

Abstract
Phagocytes are specialized cells of the immune system, designed to engulf and destroy harmful microorganisms inside the newly formed phagosome. The latter is an intracellular organelle that is transformed into a toxic environment within minutes and disappears once the pathogen is destroyed. Reactive oxygen species and reactive nitrogen species are produced inside the phagosome. Intracellular granules or lysosomes of the phagocyte fuse with the phagosome and liberate their destructive enzymes. This process of phagocytosis efficiently protects against most infections; however, some microorganisms avoid their destruction and cause severe damage. To understand such failure of phagosomal killing, we need to learn more about the actual destruction process in the phagosome. This paper summarizes methods to investigate the biochemistry of the phagosome and discusses some of their limitations. In accordance with the nature of the phagosome, the issue of localization and temporal dynamics is emphasized, and recent developments are highlighted.
Keywords: Phagocytosis, pathogen killing, ROS, pH, fluorescent probes
1. PHAGOCYTOSIS
Phagocytosis is a central mechanism of our immune response to pathogenic bacteria and certain eukaryotic pathogens. Immunologists have unravelled the fascinating complexity of adaptive immunity including specific antigen recognition and presentation leading to the production of target-specific antibodies. When the initial threat is a bacterium, the task of the immune response is to kill large numbers of identical, pathogenic bacteria that thrive in our body. Antibodies serve mainly 3 functions against bacteria: neutralization, complement activation, and opsonization to improve phagocytosis. The latter is executed mainly by macrophages and by neutrophils. A lack of neutrophils, neutropenia, is life threatening. This fact among others illustrates that we rely on the proper functioning of phagocytosis. With over 50000 entries in PubMed, the subject of phagocytosis is widely covered, and the reader is referred to excellent reviews on the subject. Nevertheless, there are good reasons to pursue research in the area:
rare but devastating genetic diseases are related to defects in phagocytosis or killing;
antimicrobial therapies encounter fast developing resistance;
some of the most dangerous bacteria have evolved strategies to avoid killing inside Phagocytes.
The present paper will focus on a particularly challenging aspect of the following issue:
what is happening inside the phagosome?
In fact, a large body of literature covers the bactericidal mechanisms of phagocytes, and the principal elements will be briefly summarized. However, the interplay between these mechanisms is poorly understood. As long as the bacterium is killed in the end, we may not worry. For those pathogens that survive, we have a fundamental interest to identify the weak spots of our phagocytic machinery.
2. MECHANISMS OF BACTERIAL KILLING
Conceptually, the known mechanisms of killing inside phagosome rely on fundamental changes in the chemistry inside the phagosome. This may be achieved by import or export of small molecules and ions. In addition, enzymes are liberated inside the phagosome or the phagosomal membrane to produce toxic compounds or directly attack the cell wall and membranes of the pathogen.
Transport of these elements is mostly achieved by membrane-bound transport mechanisms (ion channels and pumps) and by exchange with many other cellular compartments by means of membrane fusion and fission (Figure 1). The latter not only serves to import or export soluble material but also to change the lipid and protein composition of the phagosomal membrane. The exchange may be bidirectional, for example, with the endosomal pathway or unidirectional as with the neutrophil granules.
Figure 1.
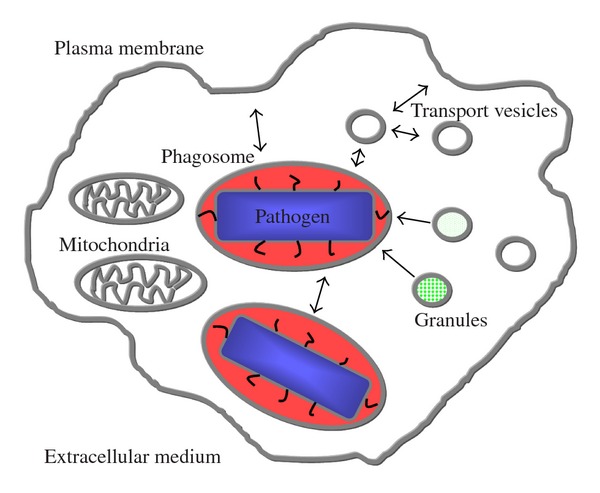
Cellular compartments in phagocytosis. The phagocyte contains multiple membrane-bound compartments, only some of them are included here. The vesicles mostly belong to the endosomal pathway. The neutrophil granules belong to several types with different content. Some of these compartments exchange membrane and liquid (arrows). The phagosome is filled with a pathogen (blue) surrounded by a limited phagosomal solution (red).
Chemicals that are important for pathogen killing can also be found in several compartments outside the phagosome. For example, mitochondria produce ROS like the phagosome, and several compartments (endosomes, lysosomes, and granules) are acidic. Granules contain ions, soluble proteins, and membrane proteins that profoundly alter the phagosomal biochemistry. Thus, the detection of these chemicals needs to be site specific. Furthermore, all the membranes contain small molecule transporters and ion channels. The free volume of the phagosome is small compared to the total cell volume. Most of the phagosomal volume is occupied by the pathogen. Phagocytes often form multiple phagosomes sequentially, and these phagosomes sometimes fuse with each other. Little is known about the exchange between multiple phagosomes. The destruction of the pathogen is based on the chemical modifications in the free phagosomal volume. The investigation of this small, rapidly changing volume is the object of this paper.
Both professional phagocytes of the immune system, macrophages and polynuclear neutrophils, are capable of killing microbes. However, the macrophage is also an antigen presenting cell. It is generally believed that bacterial killing by neutrophils is more efficient. Although the overall concept of bacterial killing is identical for both types of phagocytes, the exact mechanisms are not the same [1]. Hydrolytic enzymes that degrade proteins and polysaccharides attacking the bacterial cell wall are delivered to the phagosome. In macrophages, these enzymes are stored in lysosomes. In the neutrophils, the bactericidal proteins are stored in several thousand intracellular granules [2]. Delivery of the granules to the phagosome is faster than the phagosome maturation process in the macrophage.
Another key feature of the phagosomal killing is the production of superoxide anion (O2●−) by the NADPH oxidase. The O2●− is rapidly transformed to numerous other reactive oxygen species (ROS), mainly H2O2, hydroxyl radicals (OH●), and HOCl. Patients without a functional NADPH oxidase suffer from chronic granulomatous disease (CGD), characterized by severe recurrent infections [3]. Neutrophils produce considerably more ROS than macrophages.
Signalling out of the phagosome may influence the outcome of the phagocytic process [4]. Certain pathogens have evolved elaborate strategies to perturb the phagocyte. To cite a few examples, Mycobacterium tuberculosis inhibits macrophage calcium signalling [5] and also secretes a phosphatase to dephosphorylate phosphoinositol-3-phosphate (PI(3)P) and thereby arrest phagosome maturation [6, 7]. Leishmania delays apoptosis of neutrophils [8], and Helicobacter pylori and Francisella tularensis perturb the activation of the NADPH oxidase by two distinct mechanisms [9].
In general, the severity of infection depends on the size of the inoculum. The immune response is a race against a fast-growing population of microbes. However, persistent infections may result from a relatively small number of surviving microbes, which may adapt to a particular niche in the body. Days, weeks, or years later, these microbes may grow again, for example, when the host becomes immunocompromised, and cause a new burst of infection. Although Staphylococcus aureus is mainly an extracellular pathogen, it may survive in macrophages, neutrophils, or endothelial cells, which contributes to cause long-lasting infections [10–12]. Thus, a few surviving pathogens may be enough to continue infection. An intriguing and sophisticated extension of this concept has been described for the eukaryotic intracellular pathogen Leishmania major. The long-term hosts for Leishmania are macrophages. Upon infection, the parasite is also taken up by neutrophils and often killed. However, some parasites survive inside the neutrophil until the neutrophil enters apoptosis. The apoptotic neutrophil is recognized by macrophages via phosphatidylserine (PS) on the outer leaflet of the membrane and internalized in a noninflammatory reaction, which favours the survival of the parasite inside its final host, the macrophage [13]. This “Trojan horse” strategy is further optimized by the presence of apoptotic parasites in the inoculum [8, 14]. PS-positive parasites silently enter neutrophils and enhance the survival of nonapoptotic parasites in the same or neighbouring neutrophils. Later, the neutrophil becomes apoptotic and is internalized by macrophages. To detect and understand such strategies of survival, we need tools not only to investigate the bulk of the infectious agents but also to detect minorities capable of resisting the immune response.
3. METHODS TO INVESTIGATE PHAGOCYTOSIS
The aim of this paper is to provide an overview over major methods in the field, their rationale, and their limitations with emphasis on the dynamic aspects of phagosome maturation. References are intended as examples to illustrate important points and suggest further reading; they are definitely not a comprehensive review of all methods and all the important work that has been done by many researches. New probes are continuously developed, and some become commercially available. The reader has to choose between well-established methods with known defaults and potentially better new probes that require extensive characterization.
Ideally, the efficiency of phagocytosis is evaluated by measuring the killing of the respective pathogen. Although this seems to be straightforward, the available methods have considerable limitations. The gold standard for bacterial survival is the capacity to form colonies. Appropriate colony-counting assays are widely used for research and clinical applications [15]. The survival of nonadherent pathogens including Leishmania may be evaluated by limited dilution essays [14]. Special care is required to either separate or destroy the pathogens that have not been internalized during the experiment before counting the viable organisms. Depending on the growth rate of the microorganism, these assays take several days before readout. An elegant semiautomated killing assay for E. coli and S. aureus was based on optical density measurements of bacterial cell cultures directly in a shaking and heating (37°C) microplate reader [16]. Killing assays give little information on small subgroups within the bulk pathogen population. They reflect the final outcome of the phagocyte response. Changes in survival need to be analyzed with additional methods for individual steps of the phagocytic process. A recent international conference entitled “How dead is dead—The ins and outs of bacterial dormancy” dealt with viable but nonculturable bacteria as well as dormant bacteria (http://www.hdid2011.de). The question concerns several important pathogens including Staphylococcus aureus [17].
For many questions, assays monitoring killing directly during phagocytosis would be helpful. To start with, there is the problem to define “killing.” Many classical live/dead stains essentially probe the integrity of the cell membrane. A mildly disturbed membrane may lead to positive results even if the microbe is capable to recover and grow afterwards. As an alternative leakage of a cytoplasmic bacterial enzyme into the phagosome has been monitored as a readout for the permeabilization of the bacterial envelope [18]. Another indicator of cell survival is the capacity to produce proteins. An elegant and sensitive detection method is described by Lilius and Atosuo in this issue. The assay is based on the expression of bacterioluciferase in transformed, live bacteria. The enzyme catalyzes a chemiluminescence reaction that can be easily monitored in a plate reader providing a direct readout for the kinetics of survival.
The fluorescent protein GFP is specifically bleached by HOCl produced by myeloperoxidase in the presence of H2O2 and chloride [19]. This was used to monitor myeloperoxidase-dependent killing of S. aureus in living neutrophils [20]. HOCl has numerous targets within the phagosome and within the pathogen once it has breached the cell membrane. GFP bleaching occurs at relatively high HOCl concentrations. Thus, the bleaching of GFP in live bacteria reflects a state of advanced damage, and actually, the killing in a colony-forming assay was slightly faster than the bleaching of GFP [20]. The technique has numerous applications such as the identification of individual bacteria that resist in killing. Since GFP can be expressed in any organism that is amenable to transfection, pathogens other than S. aureus can be investigated.
Phagocytosis is often assessed by measurements of particle uptake, which have provided most of our knowledge on the formation of phagosomes. Inert particles are frequently used, such as dead bacteria, dead yeast, zymosan, a yeast cell wall preparation, or latex beads. Fluorescent particles are easily quantified by microscopy, microplate readers, or flow cytometry. A key issue is the distinction between the particles that adhere to the surface of the phagocyte and those that are internalized into a closed phagosome. A widely used and simple technique uses quenching of the fluorescence of extracellular particles. In fact, fluorescein and its derivatives are sensitive to quenching by trypan blue. The latter does not readily enter live cells and phagosomes. Thus, when trypan blue is applied after phagocytosis, internalized particles remain fluorescent, and all extracellular particles become almost nonfluorescent [21]. Particle counting in the microscope is tedious and mainly useful when very small amounts of material are available such as mouse blood neutrophils [22] or when weakly stained cells need to be identified, such as cells with low-level expression of fluorescent proteins. Flow cytometry is less time consuming and analyses much larger numbers of phagocytic events [23]. FITC-labelled particles (E. coli, S. cerevisiae) are commercially available; however, labelling of other particles can be readily achieved in a simple reaction with FITC [22].
4. METHODS TO INVESTIGATE PHAGOSOMAL BIOCHEMISTRY
To understand the killing process, we need to know what exactly happens inside the phagosome. Which are the compounds that attack the pathogen? What are the concentrations of these compounds and when do they appear? How do pathogens interfere with the delivery of these compounds into the phagosome? It is relatively straightforward to investigate the microbicidal effect of specific compounds or mixtures on bacterial cultures. However, investigation of the phagosomal environment is challenging in several respects.
First, the phagosome is a small intracellular compartment with no direct access from the outside once it is formed. Investigation of the lumen of the phagosomes requires specific targeting of the probes or isolation of phagosomes.
Second, phagosomes are formed within seconds or minutes and evolve rapidly. In a given population of phagocytes, phagocytosis is asynchronous unless specific synchronization techniques are employed. The composition of the phagosome evolves rapidly in particular by fusion and fission with endomembranes of the phagocyte. It is a truly transient organelle that eventually disappears. The killing process is believed to be complete in minutes up to 2 hours for most pathogens.
Third, the phagosome is a small moving target particularly challenging for live cell microscopy. The phagosome moves within the phagocyte, and the phagocyte moves and often continues to internalize more preys. Most of the phagosomal volume is occupied by the pathogen leaving a small liquid filled space between the pathogen and the phagosomal membrane. Very high concentrations of compounds including host proteins may be reached in this space.
5. REACTIVE OXYGEN SPECIES
The detection of phagosomal ROS production has received considerable attention, and numerous methods have been employed. One has to distinguish between measurements of overall ROS production during phagocytosis and techniques for localized detection within the phagosome. Some but not all ROS diffuse across membranes, so do some but not all reagents for ROS detection. Nitrobluetetrazolium (NBT) forms a dark precipitate upon reduction by superoxide anion and is widely used for screening NADPH oxidase activity in patient blood samples. Although NBT is membrane permeable, the precipitate will form close to the source of ROS. When NBT is added during phagocytosis, the precipitate will mainly form in the phagosome and can be readily observed by bright field microscopy. The method is rapid, requires few cells, and gives information about the number of NBT-positive cells/phagosomes in the sample [22, 24]. NBT does not measure how much ROS were produced, and it is not suitable for kinetic measurements on the scale of minutes. Chemiluminescence is a very sensitive method, and several compounds react with ROS and yield a chemiluminescence signal. The reaction is instantaneous, and the signal reflects the quantity of ROS present at a given time. Luminol has been widely used for ROS detection. It diffuses across membranes and therefore provides a signal of overall ROS production. Isoluminol is not membrane permeable. The difference between the luminol signal and the isoluminol signal reflects intracellular ROS production [25]. A more direct measure of phagosomal ROS is obtained by covalent attachment of luminol to microbeads prior to phagocytosis. Thereby, the compound is strictly localized to the particle, and the chemiluminescence signal reflects phagosomal ROS production. The technique has been successfully used to analyze ROS production in dendritic cells [26]. The influence of the harsher phagosomal environment in neutrophils on the luminol signal has not been investigated. Chemiluminescence detection requires a luminometer or an appropriate microplate reader. Only few very sensitive and sophisticated light microscopes detect chemiluminescence, and spatial resolution is rather limited.
Fluorescence is currently the first choice to obtain single-cell and spatial information on ROS production. Several ROS-sensitive fluorescent dyes have been developed, most of them derived from fluorescein or rhodamine. It is fair to say that none of these dyes is perfect. The history of 2′,7′-dichlorodihydrofluorescein (DCFH2) perfectly illustrates the difficulties. This dye has been used for 40 years in hundreds of publications. DCFH2 is nonfluorescent and becomes irreversibly oxidized to green fluorescent 2′,7′-dichlorofluorescein (DCF). The oxidation is not specific for a particular ROS, and the actual mechanism of oxidation is complex as reviewed recently [27, 28]. H2O2 slowly oxidizes DCFH2, and the reaction is much faster in the presence of a peroxidase such as horseradish peroxidase (HRP). The hydroxyl radical (OH●) rapidly oxidizes DCFH2, whereas superoxide anion (O2●−) is a poor reactant. It has recently become clear that hypochlorous acid (HOCl) oxidizes DCFH2 to a new product named Xfluo with slightly different spectral properties than DCF [29]. Peroxynitrite (ONOO−), which is formed from NO● and O2●−, also reacts with DCFH2, thus DCF fluorescence in phagocytes may depend on the activity of NADPH oxidase as well as NO synthase [30]. When comparing kinetic luminescence signals (see above) with fluorescence-based methods, the luminescence is comparable to the slope of the fluorescence signal.
What are the advantages of DCFH2? DCF yields a strong fluorescence signal. The excitation and emission spectra of DCF are close to those of fluorescein and thereby compatible with all standard equipment for fluorescence detection. DCFH2 is commercially available in several forms, (i) a free compound, (ii) a membrane permeable diacetate (DCFH2-DA) for cell loading, which enters cells and becomes trapped after hydrolysis by cellular esterases, or (iii) an amine reactive derivative, 2′,7′-dichlorodihydrofluorescein diacetate, succinimidyl ester for covalent labelling of proteins or particles.
When phagocytes are loaded with DCFH2-DA and stimulated with phagocytic particles, the presence of ROS in the cytosol can be detected by flow cytometry [31]. The oxidation of cytosolic DCFH2 during phagocytosis is probably due to H2O2 that diffuses across the phagosomal membrane. This diffusion takes time, and in the presence of myeloperoxidase which consumes H2O2, an estimated 6% of the phagosomal H2O2 production will leak out of the phagosome [32]. The cellular antioxidant defence will further dampen the signal, while other cellular sources of ROS such as mitochondria may also generate a DCF signal. Nevertheless, the technique provides information on the single cell level and the general time course of ROS production [33].
To investigate the phagosomal ROS production, the dye should be restricted to the phagosome, which can be achieved by covalent labelling with DCFH2-DA succinimidyl ester. This has been first used to label BSA, which was then mixed with anti-BSA antibodies and internalized as antigen-antibody immune complexes [34]. These reagents are now commercialized as Fc OxyBURST Green reagent (Molecular Probes/Invitrogen). Later on, larger particles such as bacteria, beads [35], zymosan [36], or heat-killed Saccharomyces cerevisiae [23] were labelled. A recent publication also reports successful labelling of live bacteria with DCFH2-DA succinimidyl ester [37]. DCFH2-labelled particles are not only suitable for flow cytometry but also for microscopy including video microscopy with living cells. Thus, the kinetics of phagosomal ROS production can be analyzed on the level of individual phagosomes. The different reports agree that ROS production starts upon phagosome formation and proceeds with a sigmoid curve for more than 10 min. Following individual phagosomes with high time resolution is difficult as the phagosome moves inside the cell in all directions and the phagocyte moves in the microscope field. Analysis of the particle fluorescence at each time point is time consuming. When DCF fluorescence from multiple phagosomes shall be compared, the time axis for each phagosome has to be synchronized to a common reference point such as the closure of the phagosome. Inhibition of the phagocyte NADPH oxidase by DPI stops fluorescence increase, and phagocytosis by cells that lack a functional NADPH oxidase yields a very small DCF signal [29]. Because DCFH2 is sensitive to photo-oxidation, the total light exposure during microscopy needs to be limited. Noninternalized particles in the same video film can serve as a control for photo-oxidation. Excitation and emission spectra of DCF largely overlap the spectra of GFP and YFP. For a combined use of fluorescent proteins and DCFH2, CFP and mCherry are better choices provided the measuring instrument offers good spectral resolution (see below).
Measurements of oxygen consumption by neutrophils during phagocytosis suggested that the duration of ROS production ranges from 60 s [38] to over 30 min [39]. Internalized DCFH2-labelled particles often show an increase of DCF fluorescence for 10 to 30 min [29, 35]. Furthermore, at least two types of kinetics are detectable in neutrophil-like PLB985 cells upon phagocytosis of DCFH2-labelled yeast [29]. DCFH2-labelled particles would still underestimate the total duration of ROS production if the dye became completely oxidized. To determine the maximal fluorescence of the particles, they were oxidized in vitro with H2O2 and HRP and then measured with the same microscope settings. Experiments with DCFH2-labelled yeast suggest that the dye is not fully oxidized within 1 h in PLB985 cells. However, other particles may contain much less dye and reach saturation earlier [29]. The issue is further complicated by the fact that the amount of label on each particle varies and follows a Gaussian distribution. Thus, some particles may reach saturation, while others still have unoxidized DCFH2. Taken together, DCFH2-labelled particles may be currently the best choice to investigate the kinetics of phagosomal ROS production; however, a number of controls are required to make sure that the entire experimental time course actually reflects ROS production. Furthermore, the duration of phagosomal ROS production certainly depends on the cell type and on the type of ingested particle.
Dihydro-2′,4,5,6,7,7′-hexafluorofluorescein (H2HFF) is another ROS-sensitive fluorescein derivative. It is insensitive to pH from 4.5 to 7 [40]. Unfortunately, only the free dye and the BSA-coupled variant (H2HFF-OxyBURST) are commercially available. The latter has been successfully tested for phagosomal ROS production. Silica beads were labelled with H2HFF-OxyBURST by using cyanamide as a crosslinker with 12 h incubation under argon. In a second reaction, the same beads were labelled with Alexa-594 succinimidylester to obtain a ROS-insensitive reference signal. The ratio of OxyBURST fluorescence over Alexa 594 fluorescence provided a robust readout for phagosomal ROS. Like DCFH2, H2HFF is oxidized by H2O2 catalyzed by peroxidise; however, other ROS may also oxidize this dye [40].
As outlined above, DCFH2 is not specific. Presumably, most of the oxidation within the phagosome from neutrophil-like cells is due to H2O2 in conjunction with a peroxidase, which is probably MPO in neutrophil phagosomes. However, MPO will also produce HOCl which oxidizes DCFH2 to Xfluo. Furthermore, at high concentrations, HOCl is also capable to bleach DCF as well as Xfluo. Excitation spectra of DCFH2-particles inside phagosomes suggest that DCF and Xfluo are present in the same phagosome [29]. It remains to be seen whether the spectroscopic properties of the two dyes allow quantitative analysis of phagosomal HOCl production. The specificity of most ROS detection methods has been subject to debates beyond the scope of this paper. Although the ideal probe would be perfectly specific for one type of ROS, the most specific probe is not necessarily the best choice for every experiment. Probes that react with more than one type of ROS may have the advantage to be more sensitive to the overall response. Obviously, they are also more sensitive to other sources of ROS or interference from other chemicals that react with ROS.
Dihydrorhodamine (DHR) is oxidized by H2O2 to red fluorescent rhodamine 123 and is the most sensitive probe for ROS detection with flow cytometry [31]. DHR and rhodamine 123 are lipid soluble and readily enter cells and intracellular organelle such as the phagosome. Rhodamine 123 shows a tendency to accumulate in mitochondria which may themselves contribute to DHR oxidation [41]. Therefore, rhodamine 123 fluorescence only partially reflects phagosomal ROS production. With video microscopy, the phagosome may be selected as a region of interest and local rhodamine fluorescence analyzed to investigate phagosomal ROS production [42].
A new strategy for targeting probes into phagosomes arises from recent variants for fluorescent proteins (FPs). The fluorescence of certain FPs, such as EGFP, is reversibly reduced at low pH and irreversibly bleached by HOCl. Macrophages do not produce HOCl, and their phagosomes become acidic and will quench EGFP fluorescence. The reversibility can be tested by neutralizing the phagosomal pH with a proton ionophore at the end of the experiment. In contrast, neutrophil phagosomes are less acidic but contain HOCl, which will bleach the FP. When expressed by microorganisms, the FP is directly targeted to the phagosome [20]. Alternatively, GPI-anchored GFP was expressed in the outer leaflet of the macrophage plasma membrane. Upon phagocytosis, some but not all of this GFP entered the phagosome and was quenched during acidification [41]. Other endosomal compartments also acquired GPI-GFP, and therefore careful selection of the region of interest was required for image analysis.
In recent years, a number of FPs with improved sensitivity to ROS have been created (reviewed in [43]). roGFP2 appears to be the most promising member of this family. The oxidation-induced shift of the roGFP excitation spectrum is reversible and equilibrates slowly (minutes) with the redox potential of its environment, which is a fundamental difference compared to dyes like DCFH2 that become irreversibly oxidized. DCF fluorescence reflects the cumulative exposure to ROS, whereas FPs reflect a redox equilibrium. Fusion constructs between roGFP and other redox-sensitive proteins such as glutaredoxins showed improved specificity and sensitivity [44]. An alternative approach has led to the protein HyPer [45]. Here, cpYFP, which is not redox sensitive but particularly sensitive to conformational changes, was fused to a redox-sensitive protein from E. coli, OxyR. The fusion protein undergoes a reversible shift in the excitation spectrum in the presence of submicromolar H2O2. Until now, roGFP constructs and HyPer have not been successfully applied to phagosomal ROS production. They may actually be too sensitive and therefore rapidly saturate inside a phagosome. The time required for their spectral shift and the conditions of reversibility may be further limitations. Variants with different redox potential are under construction and could open new roads to investigate phagosomes.
ROS production is not an isolated event during phagosome maturation but coincides with changes in pH, granule fusion, and ion flux. Ideally, the measurement of phagosomal ROS production should be coupled to the quantitative detection of other events in and around the phagosomes. Video imaging of DCFH2-labelled particles has been successfully coupled to measurements of the cytosolic free calcium concentration ([Ca2+]i) with the fura-2 in parallel in the same cell. The combined measurement of 2 dynamic parameters revealed a transient rise in [Ca2+]i, which triggered the phagosomal ROS production [36]. At least 2 studies succeeded in the concomitant observation of DCF-fluorescence inside the phagosome and the accumulation of fluorescent proteins, CFP and/or mCherry, around the phagosome [35, 46]. Such simultaneous multicolour observations are not trivial since the excitation and emission spectra of DCF and various fluorescent proteins are broad and likely to overlap. Narrow excitation and emission bands for each fluorophore have to be selected and switched rapidly throughout the acquisition. The total exposure time needs to be kept below the level of photooxidation and photobleaching for each dye. DCF-particles tend to give very strong signals, while FPs tend to be expressed at low level giving weak signals. Thus, a minor contamination with DCF signal may override the signal from the fluorescent protein. If the signals cannot be adequately separated, the dyes have to be used in separate experiments. Averaging those experiments may still provide valuable insight into the parallel evolution of 2 or more probes, but some of the information on individual phagosomes is lost [47].
6. pH
The intraphagosomal pH in macrophages progressively decreases over 15 to 60 min down to pH 4 to 5, mainly due to the activity of V-ATPase, and this acidification is important for bacterial killing [48]. Neutrophil phagosomes are less acidic [1, 42], and some studies even report a transient alkalinization [49]. The difference between macrophages and neutrophils appears to be related to the much stronger NADPH oxidase activity in the latter, because the conversion of O2●− to H2O2 consumes protons. Phagosomal pH was investigated with partition dyes such as acridine orange and lysotracker [41]. These dyes preferentially accumulate in acid organelles such as lysosomes and phagosomes. However, their fluorescence intensity depends on numerous parameters and not only on the pH of their immediate environment. Therefore, they are suitable to identify acidic phagosomes but not convenient for quantitative pH measurements.
Direct particle labelling is required to target pH-sensitive dyes to the phagosome. The choice of dyes is larger than for ROS measurements. Fluorescein and many of its derivatives are pH sensitive, and fluorescein isothiocyanate (FITC) is coupled to amine and sulfhydryl groups in a simple reaction. Other pH indicators such as Carboxy-SNARF, Oregon green 488, and pHrodo are available as succinimidyl esters for particle labelling. Sulfosuccinimidyl esters may also be prepared by dissolving the carboxylic acid dye in a buffer that contains N-hydroxysulfosuccinimide (NHSS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) [50]. When used in fluorescence microscopy, these dyes are subject to common problems that interfere with quantitative evaluation, namely, photobleaching, leakage of the dye (if unbound) from the compartment of interest, and movements of the compartment in and out of the focal plane. Most of these problems can be overcome by ratio measurements similar to the pioneering work with calcium indicators [51]. Ideally the dye itself undergoes a pH-dependent spectral shift. When the fluorescence at two appropriate wavelengths is determined, the ratio of the two intensities indicates the pH and is independent of the dye concentration in the measured region. Alternatively, two dyes are coupled to the same particle, one sensitive to pH and the other insensitive. The ratio between the two is a reliable indicator of pH unless one of the dyes is much more sensitive to photobleaching or other environmental parameters. Such ratio measurements can be calibrated using ionophores in the presence of well defined pH buffers.
Labelling of live bacteria or fungi with succinimidyl ester or isothiocyanates of pH-sensitive dyes (carboxyfluorescein, fluorescein, and Oregon green) has been achieved without perturbing the survival of the pathogen. However, the labelling conditions were usually modified to reduce the exposure of the live organism to aggressive chemicals [52–54]. The labelling conditions need to be carefully controlled to maintain full viability of the pathogen. The total amount of dye on the live organism may be less than on the killed organism, which could cause problems of signal intensity or saturation. Nevertheless, this approach provides important insight into the survival mechanism of intraphagosomal bacteria. An interesting approach for detection of phagosomal pH was described by Martin-Orozco et al. [55]. They expressed GFP in live bacteria, Salmonella typhimurium, under the control of a pH-sensitive promoter. GFP was clearly detected in over 50% of internalized bacteria as early as 30 min after transfection. This is a rather indirect method to detect phagosomal acidification, and in exchange it reveals an important bacterial response to pH change.
7. CALCIUM
Calcium is required as a signalling molecule for a number of steps during phagocytosis from actin remodelling to activation of the NADPH oxidase (reviewed in [56]). Local calcium signals around the phagosome have been observed, which raised the question of the calcium source; it could be ER calcium stores or the phagosome itself. The calcium indicator fura-2 had been covalently linked to zymosan particles, which were then internalized by neutrophils. Ratiometric measurements of fura-2 fluorescence in individual phagosomes showed a slow decline of the phagosomal calcium concentration [57]. Thus, the phagosome appears to liberate into the cytosol the calcium that was trapped during phagosome formation. The responsible calcium channel was sensitive to econazole, an inhibitor of store-operated calcium entry, but BTP2, another inhibitor of store-operated calcium entry, failed to affect phagosomal ROS production [23, 57]. The calcium channel protein Orai1 and its ER-based regulator stim1 are involved in phagocytosis and phagosomal ROS production. However, these channels could operate in the phagosomal membrane or only in the plasma membrane [58]. Localisation of Orai1, 2, and 3 channels in phagosomes was recently reported [59]. Given the importance of cytosolic calcium for the regulation of phagocytosis, the role of calcium release from the phagosome deserves further investigation. We could also envision a role of calcium inside the phagosome, for example, eukaryotic pathogens may require calcium for their signalling mechanisms.
8. MAGNESIUM
Dictyostelium discoideum is an amoeba that feeds on bacteria and uses phagocytosis of bacteria for internalization and killing. In contrast to mammalian phagocytes, Dictyostelium is amenable for genetic screens. Taking advantage of this possibility, Lelong et al. just described a magnesium-dependent killing effect for certain Gram-negative bacteria (Klebsiella) [60]. The mechanism is not yet known, but the discovery raises the question of the actual Mg2+ concentration inside the phagosome. Previous indications that magnesium depletion of the phagosome would favour Salmonella growth led to direct Mg2+ measurements in the phagosome. The Mg2+-sensitive dye coumarin 343 was embedded into nanoparticles together with an Mg2+-insensitive reference dye. The nanoparticles were incubated together with Salmonella and internalized by macrophages. The experiment revealed a rather stable phagosomal Mg2+ concentration of about 1 mM [55]. Other commercial magnesium indicators such as mag-fura 2 have a Kd for Ca2+ below 100 μM. They are sufficiently specific of Mg2+ measurements at low intracellular calcium concentrations; however, the much higher phagosomal calcium concentration, initially above 1 mM, would be too high to detect changes in Mg2+ with these dyes.
9. POTASSIUM
Almost 10 years ago, Reeves et al. published a paper about the activation of neutrophil proteases by K+ flux into the phagosome [61]. This paper spurred intense debates and numerous papers which will not be summarized here. A second paper from the same group proposed that large conductance potassium channels (BK channels) were responsible for massive potassium influx into the phagosome. This paper was highly criticized by several groups [62–64] and ultimately retracted. The debate on BK channels somewhat overshadowed the question, whether the phagosomal potassium level changes and for what purpose. Part of the difficulties with potassium is due to the methods to study dynamic changes of K+ inside phagosomes. K+ electrodes or quantitative 86Rb flux measurements detect the average response of a large number of cells [61]. X-ray microanalysis has a high spatial resolution but requires fixed tissues. Fluorescent potassium indicators for microscopy or flow cytometry are rare. The fluorescent potassium indicator PBFI may not be sufficiently specific for phagosomal K+ measurement, because it is sensitive to the sodium concentration, to pH, and to the viscosity of the medium, all of which are likely to change during phagocyte maturation. There is more to learn about phagosomal potassium as illustrated by a recent report on a mycobacterial K+ transporter, Kef [65]. Butler et al. reported that mycobacteria pump K+ into the phagosome and thereby increase ROS production by the host macrophage; in the absence of the transporter, more mycobacteria survive.
10. CHLORIDE
There is growing interest in the role of chloride in the phagosome. At least two different chloride transporters, CFTR and ClC-3, are expressed in neutrophils and involved in phagocytosis [66, 67]. Surprisingly, Listeria monocytogenes exploits CFTR-mediated Cl−-transport to escape from the phagosome [68]. On the other hand, killing of Pseudomonas aeruginosa by neutrophils depends on extracellular chloride. Large amounts of Cl− are required for the production of HOCl by myeloperoxidase, and additional peroxidase-independent roles of chloride were also identified [69]. Phagosomal Cl− concentration was measured with the chloride-sensitive dye 6-methoxyquinoline attached to zymosan together with a chloride-insensitive reference dye [70]. As the authors pointed out, the dye is sensitive to oxidation by myeloperoxidase; therefore, this enzyme had to be inhibited throughout the measurement. Thus, the most important Cl− consumer had to be neutralized suggesting that the Cl− concentration in fully functional phagosomes would be lower than detected by this method. This example illustrates the difficulties of probing the harsh environment inside the phagosome. Chloride-sensitive FPs have been developed, including the recent ClopHensor [71]. It remains to be seen whether these FPs are suitable for the phagosome.
11. ENZYMES
Neutrophil granules and macrophage lysosomes contain a wide array of enzymes [2] involved in bacteria killing. To understand their activity inside the phagosome, one may distinguish several steps. The first step is the fusion of the granule/lysosome with the phagosome delivering its content into the phagosome. However, the enzyme may be trapped in the intragranular matrix and has to be liberated first. The enzyme may be regulated, for example, by specific inhibitors, and its activity could depend on the phagosomal environment (pH) and the availability of substrate (ROS). We can assume that enzymes from one type of granule arrive together in the phagosome, but their activity may follow a quite different time course. The arrival of various proteins in the phagosome has been widely studied at low time resolution by immunofluorescence and electron microscopy. Kinetic measurements of phagosomal activity are at their beginning.
A dynamic assay for phagosome-lysosome fusion based on fluorescence resonance energy transfer (FRET) has been developed by Yates et al. [72]. Macrophages were loaded with Alexa Fluor 594 hydrazide by pinocytosis staining the endosomal and lysosomal compartment. The cells were then exposed to opsonized silica beads previously labelled with Alexa Fluor 488 succinimidylester. The FRET signal reflected fusion of Alexa Fluor 594 containing lysosomes with phagosomes containing Alexa Fluor 488 beads, effectively measuring the concentration of lysosomal cargo inside the phagosome [73]. The signal increased rapidly over the first 10 min of phagocytosis and reached a steady state after 90 min.
Activity of hydrolytic enzymes within the phagosome can be monitored when particles are labelled with an appropriate substrate that changes its fluorescence properties upon enzyme-catalyzed cleavage. For detecting protease activity, Bodipy dyes were attached to BSA to such a high degree that most of the Bodipy fluorescence was self-quenched. Bodipy BSA or commercial DQ-BSA (green or red) was either directly opsonized with anti-BSA antibodies or attached to particles prior to phagocytosis. Cleavage of BSA relieves the self-quenching and liberates fluorescent Bodipy peptides inside the phagosome [74, 75]. A similar strategy was used with the nonfluorescent substrate (Biotin-LC-Phe-Arg)2-Rhodamine110 that liberates fluorescent rhodamine upon cleavage [72]. Parallel labelling with Alexa Fluor 594 gives a protease-independent signal reporting the number of internalized particles. The assay revealed continuous protease activity for at least 1 h in macrophages. The method is limited by the amount of substrate and may not reveal the real end of protease activity. In its present form, the assay is not specific for one particular protease.
Following the same principle, the group of DG Russell also developed dynamic assays for phagosomal lipase activity and β-galactosidase activity [73, 75]. In both cases, the substrate was lipophilic and therefore was attached to silica beads that had been coated with a mixed lipid monolayer. Again, the assays are substrate limited; thus, the observed kinetics depend on the amount of substrate relative to the amount of active enzyme. At this stage, they give an overall idea of the onset and the first hour of enzyme activity in a macrophage phagosome, and they provide a tool to compare different activation states of the macrophage [75].
Electron microscopy has been intensively used to characterize phagosomes. The technique provides unique advantages to examine the size of the phagosome, the space between the pathogen and the phagosomal membrane, the integrity of the pathogen, and the integrity of the phagosomal membrane. Staining techniques provide further insight into the composition of the phagosome. Nevertheless, biological material needs to be fixed before examination by electron microscopy. Therefore, the method has an intrinsic low time resolution and is best suitable for few fixed time points.
12. INSTRUMENTATION
Fluorescent dyes are conveniently analyzed in a sensitive spectrofluorimeter, often equipped with temperature control, stirring of the cuvette and offering the possibility to inject chemicals into the cuvette during the measurement. Microplate readers offer a much higher sample throughput and reduced sample volume. Except for the most sophisticated machines, their time resolution is limited. Flow cytometry provides information on each cell in the sample and permits the detection of minor events. Multiparameter experiments are often possible. Time resolution is quite variable between applications and instruments. Flow cytometry from microplates is likely to develop and create new opportunities for high-throughput experiments. Microscopy offers the unique advantage of subcellular localization including the possibility to follow in parallel multiple phagosomes in the same cell. Time resolution can be better than 1 s. Conventional microscopy is labour intensive and clearly “low throughput.” In fact, analysis of video films takes as much time as the actual experiment. Improved software for phagosome tracking and analysis is likely to accelerate the analysis process. Automatic imaging and image analysis systems are currently developed [76]. Together with the use of microplate readers and flow cytometers, we are likely to see faster sample turnover in the future [73]. For a long time, excitation and emission spectra of the fluorophores were only recordable in spectrofluorimeters. In recent years, microscopes with high spectral resolution become available for excitation as well as emission spectra [77]. Spectral analysis may reveal changes that remain unnoticed at fixed wavelength [29].
13. PROBE LIMITATIONS
The probes may interfere with the chemistry of the phagosome or may divert the phagosomal reactions. Classical examples for this phenomenon are calcium dyes which are also calcium buffers. Dyes with low efficiency (fluorescence intensity) need to be used at higher concentrations to obtain exploitable signals and may then bind a significant quantity of calcium. This has been a drawback of the first generation of calcium indicators, namely, quin-2 [78]. On the other hand, the buffering capability may be used to change the free calcium concentration deliberately and to suppress physiological calcium signals. For example, the calcium dependence of degranulation and superoxide generation by neutrophils has been investigated by charging the cells with different concentrations of quin-2 [79]. More recent calcium indicators such as fura-2 are more efficient and give strong fluorescent signals at micromolar concentrations. The buffering capacity of fura-2 has been useful to analyse transient calcium signals in neutrophils [80]. Thus, probes that reversibly bind their ligand may have buffering effects that need to be taken into account. On the other hand, probes that react with their ligand such as ROS detecting probes will change the actual ligand concentration. If the ligand is a substrate of other phagosomal reactions, the latter may suffer from the lack of substrate due to the interaction with the probe. These problems may be insignificant as long as there is much more ligand than probe. In any case, control experiments to estimate the nature of the probe-ligand interaction may be required. High-affinity probes have a stronger influence than probes with lower affinity, and they also saturate more rapidly. Therefore, the sensitivity of the probe needs to be in the range of the actual concentrations of the ligand.
For accurate dynamic measurements, the kinetics of the probe-ligand interaction needs to be faster than the changes in ligand concentration. This is not the case for all known dyes and becomes even more critical for reversible changes.
In many cases, we wish to compare two physiological situations such as the phagocytosis of a pathogen with a nonpathogenic species. Absolute quantitative measurements are not always required to investigate whether the pathogen changes phagosomal pH, for example. However, calibrated measurements provide a much better appreciation of the effect. Calibration is often feasible with pH and Ca2+ measurements, but for most other investigations, calibration methods are rarely available. We often do not even know whether the probe responds to the ligand in a linear fashion. Calibrated measurements will be required to test and refine mathematical methods such as the one for phagosomal ROS production [32].
14. NEW DIRECTIONS
We can hope for substantial improvements of organic fluorophores notably new blue or red dyes that can be combined with the dominating green fluorophores. We are also likely to see new variants of fluorescent proteins. Instrumentation, notably microscopy and flow cytometry, continuously improves. Techniques like FRET, FLIM, or measurement of anisotropy are barely exploited in the context of phagocytosis. There is a whole new field of nanoparticles, which is not discussed here. In fact, some promising applications have been published for the detection of H+, Ca2+, Mg2+, ROS, and others [81].
15. WHAT HAPPENS AROUND THE PHAGOSOME?
Quite often, the purpose of investigating the biochemistry of the phagosome is to understand the regulatory mechanisms and potential interference from the pathogen. The regulatory mechanisms of the phagocyte are mostly localized on the cytoplasmic surface of the phagosome. Examples concern the assembly of the NADPH oxidase, vesicle fission and fusion, the activation of GTP-binding proteins, or lipid modifications of the phagosomal membrane. Real-time measurements of these events are now possible with FP-tagged proteins. The accumulation of cytosolic subunits of the NADPH oxidase at the phagosome has been investigated by video microscopy with FP-tagged protein [46, 47, 82, 83]. The activation state of 3 closely related GTP-binding proteins, cdc42, rac1, and rac2, has been explored in a sophisticated FRET imaging study [84]. The lipid composition of the phagosome changes during maturation, transiently providing anchoring sites for numerous proteins, which are important for pathogen destruction [7]. These changes become visible with the help of FP-tagged protein domains that specifically attach to certain lipids [85]. For example, the use of PH domains with distinct lipid specificities revealed the temporary presence of 3 distinct polyphosphoinositides on the phagosomal membrane [86]. The next challenge is to connect the events on the phagosomal membrane to the changes inside the phagosome. Ideally, this should be done in the same experiment with compatible detection methods [35]. Alternatively, parallel studies under similar experimental conditions may be performed [47].
16. OTHER APPLICATIONS
The methods described here are selected for their use in professional phagocytes, namely, macrophages and neutrophils. They may also be useful for other intracellular organelles that contain bacteria. Dendritic cells internalize pathogens for the prime purpose of presenting antigen. The process involves NADPH oxidase and other phagosomal compounds, but distinct features of the dendritic cell phagosome have been identified [26]. Dictyostelium discoideum internalizes bacteria for feeding, which involves killing and degradation of the bacterium inside the phagosome [60]. A variety of bacteria invade mammalian cells, presumably to seek a niche for survival and escape from the immune system [4, 87]. In some cases, the bacteria enter a phagosome-like vacuole. The luminal environment of these vacuoles is poorly understood. In each of these internalization processes, the biochemistry of the vacuole (pH, ROS, proteases, etc.) is likely to evolve over minutes and hours and likely affects the outcome of the internalisation (destruction, survival, etc.). Some of the above mentioned dyes face saturation inside neutrophil or macrophage phagosomes. This should be much less of a problem in phagosomes from nonprofessional phagocytes. Furthermore, dyes that are destroyed in a neutrophil phagosome may be sufficiently stable in a less aggressive vacuole.
17. KEY FEATURES OF THE IDEAL DYE
Most of the methods mentioned here rely on fluorescent probes. The ideal probe has a high selectivity towards one parameter (pH, ROS, etc.) and should not be affected by any other compound in the phagosome. Complete insensitivity towards H+ is difficult to achieve, in particular over the entire range of phagosomal pH from pH4 to pH8 spanning 4 orders of magnitude in proton concentration. Insensitivity towards ROS is equally challenging as these compounds by definition are highly reactive. Control experiments in the absence of ROS production using NADPH oxidase inhibitors, namely DPI, or NOX-deficient cells are required to assess the robustness of the probe. Sensitivity and saturation are adapted to the relevant range inside phagosomes. Spatial and temporal resolution depends on the intensity of the dye as well as the imaging equipment. Photobleaching and photoactivation need to be avoided. The dye should be properly targeted and not leak out of the phagosome. Calibration is a substantial advantage. Multiple dyes in the same experiment are required to analyse the correlation of two or more parameters. This depends primarily on the excitation and emission spectra of the dyes. Finally, the dye should be compatible with live organisms.
No dye fulfils perfectly all the requirements. The existing methods have brought much insight into the biochemistry of the phagosome. New dyes are under development to improve the dynamics, the resolution, and the specificity of the methods. The awareness that infectious diseases are a continuous threat to human and animal health will motivate new efforts to understand the pathology of the infectious agents.
ACKNOWLEDGMENT
The author wishs to thank his colleagues for numerous discussions on the subject of this paper, with special thanks to Sophie Dupré, Marie Erard, Marie-Cécile Faure, Dhafer Laouini, Laurence Mery, Jean-Claude Sulpice, and Asma Tlili.
References
- 1.Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. Journal of Leukocyte Biology. 2011;90(2):271–284. doi: 10.1189/jlb.0810457. [DOI] [PubMed] [Google Scholar]
- 2.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33(5):657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 3.Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Seminars in Immunopathology. 2008;30(3):209–235. doi: 10.1007/s00281-008-0121-8. [DOI] [PubMed] [Google Scholar]
- 4.Bhavsar AP, Guttman JA, Finlay BB. Manipulation of host-cell pathways by bacterial pathogens. Nature. 2007;449(7164):827–834. doi: 10.1038/nature06247. [DOI] [PubMed] [Google Scholar]
- 5.Kusner DJ. Mechanisms of mycobacterial persistence in tuberculosis. Clinical Immunology. 2005;114(3):239–247. doi: 10.1016/j.clim.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Hilbi H. Modulation of phosphoinositide metabolism by pathogenic bacteria. Cellular Microbiology. 2006;8(11):1697–1706. doi: 10.1111/j.1462-5822.2006.00793.x. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg BE, Grinstein S. Pathogen destruction versus intracellular survival: the role of lipids as phagosomal fate determinants. Journal of Clinical Investigation. 2008;118(6):2002–2011. doi: 10.1172/JCI35433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zandbergen G, Solbach W, Laskay T. Apoptosis driven infection. Autoimmunity. 2007;40(4):349–352. doi: 10.1080/08916930701356960. [DOI] [PubMed] [Google Scholar]
- 9.Allen LAH, McCaffrey RL. To activate or not to activate: distinct strategies used by Helicobacter pylori and Francisella tularensis to modulate the NADPH oxidase and survive in human neutrophils. Immunological Reviews. 2007;219(1):103–117. doi: 10.1111/j.1600-065X.2007.00544.x. [DOI] [PubMed] [Google Scholar]
- 10.Kubica M, Guzik K, Koziel J, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001409. Article ID e1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. Journal of Immunology. 2000;164(7):3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 12.Sinha B, Herrmann M. Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Thrombosis and Haemostasis. 2005;94(2):266–277. doi: 10.1160/TH05-04-0235. [DOI] [PubMed] [Google Scholar]
- 13.Laskay T, van Zandbergen G, Solbach W. Neutrophil granulocytes—trojan horses for Leishmania major and other intracellular microbes? Trends in Microbiology. 2003;11(5):210–214. doi: 10.1016/s0966-842x(03)00075-1. [DOI] [PubMed] [Google Scholar]
- 14.van Zandbergen G, Bollinger A, Wenzel A, et al. Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(37):13837–13842. doi: 10.1073/pnas.0600843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dacheux D, Attree I, Schneider C, Toussaint B. Cell death of human polymorphonuclear neutrophils induced by a Pseudomonas aeruginosa cystic fibrosis isolate requires a functional type III secretion system. Infection and Immunity. 1999;67(11):6164–6167. doi: 10.1128/iai.67.11.6164-6167.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rada BK, Geiszt M, Káldi K, Timár C, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104(9):2947–2953. doi: 10.1182/blood-2004-03-1005. [DOI] [PubMed] [Google Scholar]
- 17.Sinha B, Fraunholz M. Staphylococcus aureus host cell invasion and post-invasion events. International Journal of Medical Microbiology. 2010;300(2-3):170–175. doi: 10.1016/j.ijmm.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Hamers MN, Bot AAM, Weening RS. Kinetics and mechanism of the bactericidal action of human neutrophils against Escherichia coli. Blood. 1984;64(3):635–641. [PubMed] [Google Scholar]
- 19.Palazzolo AM, Suquet C, Konkel ME, Hurst JK. Green fluorescent protein-expressing Escherichia coli as a selective probe for HOCl generation within neutrophils. Biochemistry. 2005;44(18):6910–6919. doi: 10.1021/bi047342s. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz J, Leidal KG, Femling JK, Weiss JP, Nauseef WM. Neutrophil bleaching of GFP-expressing staphylococci: probing the intraphagosomal fate of individual bacteria1. Journal of Immunology. 2009;183(4):2632–2641. doi: 10.4049/jimmunol.0804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hed J, Stendahl O. Differences in the ingestion mechanisms of IgG and C3b particles in phagocytosis by neutrophils. Immunology. 1982;45(4):727–736. [PMC free article] [PubMed] [Google Scholar]
- 22.Serrander L, Skarman P, Rasmussen B, et al. Selective inhibition of IgG-mediated phagocytosis in gelsolin-deficient murine neutrophils. Journal of Immunology. 2000;165(5):2451–2457. doi: 10.4049/jimmunol.165.5.2451. [DOI] [PubMed] [Google Scholar]
- 23.Steinckwich N, Frippiat JP, Stasia MJ, et al. Potent inhibition of store-operated Ca2+ influx and superoxide production in HL60 cells and polymorphonuclear neutrophils by the pyrazole derivative BTP2. Journal of Leukocyte Biology. 2007;81(4):1054–1064. doi: 10.1189/jlb.0406248. [DOI] [PubMed] [Google Scholar]
- 24.Gallois A, Klein JR, Allen LAH, Jones BD, Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. Journal of Immunology. 2001;166(9):5741–5748. doi: 10.4049/jimmunol.166.9.5741. [DOI] [PubMed] [Google Scholar]
- 25.Lundqvist H, Dahlgren C. Isoluminol-enhanced chemiluminescence: a sensitive method to study the release of superoxide anion from human neutrophils. Free Radical Biology and Medicine. 1996;20(6):785–792. doi: 10.1016/0891-5849(95)02189-2. [DOI] [PubMed] [Google Scholar]
- 26.Savina A, Jancic C, Hugues S, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126(1):205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Zhong Z, Xu Z, Chen L, Wang Y. 2′,7′-dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radical Research. 2010;44(6):587–604. doi: 10.3109/10715761003709802. [DOI] [PubMed] [Google Scholar]
- 28.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biology and Medicine. 2007;43(7):995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 29.Tlili A, Dupré-Crochet S, Erard M, Nüße O. Kinetic analysis of phagosomal production of reactive oxygen species. Free Radical Biology and Medicine. 2011;50(3):438–447. doi: 10.1016/j.freeradbiomed.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 30.Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide. 1997;1(2):145–157. doi: 10.1006/niox.1996.0113. [DOI] [PubMed] [Google Scholar]
- 31.Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. Journal of Immunological Methods. 1995;178(1):89–97. doi: 10.1016/0022-1759(94)00247-t. [DOI] [PubMed] [Google Scholar]
- 32.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. Journal of Biological Chemistry. 2006;281(52):39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 33.Swindle EJ, Coleman JW, DeLeo FR, Metcalfe DD. FcεRI- and Fcγ receptor-mediated production of reactive oxygen species by mast cells is lipoxygenase- and cyclooxygenase-dependent and NADPH oxidase-independent. Journal of Immunology. 2007;179(10):7059–7071. doi: 10.4049/jimmunol.179.10.7059. [DOI] [PubMed] [Google Scholar]
- 34.Ryan TC, Weil GJ, Newburger PE, Haugland R, Simons ER. Measurement of superoxide release in the phagovacuoles of immune complex-stimulated human neutrophils. Journal of Immunological Methods. 1990;130(2):223–233. doi: 10.1016/0022-1759(90)90052-w. [DOI] [PubMed] [Google Scholar]
- 35.Kamen LA, Levinsohn J, Cadwallader A, Tridandapani S, Swanson JA. SHIP-1 increases early oxidative burst and regulates phagosome maturation in macrophages. Journal of Immunology. 2008;180(11):7497–7505. doi: 10.4049/jimmunol.180.11.7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dewitt S, Laffafian I, Hallett MB. Phagosomal oxidative activity during β2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+ signal: Ca2+ controls time not space. Journal of Cell Science. 2003;116(14):2857–2865. doi: 10.1242/jcs.00499. [DOI] [PubMed] [Google Scholar]
- 37.Bernardo J, Long HJ, Simons ER. Initial cytoplasmic and phagosomal consequences of human neutrophil exposure to Staphylococcus epidermidis. Cytometry A. 2010;77(3):243–252. doi: 10.1002/cyto.a.20827. [DOI] [PubMed] [Google Scholar]
- 38.Segal AW, Coade SB. Kinetics of oxygen consumption by phagocytosing human neutrophils. Biochemical and Biophysical Research Communications. 1978;84(3):611–617. doi: 10.1016/0006-291x(78)90749-0. [DOI] [PubMed] [Google Scholar]
- 39.Reiss M, Roos D. Differences in oxygen metabolism of phagocytosing monocytes and neutrophils. Journal of Clinical Investigation. 1978;61(2):480–488. doi: 10.1172/JCI108959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.VanderVen BC, Yates RM, Russell DG. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic. 2009;10(4):372–378. doi: 10.1111/j.1600-0854.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeung T, Touret N, Grinstein S. Quantitative fluorescence microscopy to probe intracellular microenvironments. Current Opinion in Microbiology. 2005;8(3):350–358. doi: 10.1016/j.mib.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Jankowski A, Scott CC, Grinstein S. Determinants of the phagosomal pH in neutrophils. Journal of Biological Chemistry. 2002;277(8):6059–6066. doi: 10.1074/jbc.M110059200. [DOI] [PubMed] [Google Scholar]
- 43.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxidants and Redox Signaling. 2010;13(5):621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 44.Gutscher M, Pauleau AL, Marty L, et al. Real-time imaging of the intracellular glutathione redox potential. Nature Methods. 2008;5(6):553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 45.Belousov VV, Fradkov AF, Lukyanov KA, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 46.Li XJ, Tian W, Stull ND, Grinstein S, Atkinson S, Dinauer MC. A fluorescently tagged C-terminal fragment of p47phox detects NADPH oxidase dynamics during phagocytosis. Molecular Biology of the Cell. 2009;20(5):1520–1532. doi: 10.1091/mbc.E08-06-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tlili A, Erard M, Faure M-C, et al. Stable accumulation of p67phox at the phagosomal membrane and ROS production within the phagosome. doi: 10.1189/jlb.1210701. Journal of Leukocyte Biology. In press. [DOI] [PubMed] [Google Scholar]
- 48.Haas A. The phagosome: compartment with a license to kill. Traffic. 2007;8(4):311–330. doi: 10.1111/j.1600-0854.2006.00531.x. [DOI] [PubMed] [Google Scholar]
- 49.Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290(5805):406–409. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 50.Molecular Probes Handbook: a Guide to Fluorescent Probes and Labeling Technologies. 2011. [Google Scholar]
- 51.Jaconi MEE, Lew DP, Carpentier JL, Magnusson KE, Sjogren M, Stendahl O. Cytosolic free calcium elevation mediates the phagosome-lysosome fusion during phagocytosis in human neutrophils. Journal of Cell Biology. 1990;110(5):1555–1564. doi: 10.1083/jcb.110.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oh YK, Straubinger RM. Intracellular fate of Mycobacterium avium: use of dual-label spectrofluorometry to investigate the influence of bacterial viability opsonization on phagosomal pH phagosome-lysosome interaction. Infection and Immunity. 1996;64(1):319–325. doi: 10.1128/iai.64.1.319-325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER. Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infection and Immunity. 1999;67(2):885–890. doi: 10.1128/iai.67.2.885-890.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinha B, François PP, Nüße O, et al. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin α5β1. Cellular Microbiology. 1999;1(2):101–117. doi: 10.1046/j.1462-5822.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 55.Martin-Orozco N, Touret N, Zaharik ML, et al. Visualization of vacuolar acidification-induced transcription of genes of pathogens inside macrophages. Molecular Biology of the Cell. 2006;17(1):498–510. doi: 10.1091/mbc.E04-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nunes P, Demaurex N. The role of calcium signaling in phagocytosis. Journal of Leukocyte Biology. 2010;88(1):57–68. doi: 10.1189/jlb.0110028. [DOI] [PubMed] [Google Scholar]
- 57.Lundqvist-Gustafsson H, Gustafsson M, Dahlgren C. Dynamic Ca2+ changes in neutrophil phagosomes. A source for intracellular Ca2+ during phagolysosome formation? Cell Calcium. 2000;27(6):353–362. doi: 10.1054/ceca.2000.0130. [DOI] [PubMed] [Google Scholar]
- 58.Steinckwich N, Schenten V, Melchior C, Bréchard S, Tschirhart EJ. An essential role of STIM1, Orai1, and S100A8-A9 proteins for Ca2+ signaling and FcγR-mediated phagosomal oxidative activity. Journal of Immunology. 2011;186(4):2182–2191. doi: 10.4049/jimmunol.1001338. [DOI] [PubMed] [Google Scholar]
- 59.Nunes PDC, Shen W, Groenendyk J, Michalak M, Demaurex N. Stomal interaction molecule 1-mediated recruitment of endoplasmic recticulum membranes to nascent phagosomes. European Journal of Clinical Investigation. 2011;41, article 32 [Google Scholar]
- 60.Lelong E, Marchetti A, Guého A, et al. Role of magnesium and a phagosomal P-type ATPase in intracellular bacterial killing. Cellular Microbiology. 2011;13(2):246–258. doi: 10.1111/j.1462-5822.2010.01532.x. [DOI] [PubMed] [Google Scholar]
- 61.Reeves EP, Lu H, Jacobs HL, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416(6878):291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 62.Femling JK, Cherny VV, Morgan D, et al. The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. Journal of General Physiology. 2006;127(6):659–672. doi: 10.1085/jgp.200609504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Essin K, Salanova B, Kettritz R, et al. Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity. American Journal of Physiology. 2007;293(1):C45–C54. doi: 10.1152/ajpcell.00450.2006. [DOI] [PubMed] [Google Scholar]
- 64.DeCoursey TE. Electrophysiology of the phagocyte respiratory burst. Focus on ‘Largeconductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity’. American Journal of Physiology. 2007;293(1):C30–C32. doi: 10.1152/ajpcell.00093.2007. [DOI] [PubMed] [Google Scholar]
- 65.Butler RE, Cihlarova V, Stewart GR. Effective generation of reactive oxygen species in the mycobacterial phagosome requires K+ efflux from the bacterium. Cellular Microbiology. 2010;12(8):1186–1193. doi: 10.1111/j.1462-5822.2010.01463.x. [DOI] [PubMed] [Google Scholar]
- 66.Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS. Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. Journal of Biological Chemistry. 2006;281(18):12277–12288. doi: 10.1074/jbc.M511030200. [DOI] [PubMed] [Google Scholar]
- 67.Painter RG, Valentine VG, Lanson NA, Jr., et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. 2006;45(34):10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Radtke AL, Anderson KL, Davis MJ, DiMagno MJ, Swanson JA, O'Riordan MX. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1633–1638. doi: 10.1073/pnas.1013262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Painter RG, Bonvillain RW, Valentine VG, et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. Journal of Leukocyte Biology. 2008;83(6):1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G. CFTR-mediated halide transport in phagosomes of human neutrophils. Journal of Leukocyte Biology. 2010;87(5):933–942. doi: 10.1189/jlb.1009655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F. Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nature Methods. 2010;7(7):516–518. doi: 10.1038/nmeth.1471. [DOI] [PubMed] [Google Scholar]
- 72.Yates RM, Hermetter A, Russell DG. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic. 2005;6(5):413–420. doi: 10.1111/j.1600-0854.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 73.Russell DG, Vanderven BC, Glennie S, Mwandumba H, Heyderman RS. The macrophage marches on its phagosome: dynamic assays of phagosome function. Nature Reviews Immunology. 2009;9(8):594–600. doi: 10.1038/nri2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seetoo KF, Schonhorn JE, Gewirtz AT, et al. A cytosolic calcium transient is not necessary for degranulation or oxidative burst in immune complex-stimulated neutrophils. Journal of Leukocyte Biology. 1997;62(3):329–340. doi: 10.1002/jlb.62.3.329. [DOI] [PubMed] [Google Scholar]
- 75.Yates RM, Hermetter A, Taylor GA, Russell DG. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic. 2007;8(3):241–250. doi: 10.1111/j.1600-0854.2006.00528.x. [DOI] [PubMed] [Google Scholar]
- 76.Steinberg BE, Scott CC, Grinstein S. High-throughput assays of phagocytosis, phagosome maturation, and bacterial invasion. American Journal of Physiology. 2007;292(2):C945–C952. doi: 10.1152/ajpcell.00358.2006. [DOI] [PubMed] [Google Scholar]
- 77.Garini Y, Young IT, McNamara G. Spectral imaging: principles and applications. Cytometry A. 2006;69(8):735–747. doi: 10.1002/cyto.a.20311. [DOI] [PubMed] [Google Scholar]
- 78.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- 79.Lew PD, Wollheim CB, Waldvogel FA, Pozzan T. Modulation of cytosolic-free calcium transients by changes in intracellular calcium-buffering capacity: correlation with exocytosis and O2-production in human neutrophils. Journal of Cell Biology. 1984;99(4):1212–1220. doi: 10.1083/jcb.99.4.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nüße O, Lindau M. The calcium signal in human neutrophils and its relation to exocytosis investigated by patch-clamp capacitance and Fura-2 measurements. Cell Calcium. 1993;14(4):255–269. doi: 10.1016/0143-4160(93)90047-a. [DOI] [PubMed] [Google Scholar]
- 81.Lee YEK, Smith R, Kopelman R. Nanoparticle PEBBLE sensors in live cells and in vivo. Annual Review of Analytical Chemistry. 2009;2:57–76. doi: 10.1146/annurev.anchem.1.031207.112823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Bruggen R, Anthony E, Fernandez-Borja M, Roos D. Continuous Translocation of Rac2 and the NADPH Oxidase Component p67 phox during Phagocytosis. Journal of Biological Chemistry. 2004;279(10):9097–9102. doi: 10.1074/jbc.M309284200. [DOI] [PubMed] [Google Scholar]
- 83.Ueyama T, Tatsuno T, Kawasaki T, et al. A regulated adaptor function of p40phox: distinct p67 phox membrane targeting by p40phox and by p47 phox. Molecular Biology of the Cell. 2007;18(2):441–454. doi: 10.1091/mbc.E06-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Molecular Biology of the Cell. 2004;15(8):3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grinstein S. Imaging signal transduction during phagocytosis: phospholipids, surface charge, and electrostatic interactions. American Journal of Physiology. 2010;299(5):C876–C881. doi: 10.1152/ajpcell.00342.2010. [DOI] [PubMed] [Google Scholar]
- 86.Kamen LA, Levinsohn J, Swanson JA. Differential association of phosphatidylinositol 3-kinase, SHIP-1, and PTEN with forming phagosomes. Molecular Biology of the Cell. 2007;18(7):2463–2472. doi: 10.1091/mbc.E07-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bobard A, Mellouk N, Enninga J. Spotting the right location-imaging approaches to resolve the intracellular localization of invasive pathogens. Biochimica et Biophysica Acta. 2011;1810(3):297–307. doi: 10.1016/j.bbagen.2010.10.008. [DOI] [PubMed] [Google Scholar]