

Abstract
We report a very rare case of HEREDITARY ANGIOEDEMA, presenting as recurrent acute abdomen. A 22 yr old Maharastrian male, law college student, got admitted for about fifteen times in previous three years for severe, acute onset, upper abdominal pain, vomiting, distention and acute exudative inflammatory ascites .The whole episode used to subside spontaneously within 2–3 days with or without conservative general management .He underwent various investigations from far basic type, to advanced and invasive type with each recurrence but without definitive diagnosis. He also underwent unnecessary appendectomy. The authors did a review of his previous records, but didn't find any definite surgical or medical cause for his acute abdomen. Obviously it was something rare. Authors did search for it in various surgical and medical literature and searched extensively on internet for rare causes of abdominal pain which guided them for further appropriate investigations and diagnose him as a case of HEREDITARY ANGIOEDEMA ,as his clinical features and C1-INH,C3-C4 levels were strongly in favour of it . It goes without saying that the internet has become a standard accessory to conventional literature for cases with diagnostic dilemmas and for treatment options as well.
Keywords: Hereditary angioedema, Hereditory angioneurotic oedema, Recurrent abdominal pain, Acquired angioedema
Introduction
Hereditary angioedema is a very rare disease, where patient has very low levels of C4 and C1 esterase inhibitor which is necessary for compliment and kallikrenin - kinnin pathway to work. This occurs in 1in 1, 50000 population. Hitherto 20 families have been reported from Japan and few cases from India. Our case was difficult to diagnose as recurrent abdominal pain, ascites, pleural effusion was the presentation and cutaneous presentation was late after we diagnosed the case.
Case Report
A 22 yr old male came with history of severe pain in epigastric area and severe vomiting once in early morning. On examination his pulse was 90/min, BP was 120/80mmof Hg, was afebrile and there was no pallor. Per abdominal examination was showing tenderness in epigastric region and part of umbilical region with minimal guarding. Rest of abdomen was soft and normal. Other systems were normal. His HB was 10.7 gms %, TC 4400, ESR 25, and urine exam was normal. Serum amylase, serum lipase, serum proteins, LFT, BSL, were normal. Abdominal ultrasound was showing oedematous small bowel, ascites, minimal pleural effusion but pancreas, kidneys and liver were normal. Patient became clinically sound after 48 hrs, with conservative routine management .On interrogation, he gave history of similar episodes about fourteen times in previous three years for which he was admitted each time and treated with antibiotics and i/v fluids. Review of his records showed that he was evaluated with laboratory investigations, and abdominal ultrasound multiple times (about 6 to 7 times), which were unremarkable except for mild bowel wall oedema and moderate ascites. Ascitic fluid was of acute inflammatory exudative character with normal amylase. Gastroscopy, Colonoscopy and Barium meal follow through were normal. During his fourth admission he was started empirically on anti-tubercular treatment by a physician whom he consulted but his episode did recur. During his sixth admission he underwent appendectomy as he consulted a surgeon this time and his ultrasound was showing bowel wall oedema and crowding of loops in right iliac fossa suggestive of appendicitis.
Some of his routine investigations as mentioned above were repeated and authors performed some new investigations like abdominal CT scan with contrast which again revealed upper GI tract oedema with ascites and left sided pleural effusion. Ascitic fluid exam revealed similar results as on previous occasions. His coagulation profile was normal and anti-nuclear antibodies were negative. Due to difficulty in conclusive diagnosis authors searched in medical and surgical literature and on the internet for rare causes of recurrent abdominal pain and got few similar case reports which were ultimately diagnosed as hereditary angioedema. So he was evaluated, on that line and his blood sample was sent for C3, C4 and CI INH [complement 1 esterase inhibitor] at Super speciality Ranbaxy Laboratory and his reports were as follows- C3-104.3(control = 82-160 mg/dl), C4-less than 3.4(control = 15-43 mg/dl) and C1esterase inhibitor functional assay–less than 1(control=>67is normal and below 41 is abnormal). These were highly suggestive of hereditary angioedema.
Retrospectively when he was asked about family history, his mother had suffered from similar complaints repeatedly although of milder intensity and one day she died while was being brought to hospital for severe acute breathlessness with stridor and cyanosis [as inferred from the history given by the patient] most probably due to laryngeal oedema. This aspect of the case made our diagnosis certain. The rest of family members could not be investigated due to financial constraints.
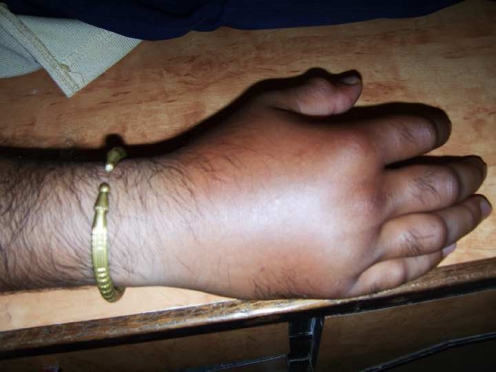
Besides this our patient also has a tendency to develop severe nonpruritic, non pitting oedema of limbs or any involved body part in response to trivial trauma. He did develop the same after the case was diagnosed [as shown in the photograph below] which again is in strong favour of our diagnosis. The patient is being treated with danazol and the period between acute attacks has prolonged. He has been given a card mentioning his disease and treatment required during emergency like laryngeal oedema. Even though no definite treatment is available, now, we are avoiding repeated investigations and the patient receives only a supportive line of treatment during acute attack.
Discussion
HEREDITORY ANGIOEDEMA (HAE), if we see, common practicing consultant is unaware of this term as it is a very rare disease, but as it has varied presentations mimicking many other acute conditions patient has to suffer for that .Hereditary angioedema is an autosomal dominant condition where C1 inhibitor gene (C1INH) located on chromosome 11 in P11q13 is abnormal or lacking and due to that there is dysregulation of complement kallikrenin – kinnin system [8]. The actual factor or factors responsible for the oedema formation remain somewhat controversial. Researchers have demonstrated activation of the kinnin system and increased bradykinin concentration associated with clinical flares. Bradykinin is an important inflammatory mediator that causes neutrophil chemotaxis, capillary dilation, and smooth muscle relaxation, and it has been linked to other forms of angioedema. In an animal model of C1-INH deficiency, bradykinin antagonists prevent capillary leakage. Others implicate C2 kinin, a metabolite of C2b, as the active agent in the presence of plasmin, but actual cause for oedema remains controversial [8].
There are three types of HAE –Type I- seen in 85% of cases here C1 INH quantitative levels are low and which is responsible for angioedema. Type II-seen in 15% of cases, C1 INH concentration is normal. As even though normal protein is there, it is insensitive. That dysfunctional protein has got abnormal action. TypeIII –resemble HAE but occur in females only.
There are different types of clinical presentation, 1) Cutaneous-where nonraised, nonitching, flares are present called erythema marginatum. 2) Abdominal –may present as recurrent abdominal pain, may land up with unnecessary explorations, as occurred in this case. 3) Extremity oedema – nonpruritic angioedema is present over upper or lower limb 4) Laryngeal oedema– worst presentation, if patient does’ not get immediate treatment mortality is 100% .Rarely patient may present as scrotal oedema. Attacks of these symptoms are usually precipitated by infection, trauma, and emotional stress [3].
Hereditary angioedema differs from acquired angioedema that oedema is nonpitting and nonpruritic and runs in family.
INVESTIGATIONS- blood levels of C4 [6], C3, and C1 esterase inhibitor. Depending on type, levels may very. But in commonest type there are low levels of C1 esterase inhibitor and low levels of C4 [6]. Usually levels should be done during attacks.
TREATMENT –Treatment depends on- for acute prophylaxis, or for maintenance therapy. Acute laryngeal oedema needs urgent attention and should be treated with vapour treated C1 esterase inhibitor concentrate [2], fresh frozen plasma or if both are not available then tracheotomy [1]. Other conditions are self limiting but danazol, stanolol have been tried for acute condition, or for prevention of attacks [5, 7]. Aminoepsolic acid has been tried but failed [4]. There is no role of steroids, antihistamines, adrenaline locally or nor epinephrine.
Summary
Though HEREDITARY ANGIOEDEMA is very rare, it needs to be taken as one of the differential diagnosis for recurrent abdominal pain, when other causes like acute gastritis, acute pancreatitis, Familial Mediterranean fever, bleeding disorders, Henoch Shonlein purpura are ruled out. And even though C1 esterase inhibitor is expensive investigation, at least we should do C3 and C4 levels and if levels of C4 are low we should investigate further for C1 esterase inhibitor during acute attack. Our case presented as abdominal pain all the times and cutaneous presentation was after establishment of the diagnosis, so it was really very difficult to diagnose this case.
Contributor Information
Madhura Milind Killedar, Email: mdhurakilledar@gmail.com.
Anand S. Malani, Email: dranandsm@yahoo.com
References
- 1.Megerian CA, Arnold JE, Berger M. Angioedema. 5 years experience with a review of the disorder’s presentation and treatment. Laryngoscope. 1992;102:256–260. doi: 10.1288/00005537-199203000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Waytes AT, Rosen FS, Frank MM. Treatment of hereditary angioedema with a vapour treated C1 inhibitor concentrate. N Engl J Med. 1996;334:1630–1634. doi: 10.1056/NEJM199606203342503. [DOI] [PubMed] [Google Scholar]
- 3.Baranwal AK, Singh S, Kumar L. Hereditary angioneurotic oedema. Indian Pediatr. 1999;36:187–189. [PubMed] [Google Scholar]
- 4.Bork K, Witzke G. Long-term prophylaxis with C1-inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1-inhibitor deficiency. J Allergy Clin Immunol. 1989;83(3):677–682. doi: 10.1016/0091-6749(89)90082-1. [DOI] [PubMed] [Google Scholar]
- 5.Cicardi M, Bergamaschini L, Cugno M, et al. Long-term treatment of hereditary angioedema with attenuated androgens: a survey of a 13-year experience. J Allergy Clin Immunol. 1991;87(4):768–773. doi: 10.1016/0091-6749(91)90120-D. [DOI] [PubMed] [Google Scholar]
- 6.Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med. 1976;84(5):580–593. doi: 10.7326/0003-4819-84-5-580. [DOI] [PubMed] [Google Scholar]
- 7.Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med. 1976;295(26):1444–1448. doi: 10.1056/NEJM197612232952602. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen EW, Gran JT, Straume B, et al. Hereditary angio-oedema: new clinical observations and autoimmune screening, complement and kallikrein-kinin analyses. J Intern Med. 1996;239(2):119–130. doi: 10.1046/j.1365-2796.1996.418764000.x. [DOI] [PubMed] [Google Scholar]