboratory and bred at Yale. actOVA mice8 were acquired from Daniel Goldstein (Yale University School of Medicine [YUSM]) backcrossed to B6 and BALB/c. actH60 and B6.H60 mice (generated by D.R.)7 were bred at Yale. Kb−/− were purchased from The Jackson Laboratory and backcrossed to B6 actOVA, B6.H60, and actH60 mice to create actOVA.Kb−/−, B6.H60.Kb−/−, and actH60.Kb−/− mice, respectively.
ActOVA.Kb−/− mice were typed by flow cytometry of blood with antibodies against OVA (ab8389; Abcam) and Kb (AF6–88.5; BD Biosciences PharMingen). B6.H60.Kb−/− and actH60.Kb−/− mice were typed for Kb by flow cytometry of blood and by PCR for H60 with the following primers: actH60-forward, CAAGAGCAACCATTGCCTGATT; actH60-reverse, GCACACATCTTAGTAGCATTTTCCTCA; B6. H60-forward, GGTCCAACACTTTCTTCTCTCAGAGT; B6.H60-reverse, TACTTGTACGTGTATATTGAGTGTGAGTGT.
IFNAR1−/− mice11 were acquired from Helene Rosenberg (National Institutes of Health). CD11c-cre mice12 were obtained from Richard Flavell (YUSM). ROSA26-flox-stop-DTA mice13 were obtained from Mark Shlomchik (YUSM). These mice were intercrossed to create CD11c-cre × ROSA26-flox-stop-DTA mice (CD11c-DTA).13 Mice were typed by PCR with the following primers: CD11c-cre, CD11c promoter, ACTTGGCAGCTGTCTCCAAG; Cre ORF, GCGAACATCTTCAGGTTCTG; for ROSA26, R26R1 (5-AAAGTCGCTCTGAGTTGTTAT-3′), R26R2 (5′-GCGAAGAGTTTGTCCTCAACc-3′), and R26R3(5′-GGAGCGGGAGAAATGGATATG-3′). The depletion of CD11c+ cells was confirmed by flow cytometry of splenocytes when mice were harvested for use as BM donors.
Cell separations
All donor BM was depleted of T cells using anti–Thy1.2 microbeads and the AutoMACs (Miltenyi Biotec). CD8 or CD4 cells were purified from lymph nodes (LNs) by negative selection as previously described.14 In some experiments, bead-purified CD8+ T cells were stained with anti–CD62L (Mel-14; BD Biosciences PharMingen), anti-CD44 (IM7; BD Biosciences PharMingen), and sorted into CD62L+CD44− naive and CD44+ memory populations using a FACSAria (BD Immunocytometry Systems).
Bone marrow transplantation
All transplantations were performed according to protocols approved by the Yale University Institutional Animal Care and Use Committee. For experiments in the C3H.SW→B6 strain pairing, B6 background mice were irradiated with 1000 cGy and reconstituted with 5 to 7 × 106 C3H.SW BM cells with or without additional T cells. For experiments in the B6→BALB/c strain pairing, BALB/c background mice received 900 cGy and were reconstituted with 107 B6 background BM with or without B6 T cells. For experiments in the BALB/c→B6 actOVA strain pairing, hosts received 200 μg intraperitoneally of anti-NK1.1 (PK136; laboratory-prepared) on days −2 and −1 to diminish NK cell-mediated rejection. On day 0, mice were irradiated and reconstituted with 107 T cell–depleted BALB/c BM cells.
Tetramers
The H-2Kb/LTFNYRNL tetramer conjugated to phycoerythrin (PE) was obtained from the National Institutes of Health tetramer core facility (Atlanta, GA). The H-2Kb/SIINFEKL tetramers were laboratory-prepared as previously described.15,16 Biotinylated Kb-peptide complexes were tetramerized by the addition of allophycocyanin-conjugated streptavidin (Invitrogen).
Peptides
LTFNYRNL and SIINFEKL were synthesized by the Keck laboratory at YUSM.
Flow cytometry
Intracellular cytokine staining.
Splenocytes were cultured with 100nM SIINFEKL or LTFNYRNL for 5 hours; monensin was added for the final 2 hours. Before permeabilization, cells were incubated with ethidium monoazide to allow exclusion of dead cells. Cells were stained with antibodies against CD8, CD229.1 (Ly9.1; 30C7; BD Biosciences PharMingen), CD44, CD62L, permeabilized, and then stained with antibodies against IFN-γ (XMG1.2; BD Biosciences) or isotype controls (for the anti–cytokine antibodies). Analysis was performed on an LSRII (BD Immunocytometry Systems).
The 25-D1 hybridoma,17 which produces an antibody that recognizes SIINFEKL-Kb, was obtained from Peter Cresswell (YUSM; lab prepared) and was biotin-conjugated.
BrdU labeling and staining.
The in vivo proliferation of CD8+ T cells was assessed by injecting 2 mg 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) intraperitoneally 2 hours before death. Splenocytes were stained with antibodies against CD229.1, CD8, and SIINFEKL or LTFNYRNL-tetramers. Samples were stained for BrdU (FITC BrdU Flow Kit; BD Biosciences PharMingen) according to the manufacturer's instructions.
DEC-OVA memory cell generation
To generate SIINFEKL-reactive CD8 memory cells, C3H.SW mice were injected with 50 μg anti–DEC205-OVA18 (laboratory-prepared) and 50 μg anti–CD40 (FGK45; laboratory-prepared). Memory cells were harvested 2 months later.
In vivo CTL assay
Mice were transplanted as described in the text and were infused with LTFNYRNL- or SIINFEKL-pulsed splenocytes, labeled with CFSE or cyan (Invitrogen) respectively. One day after injection, splenocytes were harvested and CFSE+ and cyan+ cells were enumerated.
Confocal microscopy
Spleens were digested with collagenase and cells were stained with anti–CD11c-PE (HL3; BD Biosciences PharMingen) for 20 minutes, washed once, and resuspended in sort buffer. The sorted CD11c+ cells were stained with anti–Kd-biotin (SF1–1.1; BD Biosciences PharMingen), anti–Kb-allophycocyanin (AF6-88.5; BioLegend), followed by streptavidin-Alexa488 (Invitrogen). The stained cells were dropped onto slides and imaged by confocal microscopy (LSM Confocal Microscope, Carl Zeiss). Images were assembled using ImageJ software (National Institutes of Health).
Amnis ImageStream analysis
Splenocytes were stained with antibodies against Kd (FITC; SF1-1.1, BD Biosciences PharMingen), Kb (PE; AF6-88.5, BD Biosciences PharMingen), CD11c (Pacific Blue; N418, eBioscience), or CD8 (Pacific Blue; TIB105, laboratory-conjugated). Cell nuclei were stained with DRAQ5 (eBioscience), and dead cells were excluded with 7-amino-actinomycin D (7-AAD; eBioscience). Analysis was performed using the ImageStream instrument with 40× magnification (Amnis). The data were analyzed with IDEAS Version 4.0 software (Amnis).
OT-1 proliferation experiments
At day 6 after transplantation, BALB/c→B6actOVA mice were killed. Collagenase-digested splenocytes were stained with anti–CD11c-PE, anti–Kd-FITC, anti–Kb-allophycocyanin, and CD11c+Kb+Kd+ cells were sort-purified. The sorted cells were cultured with 105 CFSE-labeled OT1 cells. Cultures were harvested 3 days later, and CFSE dilution of OT-1 cells was assessed by flow cytometry.
Statistics
Differences in cell numbers or percentages were determined by Mann-Whitney U test. Comparisons are 2-tailed unless noted to be otherwise.
Results
Donor CD8 cells are cross-primed against H60 and OVA
To measure cross-priming, B6.H60 (congenic for H60; H60 expression is mostly limited to hematopoietic cells),19 B6.H60.Kb−/−, actH60.Kb−/− (H60 driven by an actin promoter; expression is ubiquitous but at a low level), and control Kb−/− mice were irradiated and reconstituted with C3H.SW (H-2b; H60−) BM and 2.4 × 106 CD8+ and 105 CD4+ polyclonal C3H.SW T cells. CD4 cells were infused as priming to H60 is optimal with CD4 help20–22 (and data not shown). Mice were killed on days 8, 14, 22, and 38 after transplantation and donor (Ly9.1+) H60-reactive CD8 cells were enumerated using the LTFNYRNL:Kb tetramer20 (TetH60). TetH60+ cells were readily found in spleen, BM, liver, and LN (Figure 1A, representative flow cytometry). TetH60+ cells were detectable by day 8 in all groups except for Kb−/− negative control mice, with a peak percentage in the spleen at day 14 after transplantation (Figure 1B). At day 14, we injected transplanted actH60.Kb−/− mice with a single pulse of BrdU 2 hours before sacrifice to determine the fraction of cycling TetH60+ cells. Between 21% and 50% of TetH60+ cells were in division, indicative of active cross-presentation and subsequent activation of LTFNYRNL-reactive CD8 cells (Figure 1C).
Figure 1.

LTFNYRNL derived from H60 is efficiently cross-presented. (A-D) Kb−/−, B6.H60, B6.H60.Kb−/−, and actH60 mice were irradiated and reconstituted with 6 × 106 BM cells, 2.4 × 106 CD8 cells, and 105 CD4 cells from C3H.SW (Ly9.1+) mice. (A) Representative flow cytometry at day 14 after BMT. (B) Percentage of Ly9.1+CD8+ cells in spleen (top panel) and liver (botom panel) that were TetH60+ (3 mice per group at each time point); bars represent SE. (C) Representative BrDU staining of Ly9.1+CD8+TetH60+ cells in actH60.Kb−/− recipients after a single BrDU pulse 2 hours before death at day 14. Each plot is from an individual mouse. (D) Representative staining for intracellular IFN-γ in LTFNYRNL-stimulated splenocytes harvested at day 14 (gated on live Ly9.1+CD8+ cells). (E-F) Mice were transplanted as in panels A through D; and on days 14 and 28, recipients were injected with LTFNYRNL- or SIINFEKL-pulsed C3H.SW splenocytes labeled with CFSE or cyan, respectively. Eighteen hours later, splenocytes were analyzed for the presence of CFSE+ and cyan+ cells. Representative flow cytometry at day 14 (E) and the ratios of cyan+/CFSE+ cells (F; each symbol is from an individual mouse). CD8 cells in actOVA.Kb−/− mice transplanted as in panels A to D mount a strong response to SIINFEKL (G). Shown is representative flow cytometry of spleen, LN, and blood cells at day 14 (gated on donor CD8+ cells). Data are representative of at least 2 experiments with similar results.
We also tested the function of cross-primed H60-reactive cells. They produced IFN-γ when restimulated with LTFNYRNL (Figure 1D), and the fraction of IFN-γ+ cells was proportional to the frequency of TetH60+ cells. To assess their cytolytic activity, at days 14 and 28, we infused C3H.SW splenocytes pulsed with LTFNYRNL (CFSE-labeled) or as a control splenocytes pulsed with SIINFEKL (cyan-labeled). Eighteen hours later, mice were killed and the ratios of cyan+/CFSE+ cells were determined (Figure 1E-F). LTFNYRNL-pulsed cells were selectively killed in B6.H60, B6.H60.Kb−/−, and actH60.Kb−/− but not in control Kb−/− mice.
Donor CD8 cells in similarly transplanted B6 actOVA.Kb−/− mice8 mounted strong responses to SIINFEKL (Figure 1G). Thus, epitopes from both H60 and OVA were efficiently cross-presented.
Both hematopoietic and nonhematopoietic antigens are cross-presented
That cross-primed CD8 cells could be detected early after transplantation suggested that hematopoietic antigens derived from apoptotic cells induced by irradiation were a key source of antigen. To test this, we used the actOVA system and compared the generation of SIINFEKL-reactive cells in actOVA.Kb−/− mice with that in retransplanted actOVA.Kb−/−→Kb−/− (hematopoietic OVA only) and Kb−/−→actOVA.Kb−/− (nonhematopoietic OVA) BM chimeric mice. We saw impressive and early cross-priming in both sets of chimeras (representative flow cytometry, Figure 2A; total number of TetSIINFEKL+ cells, Figure 2B). The tetramer data were paralleled by the fraction of cells that produced IFN-γ when restimulated with SIINFEKL peptide (Figure 2D). Thus, both hematopoietic and nonhematopoietic antigens were productively cross-presented.
Figure 2.

CD8 cells are cross-primed whether antigen is restricted to hematopoietic or nonhematopoietic cells. Kb−/−, actOVA, actOVA.Kb−/−, actOVA.Kb−/−→Kb−/−, and Kb−/−→actOVA.Kb−/− mice were irradiated and reconstituted with 6 × 106 BM cells, 2.4 × 106 CD8 cells, and 105 CD4 cells from C3H.SW mice. (A) Representative flow cytometry from day 13, gated on Ly9.1+CD8+ cells. (B) Total number of TetSIINFEKL+ cells per spleen on days 13 and 35. P = .007, comparing Kb−/− recipients versus actOVA.Kb−/− recipients. P < .0001, comparing actOVA versus actOVA.Kb−/− mice. P = .05, comparing Kb−/− recipients versus other groups except actOVA recipients (by one-tailed Mann-Whitney). Data from actOVA and actOVA.Kb−/− recipients are combined from 2 experiments with similar results. (C) Percentage of CD8 cells that were TetSIINFEKL+ in blood at day 13 (data combined from 3 experiments; P < .0001). Ly9.1+CD8+ splenocytes from mice transplanted as in panel A produced IFN-γ when restimulated with SIINFEKL at day 13 after BMT.
Tissue distribution of antigen with Kb, but not antigen alone, controls the magnitude of the T-cell response
Unexpectedly, TetSIINFEKL+ cells were more numerous in actOVA.Kb−/− than in actOVA recipients (Figure 2A-C). TetH60+ cells were also more numerous in actH60.Kb−/− mice than in actH60 mice (data not shown). These data suggested that either exclusive cross-priming was more efficient than was direct priming or that ubiquitous expression of an antigen with its presenting MHC class I molecule blunted the T-cell response. To distinguish these, we compared the accumulation of TetH60+ cells in retransplanted actH60→actH60, actH60→actH60.Kb−/−, and actH60.Kb−/−→actH60 BM chimeras (Figure 3A, representative flow cytometry; Figure 3B, total number of TetH60+ CD8 cells). As was the case in nonchimeric actH60 and actOVA mice, the response against H60 was blunted in actH60→actH60 recipients relative to that in actH60Kb−/−→actH60.Kb−/− mice. However, relative to actH60→actH60 mice, the number of TetH60+ cells was increased in actH60→actH60.Kb−/− recipients. Thus, even though hematopoietic LTFNYRNL could be directly presented in both actH60→actH60 and actH60→actH60.Kb−/− mice, when the presenting Kb was present in nonhematopoietic tissues, the accumulation of TetH60+ cells was reduced.
Figure 3.

Widespread expression of Kb-LTFNYRNL, but not LTFNYRNL without Kb, blunts the accumulation of TetH60+ cells. Kb−/−, actH60→actH60, actH60.Kb−/−→actH60.Kb−/−, actH60→actH60.Kb−/−, and actH60.Kb−/−→actH60 mice were irradiated and reconstituted with 6 × 106 BM cells, 2.4 × 106 CD8 cells, and 105 CD4 cells from C3H.SW mice. (A) Representative flow cytometry from day 14 (gated on Ly9.1+CD8+ cells). (B) Total numbers of Ly9.1+CD8+ TetH60+ cells per spleen at day 14. P = .0571, comparing actH60→actH60 to actH60→actH60.Kb−/− mice.
Cross-priming does not require CD4 cells
Because responses by naive CD8 cells against directly presented SIINFEKL and LTFNYRNL depend on CD4 help20,21 (and data not shown), we could not reliably use the cross-priming of naive CD8 cells to determine the degree to which cross-presentation itself requires CD4 cells. However, SIINFEKL-reactive CD8 memory cells can be directly restimulated after transfer to actOVA mice in vivo without CD4 help (not shown). We therefore created SIINFEKL-reactive memory CD8 cells and tested whether they required CD4 help to be activated by cross-presenting APCs. Irradiated actOVA.Kb−/− mice were reconstituted with C3H.SW BM and 5 × 105 sort-purified CD44+CD8+ cells from OVA-vaccinated mice (containing ∼ 2.5 × 104 TetSIINFEKL+ cells). At day 13, TetSIINFEKL+ cells were nearly 100-fold more numerous in actOVA.Kb−/− than in control Kb−/− recipients (Figure 4A, representative flow cytometry; Figure 4B, total number of TetSIINFEKL+ cells per spleen) indicating that cross-presentation does not require CD4 help.
Figure 4.

SIINFEKL-specific but not spontaneous memory T cells are restimulated by cross-presenting APCs. (A-B) Kb−/− and actOVA. Kb−/− mice were irradiated and reconstituted with C3H.SW BM and 5 × 105 sort-purified CD8+CD44+ CD8 cells from C3H.SW mice vaccinated 60 days earlier with anti–DEC205-OVA. Mice were killed 13 days after transplantation. (A) Representative flow cytometry (gating on Ly9.1+CD8+ cells). (B) Number of TetSIINFEKL+ cells per spleen. P = .024. (C) Kb−/− and actOVA.Kb−/− mice were transplanted as in panel A, except that they received C3H.SW sort-purified naive CD8+CD44−CD62L+ (1 × 106) or memory CD8+CD44+ (3 × 105) cells, with or without 105 CD4+CD25− cells. Representative flow cytometry of spleen, LN, and blood is shown in panel C (gated on Ly9.1+CD8+ cells). Percentage of CD8 cells that were TetSIINFEKL+ and total number of TetSIINFEKL+ per spleen are shown in panel D. P = .0043, comparing percentage and total number of TetSIINFEKL+ cells in recipients of CD4 cells and TN versus CD4 cells and TM. P < .036, comparing percentage and total number of TetSIINFEKL+ cells in recipients of TN with or without CD4 cells. Data combined from 2 independent experiments.
Naive CD8 cells are cross-primed
Given that SIINFEKL-reactive memory cells were readily cross-primed and that there is sufficient TCR repertoire in memory CD8 cells to cause GVHD in this strain pairing,23 we considered the possibility that memory CD8 cells cross-reactive against H60 or SIINFEKL were being cross-primed and not naive CD8 cells. To test this, we transplanted actOVA.Kb−/− mice with C3H.SW BM and 106 sort-purified naive (CD62L+CD44−) or 3 × 105 memory (CD44+) CD8 cells from unmanipulated C3H.SW mice along with 105 CD4+CD25− cells. An additional group also received 106 naive CD8 cells without CD4 cells. At day 13, TetSIINFEKL+ cells were present in naive CD8 plus CD4 but not memory CD8 plus CD4 recipients (Figure 4C, representative flow cytometry; Figure 4D, percentage and total number of TetSIINFEKL+ cells). TetSIINFEKL+ cells also expanded in recipients of naive CD8 cells and no CD4 cells, although they were approximately 10-fold fewer than when CD4 cells were also infused, confirming the importance of CD4 help.
Optimal cross-presentation requires CD40L and type I IFN stimulation of conventional DCs
If mechanisms by which APCs cross-present miHAs in alloBMT could be identified, they could be targeted for GVHD treatment. One approach to investigate critical mechanisms would be to use donor BM from mice deficient in genes that might be involved in cross-presentation24–27 in the actOVA.Kb−/− or actH60.Kb−/− systems. However, appropriate gene-deficient mice were not available back-crossed to C3H.SW. We therefore determined whether CD8 cells are cross-primed against SIINFEKL when BALB/c (H-2d) mice carrying the actOVA transgene (BALB/c-OVA) are transplanted with B6 BM and CD8 cells, as most gene-modified mice are available backcrossed to B6. Because BALB/c mice are Kd+Kb−, only donor-derived B6 APCs can present SIINFEKL. BALB/c-OVA or control BALB/c mice were irradiated and reconstituted with BM and 106 CD8 and 105 CD4 cells from B6 mice. By day 14, a clear TetSIINFEKL+ population developed in BALB/c-OVA recipients (Figure 5A, representative flow cytometry; Figure 5B, time course). In some experiments, we also saw TetH60+ cells in BALB/c recipients as they are H60+ (not shown). Cross-primed CD8 cells produced IFN-γ when restimulated with SIINFEKL (not shown) and selectively killed SIINFEKL-pulsed B6 splenocytes infused on day 13 into transplanted BALB/c-OVA recipients (Figure 5C). Thus, even though most alloreactive donor T cells recognize intact host MHC, there was still substantial cross-priming by donor APCs.
Figure 5.

Optimal cross-presentation requires CD40L and type I IFN stimulation of conventional DCs. BALB/c or BALB/c-OVA mice were irradiated and reconstituted with B6 (CD45.2) BM, 106 CD8, and 105 CD4 cells from B6 CD45.1 mice. (A) Representative flow cytometry of spleen, LN, and blood on day 14 (gated on CD8+CD45.1+ cells). (B) Time course (at least 3 mice/group at each time). At day 14 after transplantation, unpulsed (cyan) or SIINFEKL-pulsed (CFSE) B6 splenocytes were infused and spleens were harvested 18 hours later. Note the selective killing of SIINFEKL-pulsed cells (C). Mice were transplanted as in panel A, except that they received donor B6 BM from WT, MHC class II−, CD11c-cre x ROSA26-flox-stop-DTA, CD40−/−, or IFNAR1−/− mice. Recipients were killed on day 14. Shown is representative TetSIINFEKL staining (D; gated on CD8+CD45.1+ cells). Percentages of CD8 cells that were TetSIINFEKL+ are shown in panel E. Data from 3 experiments combined. P = .02, comparing recipients of WT BM compared with recipients of IFNAR1−/− or CD40−/− BM.
We then used B6 gene-deficient and transgenic mice as BM donors in the B6→BALB/c-OVA model to investigate mechanisms of cross-priming (Figure 5C-E). To test whether donor CD11c+ cells were the key cross-presenting APC, we used CD11c-cre transgenic mice28 crossed to ROSA26-flox-stop-DTA mice12 (CD11c-DTA) as BM donors. When cre is expressed, a stop cassette is excised 5′ of the DTA sequence, inducing its expression and a profound deficiency of both conventional and plasmacytoid DCs13,29 (and data not shown). Strikingly, we observed no TetSIINFEKL+ cells in recipients of CD11c-DTA BM. Given that mouse plasmacytoid DCs have not been shown to cross-present,30 these data make it likely that donor conventional DCs are the key cross-presenting cell. Cross-priming of naive CD8 cells is optimal with CD4 help.27 Surprisingly, however, we found cross-priming to be intact when donor BM was MHC class II−/−. Therefore, even though the CD8 response against SIINFEKL is optimal with CD4 help (Figure 4), CD4 cells need not make direct contact with cross-presenting APCs. Because some polyclonal CD4 cells in B6→BALB/c recipients would be activated by fully allogeneic BALB/c APCs, we reasoned that CD4 cells could be delivering signals in-trans to donor B6 DCs without direct TCR/MHC class II contacts. CD40L induced in alloreactive donor CD4 cells seemed a likely candidate molecule.24 We therefore tested whether cross-priming would be reduced if donor BM was CD40-deficient. Indeed, in all but one recipients of CD40−/− BM, we saw no TetSIINFEKL+ CD8 cells. Thus, it is likely that CD4 cells primed by host APCs activate donor APCs via CD40L in a manner that does not require them to make TCR contacts with MHC class II on the donor cross-presenting APCs. Type I IFNs have also been shown to promote cross-presentation.31 We therefore used the same system to determine whether cross-presentation would be reduced if donor BM was deficient in the type I IFN receptor (IFNAR1−/−). Although not completely eliminated, cross-presentation was significantly reduced in recipients of IFNAR1−/− donor BM. In sum, these data indicate that donor CD11c+ DCs are the key cell type that cross-primes donor CD8 cells and that for optimal cross-priming they need to be stimulated by both CD40L and type I IFNs.
Donor cells are “cross-dressed” with host-derived transmembrane proteins
After observing cross-priming in the C3H.SW→actOVA.Kb−/− experiments, we wanted to determine which donor cells cross-presented SIINFEKL. To do so, we stained donor-derived cells in transplanted C3H.SW→actOVA and C3H.SW→actOVA.Kb−/− mice with the 25-D1 antibody, which recognizes Kb-SIINFEKL complexes.17 We first focused on donor Ly9.1+ (donor-specific) CD11c+ cells, and to our surprise, we only observed significant 25-D1 staining in actOVA and not actOVA.Kb−/− recipients (Figure 6A), even though cross-priming occurred in the latter. The majority of Ly9.1+CD11c+ cells bound 25-D1 such that there was a shift in the entire population, rather than distinct positive and negative subsets. This was unlikely to be the result of doublets of C3H.SW and actOVA cells as we used narrow forward scatter and side scatter width gates to exclude these. In contrast, staining in actOVA.Kb−/− recipients was similar to that in Kb−/− controls. 25-D1 staining of donor cells in actOVA recipients was not limited to DCs but was also observed in CD4 and CD8 cells (Figure 6A). 25-D1 staining on donor cells in actOVA recipients was seen as early as day 3 and as late as day 11 (not shown).
Figure 6.

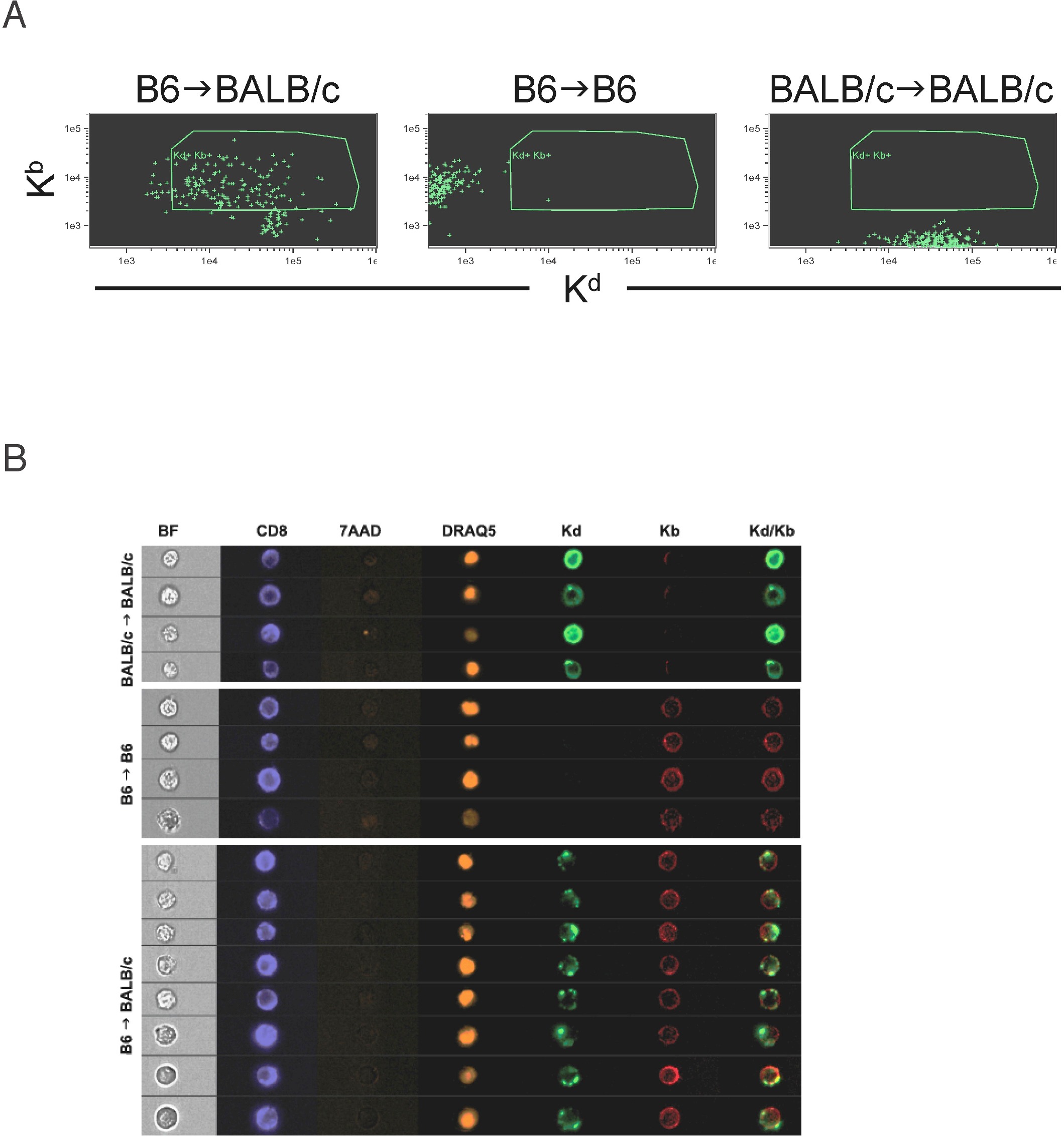
Donor cells acquire host transmembrane proteins, including MHC class I-SIINFEKL complexes. B6, actOVA, and actOVA.Kb−/− mice were irradiated and reconstituted with C3H.SW BM, 2 × 106 CD8 cells, and 1 × 105 CD4 cells. Mice were killed 5 days after transplantation. (A) Representative flow cytometry of 25-D1 (top row) and Kb staining (bottom row) of donor Ly9.1+ CD11c+, CD8+, and CD4+ cells. Data are representative of 2 independent experiments with at least 3 mice per group. (B) actOVA.Kb−/− and actOVA mice were irradiated; and 5 days later, WT CD45.1 B6 or CFSE-labeled Kb−/− splenocytes were infused intravenously. Two days later, mice were killed. Shown is 25-D1 staining of CD45.1+ (left panel) and CFSE+CD8 cells (right panel). Note the shift in 25-D1 staining, which occurred only in actOVA recipients. (C-D) BALB/c-OVA (CD45.2) mice were irradiated and reconstituted with B6 CD45.1 BM and 106 CD8 and 105 CD4 cells. On day 5, splenocytes were harvested and stained with antibodies against CD11c, Kb, Kd, CD45.1, and surface OVA. Shown is representative flow cytometry gating on CD11c+ cells. As controls, nontransplanted B6 and BALB/c mice were analyzed. Note that BALB/c-actOVA recipients of B6 CD45.1 BM had a population of CD11c+ cells with a similar intensity of Kb staining as on CD11c+ cells from control B6 mice, but with significant staining for Kd. A similar pattern was seen for CD45.2 and surface OVA (histogram). Data are representative of at least 3 independent experiments. For confocal images, mice were transplanted as in panel C, and on day 6 cells were stained with anti–CD11c-PE and CD11c+ cell were sorted. Sorted cells were stained with antibodies against Kd (biotin followed by SA-Alexa488) and anti-Kb (APC) and then dropped onto slides. Original magnifications ×400. Note cells that express both Kd and Kb (white circles). There were also cells that stained for Kb or Kd only, which are internal controls for staining specificity. No Kb+ cells were seen when cells from control BALB/c →BALB/c were imaged (not shown). For Amnis image flow analysis (E), BALB/c mice were transplanted as in panels C through D. On day 6, splenocytes were harvested and stained with antibodies against CD11c (Pacific Blue), Kb (PE), and Kd (FITC) in addition to staining with 7-AAD for live/dead exclusion and DRAQ5 to allow isolation of 2N DNA content cells and analyzed on the ImageStream instrument. Shown are representative images from > 200 KbKd double-positive CD11c+ cells analyzed in B6→BALB/c recipients and more than 200 CD11c+ cells in B6→B6 and BALB/c→BALB/c syngeneic controls. Results are representative of 2 independent experiments.
It was possible that either intact Kb-SIINFEKL complexes (present in actOVA but not in actOVA.Kb−/− mice) were being transferred and/or there was free SIINFEKL peptide exclusively in actOVA mice that “pulsed” donor Kb molecules in vivo. We therefore also stained cells in transplanted mice for Kb and observed a similar shift in Kb on donor cells in transplanted control B6 and actOVA mice, but not in either Kb−/− or actOVA.Kb−/− recipients (Figure 6A bottom panel). This suggested that the increase in 25-D1 we observed could have been the result of transfer of intact Kb-SIINFEKL complexes from host to donor. To further test this hypothesis, we infused CFSE-labeled Kb−/− or unlabeled CD45.1 wild-type splenocytes into actOVA, actOVA.Kb−/−, and B6 mice that had been irradiated 5 days earlier. We then gated on CFSE+CD8+ or CD45.1+CD8+ cells and analyzed Kb expression and 25-D1 binding. Both transferred Kb−/− and WT CD8 cells extracted from irradiated actOVA, but not actOVA.Kb−/− or B6 mice, stained with 25-D1 (Figure 6B). Thus, intact Kb-SIINFEKL complexes must have been transferred from host to donor cells.
We also analyzed donor-derived cells early after transplantation in the B6→BALB/c strain pairing (Figure 6C). BALB/c or BALB/c-OVA mice were irradiated and reconstituted with B6 CD45.1 BM. Six days later, mice were scarificed and CD11c+ cells were analyzed for their expression of Kb, Kd, CD45.1, CD45.2, and surface transmembrane OVA (Figure 6C). Most DCs in B6→BALB/c or B6→BALB/c-OVA mice expressed a similar amount of Kb as did DCs in control B6 mice, consistent with their being of B6 in origin. The majority of these Kb+ DCs expressed Kd, although its expression varied over a wide range of fluorescent intensities compared with control BALB/c mice. Expression of host-derived CD45.2 and transmembrane OVA paralleled that of Kd (Figure 6C).
Taken together, these data suggest that donor-derived cells acquired host-derived transmembrane proteins. To better characterize these hybrid cells, we analyzed sorted CD11c+ cells from spleens of B6→BALB/c mice 6 days after transplantation by confocal microscopy (Figure 6D). We observed Kb+Kd−, Kd+ Kb−, and Kb+Kd+ cells. Kd appeared to be on the cell surface, and not part of another attached cell, confirming that the donor cells staining for host-specific proteins by flow cytometry were not doublets. Overall, Kd was less uniformly distributed than was Kb. Kb+Kd+ cells were only present in B6→BALB/c mice, and not in control B6→B6 or BALB/c→BALB/c mice (not shown).
We also analyzed hybrids using the Amnis Image Stream system, which allows high-throughput multichannel fluorescent imaging of a large number of cells.32 BALB/c mice were irradiated and reconstituted with B6 BM and splenocytes containing 106 T cells. Six days later, mice were killed and splenocytes were stained with antibodies against CD11c (or CD8; both in Pacific Blue), Kb (PE), and Kd (FITC). Cells were also stained with 7-AAD to exclude dead cells and with the intravital DNA-binding dye DRAQ5. Cells were then analyzed using the ImageStream instrument. To assess the pattern of Kb and Kd staining on CD11c+ cells, we gated on 7-AAD− cells that had a DRAQ5 signal consistent with being single cells (> 90% of events). We then gated on CD11c+ cells and analyzed the intensity and distribution of Kb and Kd staining (Kb vs Kd plot, supplemental Figure 1A, available on the Blood Web site; see the Supplemental Materials link at the top of the online article; Kb vs Kd plot; Figure 6E, cell images). As observed by flow cytometry, most Kb+ cells acquired some Kd. Although the distribution of Kb was fairly uniform, in an analysis of ∼ 200 KdKb double-positive cells, Kd signal was more focal, similar to the confocal images. The majority of cells had at least a 3 areas of Kd signal, but a smaller percentage of events had a single and more intense area of staining. Similar data were obtained for CD8+ cells (supplemental Figure 1B). Of note, CD11c+ donor-derived cells in B6→B6 transplants also had focal areas of more intense Kb staining, consistent with donor B6 cells acquiring host-derived Kb.
Cross-dressed DCs can prime T cells directed against transferred host MHC class I–peptide complexes
Given that we had observed donor-derived DCs with host-derived MHC in both the C3H.SW→B6 and B6→BALB/c strain pairing, we reasoned that this could be a mechanism whereby donor cells can acquire host MHC-peptide complexes, thereby enabling them to prime donor CD8 cells. The effect would be to cross-prime donor-derived T cells, but without antigen processing characteristic of cross-presentation.33 To test this hypothesis, we determined whether donor-derived DCs that acquire host MHC-peptide complexes can prime CD8 cells specific for the host complex. To set up such a situation, B6 actOVA mice were irradiated and reconstituted with BALB/c BM. Seven days later, sort-purified splenic CD11c+Kb+ cells with the same level of Kd as was present in control BALB/c→BALB/c recipients (Figure 7A) were cultured with CFSE-labeled OT-1 transgenic T cells, which specifically and sensitively recognize Kb-SIINFEKL. Hybrid Kb+Kd+ DCs induced OT-1 division, whereas control BALB/c DCs induced none (Figure 7B). Thus, APCs cross-dressed with host MHC class I-peptide complexes were functional.
Figure 7.

Hybrid DCs are effective APCs. B6actOVA mice were irradiated and reconstituted with BALB/c BM. As controls, B6 and BALB/c mice underwent syngeneic transplants. Mice were killed on day 7 and Kb+Kd+ CD11c+ cells were sorted (A) along with CD11c+ cells from control actOVA and BALB/c mice (not shown). DCs were cultured with CFSE-labeled OT-1 cells at the DC/OT-1 ratios shown. Cultures were harvested at days 3, 4, and 5. Shown is proliferation on day 3 of culture (B); similar data were obtained after 4 and 5 days of culture (not shown). Data are representative of 2 experiments with similar results.
Discussion
GVHD can develop or persist months to years after transplantation when host hematopoiesis has been mostly if not completely replaced by donor-derived cells. If CD8 cells continue to contribute to GVHD at these late times, then they must be independent of further APC stimulation, be cross-primed by donor-derived APCs, or both. The main goals of our studies were to determine whether cross-priming occurs, and if so, to identify pathways that can be targeted to prevent it.
In the C3H.SW→B6 model, donor-naive CD8 cells mounted strong responses against SIINFEKL or LTFNYRNL when these antigens were exclusively cross-presented by donor APCs. These cross-primed cells were functional effectors that produced IFN-γ and were cytolytic in vivo. Surprisingly, the magnitude and quality of cross-priming responses were similar whether antigen was exclusively nonhematopoietic or hematopoietic, even though one might have predicted that the massive death of hematopoietic cells after irradiation would have provided a more accessible source of antigen.
Little is known about the nature of CD8 responses against nonhematopoietic miHAs in humans or mice as methods for identifying miHAs have relied on propagating donor-derived T cells, in most cases harvested from mice or humans post alloSCT, with host-derived hematopoietic cells.34 Thus, T cells reactive against exclusively nonhematopoietic antigens would not be expanded and their targets not identified. Because T cells can be cross-primed against nonhematopoietic antigens, a complete picture of anti–miHA T-cell reactivity must include an analysis of T cells that target nonhematopoietic antigens, which will require methods for miHA identification that do not rely on the propagation of T cell lines by host-derived APCs.
Cross-priming even occurred in the fully MHC-mismatched B6→BALB/c strain pairing. Although CD8 cells reactive against host antigens presented by donor APCs would not be likely to cause GVHD as host target tissues would lack the restricting MHC and CD8-mediated GVHD requires direct TCR:MHC class I contact,14 we took advantage of this system to study the mechanisms of cross-priming. Donor T cells were exclusively cross-primed by a CD11c+ cell, most likely a conventional DC,35 as mouse plasmacytoid DCs have little capacity to cross-present exogenous antigens.30 To optimally cross-prime naive CD8 cells, donor DCs must be stimulated by both CD40L and type I IFNs. The CD40L:CD40 signal did not require direct cognate recognition of MHC class II on donor APCs by CD4 cells (the most likely source of CD40L) as cross-priming was robust even when donor APCs were MHC class II−. These studies suggest that targeting type I IFNs or CD40:CD40L interactions could inhibit GVHD mediated by cross-primed CD8 cells.
Another surprising finding was how the presence and distribution of Kb-SIINFEKL or Kb-LTFNYRNL, but not the parent proteins without the restricting MHC class I, had a decisive impact on the magnitude of the T-cell response. That this reduction was seen in spleen and LN indicates that suppression probably occurs before T-cell egress into blood and then GVHD target organs. It is possible that, when target MHC-peptides are widely expressed at a high level, even on stromal, endothelial, and hematopoietic cells in secondary lymphoid tissues, reactive T cells are consumed or induced to undergo activation-induced cell death. These data suggest that antigens with a more restricted tissue distribution may generate the largest T-cell responses.
Our findings extend those of Asakura et al36 and Flutter et al.37 Flutter et al found in a delayed T-cell infusion model that alloreactive T-cell responses were diminished when antigen was widespread, and used TCR transgenic T cells to track alloreactivity.37 Asakura et al36 reported that total donor CD8 expansion and GVL were augmented when alloantigen was hematopoietically restricted. Because miHA-reactive T cells could not be specifically tracked, it was possible that they were generated equally whether alloantigen/MHC was ubiquitous or only hematopoietic, but that they were functionally impaired. Because we could enumerate polyclonal miHA-specific T cells by tetramers, we were able to show that the major effect of ubiquitous antigen: MHC class I expression was a reduction in the number of miHA-specific T cells.
We had hoped to identify donor APCs that cross-present SIINFEKL using the 25-D1 antibody. Instead, we unexpectedly discovered widespread transfer of host-derived transmembrane proteins to donor cells, including DCs. Confocal microscopy and Amnis imaging confirmed that these hybrids were single cells with relatively large but focal areas of host-derived MHC class I. Importantly, donor DCs that acquired host Kb-SIINFEKL were capable of priming OT-1 cells in vitro. These data suggest that transfer of host MHC-peptide complexes to donor APCs could enhance the early priming of alloreactive T cells, even in the fully allogeneic setting. We were unable to dissect whether such cross-dressing contributes to GVHD, as hosts that would have allogeneic MHC class I-peptide complexes to donate to donor APCs would also be able to directly prime allogeneic T cells.
Transfer of membrane and transmembrane proteins from cell to cell has been described in a number of systems.38–46 T cells in contact with targets can acquire target cell-derived MHC in a contact-dependent manner.38,39 Exosomes can also be the vehicle for transferring transmembrane proteins, and the relocated MHC can endow the recipient APC with the ability to prime T cells.41,43,45–47 Most relevant to our studies, the transfer of transmembrane proteins can be potentiated with prior freeze/thaw killing of the donor cells.42 In our studies, irradiation and cell death, physiologic in the context of alloSCT, were key as we did not see hybrid DCs in stable mixed BM chimeras (not shown), and the microscopy images suggest transfer of relatively large membrane fragments.
In conclusion, our studies make important and novel contributions to the understanding of alloimmune CD8 responses. We have established the extent and importance of cross-presentation and cross-priming against miHAs, and have identified CD40L:CD40 interactions and type I IFNs as therapeutic targets to prevent it. Our studies show that exclusively nonhematopoietic antigens are cross-presented and targeted by alloreactive CD8 cells. We also show how peptide/MHC distribution contributes to immunodominance. Finally, we have identified a novel mechanism whereby donor-derived cells can present host MHC-peptide complexes.
Supplementary Material
Acknowledgments
The authors thank the Yale Animal Resources Center for expert animal care.
This work was supported by the National Institutes of Health (HL-083072 and AI064343). W.D.S. is a recipient of a Clinical Scholar award from the Leukemia & Lymphoma Society.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.W. and H.L. designed and performed experiments, analyzed data, and wrote the paper; C.M.-M. and H.S.T. assisted with experiments; N.L. created SIINFEKL-reactive memory T cells; D.R. created B6.H60 and actH60 mice, provided technical advice, and edited the manuscript; and W.D.S. designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for X.W. is Institute of Immunology, Zhejiang University, Hangzhou, People's Republic of China.
Correspondence: Warren D. Shlomchik, Yale University School of Medicine, Cancer Center and Departments of Medicine and Immunobiology, PO Box 208032, New Haven, CT 06520; e-mail: warren.shlomchik@yale.edu.
References
- 1.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7(5):340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 2.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen- presenting cells. Science. 1999;285(5426):412–415. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 3.Merad M, Hoffmann P, Ranheim E, et al. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat Med. 2004;10(5):510–517. doi: 10.1038/nm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matte CC, Liu J, Cormier J, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med. 2004;10(9):987–992. doi: 10.1038/nm1089. [DOI] [PubMed] [Google Scholar]
- 5.Hochweller K, Wabnitz GH, Samstag Y, Suffner J, Hämmerling GJ, Garbi N. Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proc Natl Acad Sci U S A. 2010;107(13):5931–5936. doi: 10.1073/pnas.0911877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirberg J, Berns A, von Boehmer H. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med. 1997;186(8):1269–1275. doi: 10.1084/jem.186.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell PS, Chase CM, Madsen JC, et al. Coronary artery disease from isolated non-H2-determined incompatibilities in transplanted mouse hearts. Transplantation. 2011;91(8):847–852. doi: 10.1097/TP.0b013e3182122f82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3(11):1355–1362. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 9.Malarkannan S, Shih PP, Eden PA, et al. The molecular and functional characterization of a dominant minor H antigen, H60. J Immunol. 1998;161(7):3501–3509. [PubMed] [Google Scholar]
- 10.Choi EY, Christianson GJ, Yoshimura Y, et al. Real-time T-cell profiling identifies H60 as a major minor histocompatibility antigen in murine graft-versus-host disease. Blood. 2002;100(13):4259–4265. doi: 10.1182/blood-2002-05-1299. [DOI] [PubMed] [Google Scholar]
- 11.Muller U, Steinhoff U, Reis LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 12.Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis. 2005;43(3):129–135. doi: 10.1002/gene.20162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birnberg T, Bar-On L, Sapoznikov A, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity. 2008;29(6):986–997. doi: 10.1016/j.immuni.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Matte-Martone C, Liu J, Jain D, McNiff J, Shlomchik WD. CD8+ but not CD4+ T cells require cognate interactions with target tissues to mediate GVHD across only minor H antigens, whereas both CD4+ and CD8+ T cells require direct leukemic contact to mediate GVL. Blood. 2008;111(7):3884–3892. doi: 10.1182/blood-2007-11-125294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murali-Krishna K, Altman JD, Suresh M, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8(2):177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 16.Altman JD, Moss PAH, Goulder PJR, et al. Phenotypic analysis of antigen-specific T lymphocytes [published erratum appears in Science. 1998;280(5371):1821]. Science. 1996;274(5284):94–96. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 17.Porgador A, Yewdell JW, Deng YP, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide MHC class I complexes using a monoclonal antibody. Immunity. 1997;6(6):715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 18.Boscardin SB, Hafalla JC, Masilamani RF, et al. Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J Exp Med. 2006;203(3):599–606. doi: 10.1084/jem.20051639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerwenka A, Bakker AB, McClanahan T, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12(6):721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 20.Choi EY, Yoshimura Y, Christianson GJ, et al. Quantitative analysis of the immune response to mouse non-MHC transplantation antigens in vivo: the H60 histocompatibility antigen dominates over all others. J Immunol. 2001;166(7):4370–4379. doi: 10.4049/jimmunol.166.7.4370. [DOI] [PubMed] [Google Scholar]
- 21.Ryu SJ, Jung KM, Yoo HS, et al. Cognate CD4 help is essential for the reactivation and expansion of CD8 memory T cells directed against the hematopoietic cell-specific dominant minor histocompatibility antigen, H60. Blood. 2009;113(18):4273–4280. doi: 10.1182/blood-2008-09-181263. [DOI] [PubMed] [Google Scholar]
- 22.Choi EY, Christianson GJ, Yoshimura Y, et al. Immunodominance of H60 is caused by an abnormally high precursor T cell pool directed against its unique minor histocompatibility antigen peptide. Immunity. 2002;17(5):593–603. doi: 10.1016/s1074-7613(02)00428-4. [DOI] [PubMed] [Google Scholar]
- 23.Zheng H, Matte-Martone C, Jain D, McNiff J, Shlomchik WD. Central memory CD8+ T cells induce graft-versus-host disease and mediate graft-versus-leukemia. J Immunol. 2009;182(10):5938–5948. doi: 10.4049/jimmunol.0802212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Mierlo GJD, Boonman ZFHM, Dumortier HM, et al. Activation of dendritic cells that cross-present tumor-derived antigen licenses CD8+ CTL to cause tumor eradication. J Immunol. 2004;173(11):6753–6759. doi: 10.4049/jimmunol.173.11.6753. [DOI] [PubMed] [Google Scholar]
- 25.Le Bon A, Etchart N, Rossmann C, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4(10):1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 26.Kuchtey J, Chefalo PJ, Gray RC, Ramachandra L, Harding CV. Enhancement of dendritic cell antigen cross-presentation by CpG DNA involves type I IFN and stabilization of class I MHC mRNA. J Immunol. 2005;175(4):2244–2251. doi: 10.4049/jimmunol.175.4.2244. [DOI] [PubMed] [Google Scholar]
- 27.Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186(1):65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204(7):1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33(6):967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29(3):352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19(1):33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 32.McGrath KE, Bushnell TP, Palis J. Multispectral imaging of hematopoietic cells: where flow meets morphology. J Immunol Methods. 2008;336(2):91–97. doi: 10.1016/j.jim.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86(4):353–362. doi: 10.1038/icb.2008.3. [DOI] [PubMed] [Google Scholar]
- 34.Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. 2011;89(3):396–407. doi: 10.1038/icb.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung S, Unutmaz D, Wong P, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17(2):211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asakura S, Hashimoto D, Takashima S, et al. Alloantigen expression on non-hematopoietic cells reduces graft-versus-leukemia effects in mice. J Clin Invest. 2010;120(7):2370–2378. doi: 10.1172/JCI39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flutter B, Edwards N, Fallah-Arani F, et al. Nonhematopoietic antigen blocks memory programming of alloreactive CD8+ T cells and drives their eventual exhaustion in mouse models of bone marrow transplantation. J Clin Invest. 2010;120(11):3855–3868. doi: 10.1172/JCI41446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang JF, Yang Y, Sepulveda H, et al. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science. 1999;286(5441):952–954. doi: 10.1126/science.286.5441.952. [DOI] [PubMed] [Google Scholar]
- 39.Hudrisier D, Riond J, Mazarguil H, Gairin JE, Joly E. Cutting edge: CTLs rapidly capture membrane fragments from target cells in a TCR signaling-dependent manner. J Immunol. 2001;166(6):3645–3649. doi: 10.4049/jimmunol.166.6.3645. [DOI] [PubMed] [Google Scholar]
- 40.Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3(12):1156–1162. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 41.Herrera OB, Golshayan D, Tibbott R, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol. 2004;173(8):4828–4837. doi: 10.4049/jimmunol.173.8.4828. [DOI] [PubMed] [Google Scholar]
- 42.Dolan BP, Gibbs KD, Ostrand-Rosenberg S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8(+) T cells. J Immunol. 2006;177(9):6018–6024. doi: 10.4049/jimmunol.177.9.6018. [DOI] [PubMed] [Google Scholar]
- 43.Smyth LA, Herrera OB, Golshayan D, Lombardi G, Lechler RI. A novel pathway of antigen presentation by dendritic and endothelial cells: implications for allorecognition and infectious diseases. Transplantation. 2006;82(1 suppl):S15–S18. doi: 10.1097/01.tp.0000231347.06149.ca. [DOI] [PubMed] [Google Scholar]
- 44.de Heusch M, Blocklet D, Egrise D, et al. Bidirectional MHC molecule exchange between migratory and resident dendritic cells. J Leukoc Biol. 2007;82(4):861–868. doi: 10.1189/jlb.0307167. [DOI] [PubMed] [Google Scholar]
- 45.Smyth LA, Harker N, Turnbull W, et al. The relative efficiency of acquisition of MHC:peptide complexes and cross-presentation depends on dendritic cell type. J Immunol. 2008;181(5):3212–3220. doi: 10.4049/jimmunol.181.5.3212. [DOI] [PubMed] [Google Scholar]
- 46.Qu C, Nguyen VA, Merad M, Randolph GJ. MHC class I/peptide transfer between dendritic cells overcomes poor cross-presentation by monocyte-derived APCs that engulf dying cells. J Immunol. 2009;182(6):3650–3659. doi: 10.4049/jimmunol.0801532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}