

Abstract
Despite improvement of antifungal therapies over the last 30 years, the phenomenon of antifungal resistance is still of major concern in clinical practice. In the last 10 years the molecular mechanisms underlying this phenomenon were extensively unraveled. In this paper, after a brief overview of currently available antifungals, molecular mechanisms of antifungal resistance will be detailed. It appears that major mechanisms of resistance are essential due to the deregulation of antifungal resistance effector genes. This deregulation is a consequence of point mutations occurring in transcriptional regulators of these effector genes. Resistance can also follow the emergence of point mutations directly in the genes coding antifungal targets. In addition we further describe new strategies currently undertaken to discover alternative therapy targets and antifungals. Identification of new antifungals is essentially achieved by the screening of natural or synthetic chemical compound collections. Discovery of new putative antifungal targets is performed through genome-wide approaches for a better understanding of the human pathogenic fungi biology.
1. Introduction
The fungal kingdom encompasses an enormous diversity of taxa with varied ecological niches, life-cycle strategies, and morphologies. However, little is known of the true biodiversity of Kingdom Fungi. Of the 1.5 million species estimated to belong to this kingdom, only about 5% were formally classified. Many fungi are parasites for plants, animals, human, and other fungi. Plant pathogenic fungi are able to cause extensive damage and losses to agriculture and forestry including the rice blast fungus, Dutch elm disease, and chestnut blight. Some other fungi can cause serious diseases in humans, several of which may be fatal if left untreated. These include aspergillosis, candidosis, coccidioidomycosis, cryptococcosis, histoplasmosis, mycetomas, mucormycosis, and paracoccidioidomycosis. The so-called dermatophytic and keratinophilic fungi can attack eyes, nails, hair, and especially skin and cause local infections such as ringworm and athlete's foot. Fungal spores are also a cause of allergies, and fungi from different taxonomic groups can provoke allergic reactions. In this paper, after a brief presentation of the medical impact of fungal infections at the global level and a summary of clinical treatments available today for clinicians, we will review the mechanisms underlying in vitro resistance to antifungals in fungal species of major importance in human medicine. Lastly, an overview of ongoing research undertaken to develop new therapeutic strategies to fight against fungal infections will be exposed.
2. Fungal Infections, Clinical Treatments, and Incidence of Antifungal Drug Resistance
2.1. Fungal Infections
At the beginning of the 20th century, bacterial epidemics were a global and important cause of mortality. In contrast, fungal infections were almost not taken into account. Since the late 1960s when antibiotic therapies were developed, a drastic rise in fungal infections was observed, and they currently represent a global health threat. This increasing incidence of infection is influenced by the growing number of immunodeficient cases related to AIDS, cancer, old age, diabetes, cystic fibrosis, and organ transplants and other invasive surgical procedures.
These infections are caused by two types of microorganisms: primary and opportunistic pathogens. Primary pathogens are naturally able to establish an infection in the healthy population. In contrast, opportunistic pathogens, among them commensal microorganisms of the healthy population, are able to develop infectious colonization of the human body when particular criteria, such as immunosuppression, are met. Fungal pathogens can be divided into two major groups: filamentous fungi and yeasts. Most of the primary pathogens are filamentous fungi, while most of the opportunistic pathogens are yeasts and some species of filamentous fungi are increasingly identified as opportunistic pathogens. It is also important to note that fungal infections can be classified in function of the tissue infected (see Table 1).
Table 1.
Characteristics of main fungal infections worldwide.
| Body location | Pathogen type | Organ | Most frequent genus | Estimated incidence of infection* |
|---|---|---|---|---|
| superficial | primary | Skin and hair | Malassezia | ~140,000,000 cases/year |
| cutaneous | primary | Skin and nails | Trichophyton Epidermophyton Microsporum |
~1,500,000,000 cases/year |
| mucosal | opportunistic | Vagina, digestive tract, urinary tract and |
Candida | ~75,000,000 cases/year ~9,500,000 cases/year |
| eye | Aspergillus, Fusarium | ~1,000,000 cases/year | ||
| systemic | opportunistic | any organ (lungs, brain, bloodstream etc.) |
Candida
Aspergillus Cryptococcus Histoplasma Pneumocystis Coccidioidomyces and so on |
~300,000 cases/year ~350,000 cases/year ~1,000,000 cases/year ~500,000 cases/year >200,000 cases/year up to 300,000 cases/year |
*adapted from “The Fungal Research Trust. How common are fungal diseases? Fungal Research Trust 20th Anniversary meeting. London June 18th 2011.”
Superficial mycoses, such as tinea versicolor, are limited to the most external part of the skin and hair. These infections are most frequently caused by the species Malassezia globosa and M. furfur, which are estimated to be carried by 2% to 8% of the healthy population worldwide but could lead to tinea versicolor in some conditions that are still unclear [1].
Cutaneous and subcutaneous mycoses caused by dermatophytes fungi affect keratinized structures of the body. The most frequently involved dermatophyte genera are Trichophyton, Epidermophyton, and Microsporum. In most cases, cutaneous fungal infections require a challenge of immune system, and their incidence varies depending on the site of infection. For example, onychomycoses are very frequent in the global population, with an incidence varying from 5 to 25% [2].
Mucosal infections are mostly caused by opportunistic yeasts, and those belonging to the Candida genus are by far the most frequent. Vaginal, esophageal, oropharyngal, and urinary tract candidiasis are very frequent in immunocompromised patients. For example, esophageal candidiasis is associated with the entry into the clinical phase of AIDS and during the 1980s more than 80% of seropositive patients developed such an infection [3]. Fungal infections, of the eye are also classified as mucosal fungal infections, but are caused more frequently by Fusarium or Aspergillus species rather than Candida species.
Theoretically systemic mycoses may involve any part of the body, and a lot of species formerly considered as nonpathogenic are now recognized opportunistic pathogens responsible for deep-seated mycoses. These infections, with symptoms ranging from a simple fever to a severe and rapid septic shock, are very common in immunocompromised patients and are frequently associated with an elevated mortality rate. The most frequent pathogens involved in systemic fungal infections are Candida, Pneumocystis, Histoplasma, Aspergillus, Cryptococcus, Mucor, Rhizopus, and Coccidioidomyces [4–6].
2.2. Antifungal Agents
Despite extensive research dedicated to the development of new therapeutic strategies, there are only a limited number of available drugs to fight against invasive fungal infections. Indeed, only four molecular classes that target three distinct fungal metabolic pathways are currently used in clinical practice to treat essentially systemic fungal infections: fluoropyrimidine analogs, polyenes, azoles, and echinocandins. Several other classes, such as morpholines and allylamines are only used as topical agents due to either poor efficacy, or severe adverse effects when administered systemically.
2.2.1. Fluoropyrimidines
Fluoropyrimidines, of which only 5-fluorocytosine (5-FC) and 5-fluorouracil (5-FU) are used in human medicine, are synthetic structural analogs of the DNA nucleotide cytosine (Figure 1).
Figure 1.
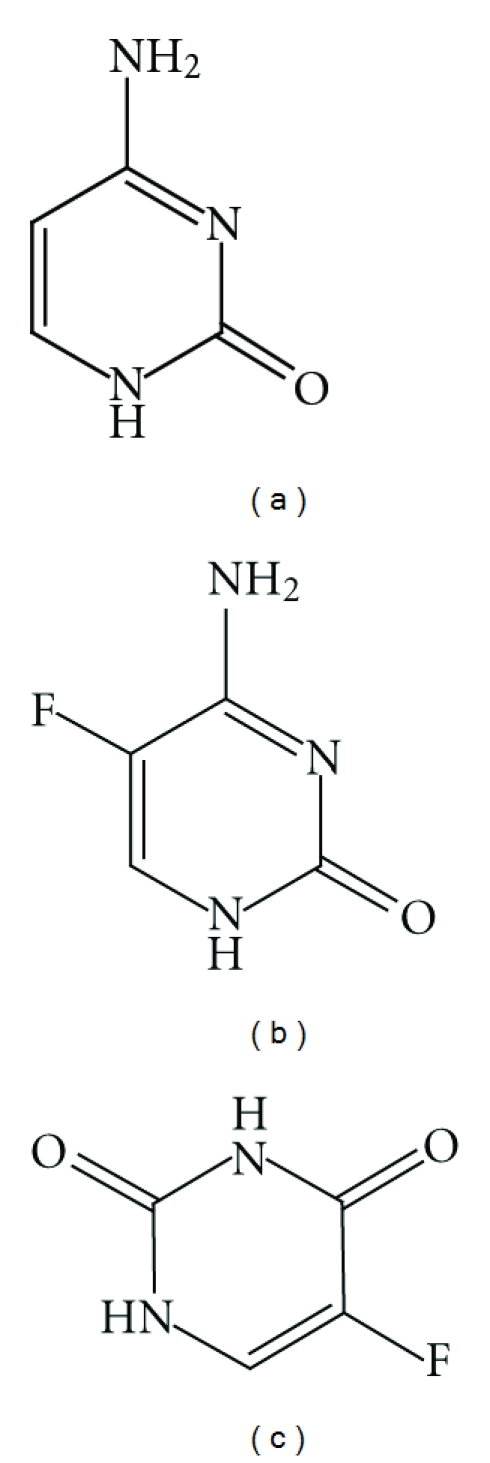
Chemical structures of cytosine (a) and of two fluoropyrimidines, 5-fluorocytosine (b), and 5-fluorouracil (c).
5-FC was synthesized in 1957 by Duschinsky et al., initially as an antitumor therapy [7]. In 1963, Grunberg and coworkers discovered its antifungal potential by means of murine models of cryptococcosis and candidiasis [8]. Several years later 5-FC was successfully used for the treatment of systemic candidiasis and of cryptococcal meningitis [9].
5-FC possesses a broad range of activity. This drug is active against Candida and Cryptococcus genera. 5FC activity on Phialophora, Cladosporium, and Aspergillus genera is much less limited. 5-FC is also active against protozoa belonging to Leishmania and Acanthamoeba genus [10].
Due to its high hydrosolubility and small size, 5-FC possesses interesting pharmacokinetic properties, since it diffuses rapidly throughout body even when orally administered [12]. Generally, it produces negligible side effects, despite some severe adverse effects, such as hepatotoxicity or bone marrow lesions [11, 13–15], occurring in rare cases [16]. Surprisingly, these side effects are identical to those observed with 5-FU treatment, despite the fact that humans do not possess a cytosine deaminase enzyme that is responsible for the conversion from 5-FC into 5-FU in fungal cells [17, 18]. Some data suggest that the intestinal microbiome could be responsible for the 5-FU production and the observed side effects [19].
Despite its numerous pharmacological advantages, the use of 5-FC in clinical practice is decreasing because of the frequent occurrence of innate or acquired resistance to this drug in fungal pathogens. Thus, with few exceptions [20], 5-FC is never used as monotherapy but always in combination with another antifungal, such as amphotericin B [21, 22]. However, the elevated renal and liver toxicities of amphotericin B, that further increase 5-FC hepatotoxicity, has led to combination therapy of 5-FC more frequently with azole drugs.
5-FC itself has no antifungal activity, and its fungistatic properties are dependent upon the conversion into 5-FU [16, 20, 23]. The drug rapidly enters the fungal cell through specific transporters, such as cytosine permeases or pyrimidine transporters [24], before it is converted into 5-FU by the cytosine deaminase [16]. 5-FU itself is converted into 5-fluorouracil monophosphate (5-FUMP) by another enzyme, uridine phosphoribosyltransferase (UPRT). 5-FUMP can then be either converted into 5-fluorouracil triphosphate, which incorporates into RNA in place of UTP and inhibits protein synthesis, or converted into 5-fluorodeoxyuridine monophosphate, which inhibits a key enzyme of DNA synthesis, the thymidylate synthase, thus inhibiting cell replication (Figure 2) [16, 25, 26].
Figure 2.
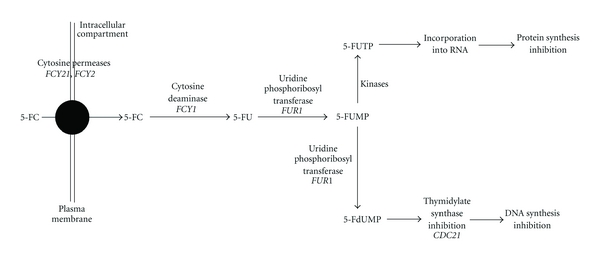
Intracellular metabolization and action mode of 5-FC in S. cerevisiae, adapted from [11]. In bold are indicated gene names of the respective enzymes. 5-FC: 5-fluorocytosine; 5-FU: 5-fluorouracil; 5-FUMP: 5-fluorouridine monophosphate; 5-FUTP: 5-fluorouridine triphosphate; 5-FdUMP: 5-fluoro deoxyribouridine monophosphate.
2.2.2. Polyenes
More than 200 molecules belonging to the chemical class of polyenes have an antifungal activity, most of them being produced by Streptomyces bacteria. However, only three possess a toxicity allowing their use in clinical practice: amphotericin B (AmB), nystatin, and natamycine.
Streptomyces bacteria synthesize polyenes through a gene cluster phylogenetically conserved within these species. This cluster contains genes coding for several polyketide synthases, ABC (ATP-binding cassette) transporters, cytochrome P450-dependent enzymes, and enzymes responsible for the synthesis and the binding of the mycosamine group [27]. Although it is possible to synthesize polyenes chemically, they are still produced from Streptomyces cultures for economic reasons.
Polyenes are cyclic amphiphilic organic molecules known as macrolides. Most of them consist of a 20 to 40 carbons macrolactone ring conjugated with a d-mycosamine group. Their amphiphilic properties are due to the presence of several conjugated doublebounds on the hydrophobic side of the macrolactone ring, and to the presence of several hydroxyl residues on the opposite, hydrophilic side (Figure 3) [28].
Figure 3.
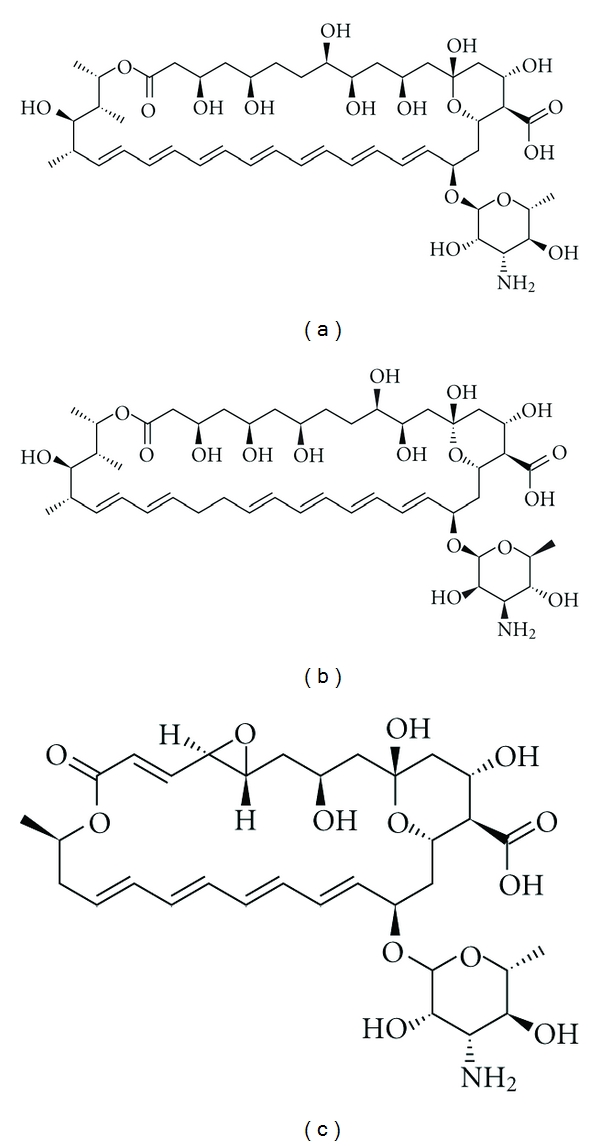
Chemical structures of amphotericin B (a), nystatin (b), and natamycin (c), three main polyene drugs.
Polyene drugs target ergosterol, the main sterol component of fungal membranes. Their amphiphilic structure allows them to bind the lipid bilayer and form pores. Magnetic nuclear resonance data suggest that eight AmB molecules bind eight ergosterol molecules through their hydrophobic moieties, with their hydrophilic sides forming a central channel of 70–100 nm in diameter (Figure 4). Pore formation promotes plasma membrane destabilization, and channels allow leakage of intracellular components such as K+ ions, responsible for cell lysis [28].
Figure 4.

3D model of pore formed by amphotericin B into lipid bilayer of the fungal plasma membrane, adapted from Baginski et al. [29]. Amphotericin B: white (H), green (C), red (O), and blue (N); ergosterol: pink.
While structural data suggest that polyenes target ergosterol, and despite the fact that their binding to ergosterol was experimentally demonstrated [29–31], controversy remains over a possible intracellular mode of action. Some research has suggested that polyene drugs are able to induce an oxidative stress (particularly in C. albicans [32, 33]) as well as their activity seems to be reduced in hypoxic conditions [34].
Polyenes possess a lower but non-negligible affinity for cholesterol, the human counterpart of ergosterol. This slight affinity for cholesterol explains the high toxicity associated with antifungals and is responsible for several side effects [28]. For this reason, only AmB is given systemically, while nystatin and natamycin are only used locally or orally. These two last molecules fortunately possess a very limited systemic activity, since their absorption trough gastrointestinal mucosa is almost nonexistent [35, 36].
For these reasons, AmB is the most used polyene antifungal for systemic infections. Due to its high hydrophobicity and poor absorption through the gastrointestinal tract, it is necessary to administer AmB intravenously [28]. However, AmB administration is accompanied with adverse effects, mostly at the level of kidneys and liver. New AmB formulations, such as liposomal AmB or lipid AmB complexes, minimize such side effects [37].
For more than 40 years, AmB was one of the goldstandards for the treatment of systemic fungal infections due to the low occurrence of acquired or innate resistance to this drug and also because of its broad range of activity [38]. Indeed, AmB is active against most yeasts and filamentous fungi. It is recommended for the treatment of infections caused by Candida, Aspergillus, Fusarium, Mucor, Rhizopus, Scedosporium, Trichosporon, Cryptococcus, and so on. AmB is also widely used to treat parasitic infections such as leishmaniasis and amibiasis [28]. Natamycin and nystatin are active against fungi belonging to the genera Cryptococcus, Candida, Aspergillus, and Fusarium. If nystatin is not used to treat molds infections, this drug is frequently used for the treatment of cutaneous, vaginal, and esophageal candidiasis, and natamycin can be used for the treatment of fungal keratosis or corneal infections [35].
2.2.3. Azoles
Azoles are by far the most commonly used antifungals in clinical practice, and consequently, they are also the mostly studied by the scientific community regarding their mode of action, their pharmacological properties, and the resistance mechanisms developed by microorganisms. Azole antifungals are also largely studied by pharmaceutical companies, who seek to enhance their efficacy and to develop the perfect antifungal.
Azoles are cyclic organic molecules which can be divided into two groups on the basis of the number of nitrogen atoms in the azole ring: the imidazoles contain two nitrogen atoms, and the triazoles contain three nitrogen atoms (Figure 5) [39].
Figure 5.
Chemical structures of the main azole antifungals, four imidazoles: clotrimazole (a), econazole (b), miconazole (c), and ketoconazole (d), two triazoles: itraconazole (e) and fluconazole (f), and three new generation triazoles: voriconazole (g), posaconazole (h), and ravuconazole (i).
Azole drugs target the ergosterol biosynthetic pathway by inhibition of a key enzyme, the lanosterol 14alpha demethylase, encoded be the ERG11 gene. This inhibition occurs through the binding of the free nitrogen atom of the azole ring to the iron atom of the heme group of the enzyme. The resulting accumulation and metabolism of 14alpha methylated sterol species leads to the synthesis of toxic compounds, which are unable to successfully replace ergosterol [40].
The first azole was synthesized in 1944 by Woolley [41], but it was not until 1958 that scientific community began to consider azoles as potential antifungal agents. In late 1960s, clotrimazole, econazole, and miconazole became available for treatment [42]. However, their use was restricted to external application due to their high toxicity when administered orally [43, 44]. In 1968, miconazole became the first antifungal available for parenteral injection, but due to its toxicity and relatively limited range among fungal species [45], its use decreased until it was no longer commercialized.
In 1981, the Food and Drug Administration (FDA) approved a new antifungal, ketoconazole, developed by Heeres and his coworkers [46]. This drug was the only antifungal available for treatment of systemic fungal infections caused by yeasts for the following ten years. However, there are several drawbacks to this drug. It is poorly absorbed when administered orally, and no ketoconazole form has ever been developed for intravenous injection. Moreover, it cannot cross the cerebrospinal barrier and is less active in immunosuppressed patients [42, 47–49]. It causes some severe side effects such as a decrease in testosterone or glucocorticoids production and liver and gastrointestinal complications [50–52]. Lastly, numerous interactions with other drugs were described. For these reasons, the triazoles were developed.
Fluconazole became available for use by clinicians in 1990 and provided many advantages over the use of imidazoles. Fluconazole is highly hydrosoluble and therefore can be easily injected intravenously. It is almost completely absorbed through the gastrointestinal tract, and it diffuses throughout the whole body, including cerebrospinal fluid [53, 54]. Fluconazole is suitable for the treatment of superficial candidiasis (oropharyngal, esophageal, or vaginal), disseminated candidiasis, cryptococcal meningitis, coccidioidomycosis, and cutaneous candidiasis. Due to its good pharmacokinetic properties as well as its broad spectrum of activity, fluconazole was the gold-standard treatment of fungal infections during the 1990s. Unfortunately, the overprescription of this drug by physicians for prophylaxis or treatment led to an increase in resistance to azole drugs. Moreover, fluconazole is almost ineffective against most molds.
Itraconazole was approved and made available by the FDA in 1992. This triazole possesses a broad spectrum of activity across fungal species comparable to this of ketoconazole and wider than fluconazole. Moreover, it is less toxic than ketoconazole and replaced it for treatment of histoplasmosis, blastomycosis, and paracoccidioidomycosis. Contrary to fluconazole, it is also used for the treatment of infections due to species belonging to the genera Aspergillus and Sporothrix [55]. However, itraconazole is hydrophobic and is thus more toxic than fluconazole. Itraconazole is only indicated for the treatment of onychomycosis, of superficial infections, and in some cases for systemic aspergillosis [56]. A new itraconazole formulation with an enhanced absorption and a decreased toxicity was approved by FDA in 1997 [57]. An injectable formulation of itraconazole was made available in 2001 [58].
Fluconazole and itraconazole are still not the perfect antifungals, since they have some nonnegligible drug interactions with such drugs that are used in chemotherapy or with AIDS treatment. These interactions can result in a decrease in azole concentration or even to an increase in toxicity [59]. Furthermore itraconazole and fluconazole are ineffective against some emerging pathogens like Scedosporium, Fusarium, and Mucorales, and resistance to azoles is increasingly reported [60].
So-called new generation triazoles have also been developed. Voriconazole and posaconazole were approved by FDA in 2002 and 2006, respectively. Ravuconazole is currently under clinical trial phase of drug development. They possess a wide range of activity, since they are active against Candida, Aspergillus, Fusarium, Penicillium, Scedosporium, Acremonium, and Trichosporon, and dimorphic fungi, dermatophytes, and Cryptococcus neoformans [61, 62]. While new generation triazoles were shown to be more effective against Candida and Aspergillus [62], compared to classical triazoles their side effects and drug interactions are similar to those observed with fluconazole and itraconazole [63]. Likewise, fungal isolates resistant to classical triazoles are also cross-resistant to new generation triazoles.
2.2.4. Echinocandins
Echinocandins constitute the only new class of antifungals made available for clinicians to fight invasive fungal infections within the past 15 years [64]. Three echinocandins were currently approved for clinical use by the FDA in United States and later by the European Agency for the Evaluation of Medicinal Products (EMEA): caspofungin in 2001 by the FDA and in 2002 by the EMEA, micafungin in 2005, and lastly anidulafungin in 2006.
Echinocandins are synthetic derivatives of lipopeptides (Figure 6). These lipopeptides are naturally produced by several fungal species: Aspergillus rugulovalvus synthesizes caspofungin B, Zalerion arboricola synthesizes pneumocandin B, and Papularia sphaerosperma synthesizes papulacandin. Echinocandins are noncompetitive inhibitors of β(1-3)-glucan synthase, an enzyme that catalyzes the polymerization of uridine diphosphate-glucose into β(1-3) glucan, one of the structural components responsible for the maintenance of fungal cell-wall integrity and rigidity [65, 66]. β(1-3)-glucan synthase consists of an activating and a catalytic subunit encoded by FKS genes. In most fungi, two FKS genes are found within the genome. It has been shown in the model organism Saccharomyces cerevisiae that FKS1 is expressed during the vegetative growth phase and FKS2 during sporulation [67]. Echinocandins are able to inhibit both isoforms of the enzyme [68]. Inhibition of β(1-3)-glucan synthase leads to cell wall destabilization and to the leakage of intracellular components, resulting in fungal cell lysis [69].
Figure 6.
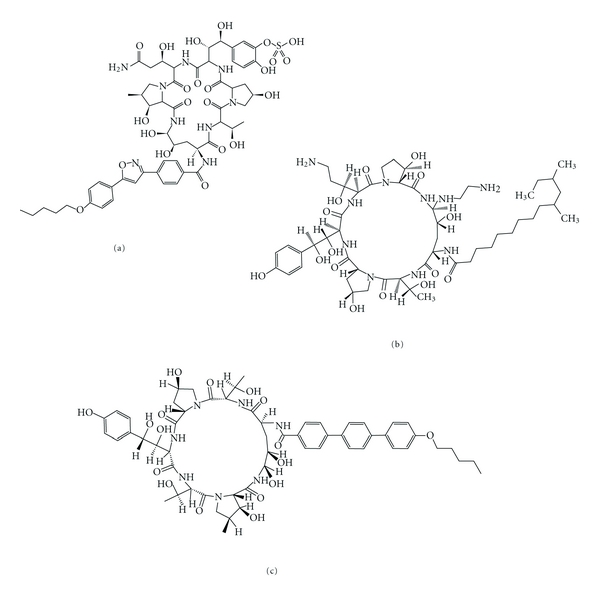
Chemical structure of the three echinocandins used in clinical practice: micafungin (a), caspofungin (b), and anidulafungin (c).
These drugs are poorly absorbed in the gastrointestinal tract because of their high molecular weights and are therefore only used intravenously. Their pharmacologic properties are one of the reasons responsible for the approval of echinocandins by the FDA and the EMEA. These molecules possess a low toxicity (very rare side effects were reported) and are slowly degraded, and a daily injection is sufficient, and contrary to other antifungals, interactions between echinocandins and other drugs are rare [64]. Combined therapy between echinocandins and AmB or another azole often leads to a synergistic effect or at least to an additive effect [70, 71].
Another reason for which the echinocandins were approved is their activity spectrum. Indeed, echinocandins are active against most fungal species, including Candida and Aspergillus. For still unclear reasons, these molecules are fungicidal in Candida but only fungistatic in Aspergillus [72, 73]. Moreover, fungicidal activity of echinocandins is species and isolate dependent within the Candida genus [74]. There exist several species within the fungal kingdom for which the echinocandins are ineffective. Such species include Cryptococcus neoformans [75] or species belonging to Trichophyton and Fusarium genera. Other species have an intermediate susceptibility to echinocandins, such as Scedosporium apiospermum, S. prolificans, and Cladophialophora bantiana [72]. However, echinocandins constitute a good alternative to fight against fungal infections and most of treatment of infections for which classical therapy with azoles or polyenes failed are successfully managed with echinocandins [64]. Therefore, caspofungin is indicated for the treatment of candidemia and invasive candidiasis, for fungal infection prophylaxis, and for the treatment of invasive aspergillosis for which itraconazole, voriconazole, or AmB is ineffective. Micafungin is used for treatment of candidemia and is particularly indicated for fungal infection prophylaxis in bone-marrow transplant patients. Anidulafungin has no particular indications, but its main advantage is its slow degradation in the body without liver or kidney involvement, thus it can be used in patients with liver and/or kidney insufficiencies [76].
What makes echinocandins unique is their fungal target. For many years, the fungal cell wall was considered to be a promising target for the development of new antifungal molecules [68]. The fungal cell wall contains elements that have no equivalents in human [77]. Its integrity is necessary for the fungal survival, since it provides a physical barrier against the host immune cells or against other microorganisms. Cell wall integrity is also responsible for osmolarity homeostasis and the maintenance of cell shape and size. Cell wall is also indispensable to essential enzymatic reactions and as an important role in cell-cell communication. The internal layer of the cell wall is composed of a β(1-3)-glucans and chitin web, in which are included some mannoproteins, while external layer is composed of mannoproteins (Figure 7) [77].
Figure 7.
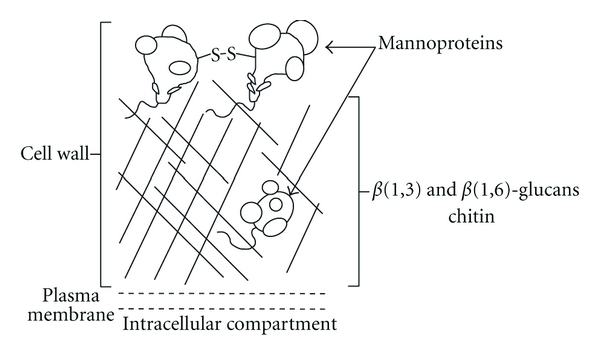
Schematic representation of S. cerevisiae cell wall, adapted from Stone et al. [69].
2.2.5. Other Antifungal Agents
Considering that the ergosterol biosynthetic pathway requires several enzymes that are unique to fungi, they constitute good targets for antifungal therapy, and three minor ergosterol biosynthesis inhibitors are used as topical antifungals. The allylamines and thiocarbamates, such as terbinafine and tolnaftate, both inhibit the ERG1-encoded enzyme, squalene epoxidase. The morpholines such as amorolfine act by inhibiting two different enzymes of the pathway, the Δ7,8-isomerase (encoded by ERG24) and the C14-reductase (encoded by ERG2). Despite their wide spectrum of activity, these antifungal agents are essentially used to treat dermatophyte infections such as tinea capitis, tinea pedis, and onychomycosis, because they do present numerous side effects.
Ciclopirox is also used as a topical antifungal agent, but its mode of action remains poorly understood in fungi [78, 79]. Another drug, griseofulvin, inhibits mitosis by interfering with microtubules function [80].
2.3. Incidence of In Vitro Resistance in Fungal Infection
The incidence of fungal infections has drastically increased over the past three decades and was simultaneously accompanied by increased acquired and innate resistance to antifungal drugs. However, antifungal resistance occurrence has to be considered independently for each antifungal class and for each fungal genus. Moreover, epidemiological data regarding incidence of resistance among fungal species is not identically distributed worldwide [81–83]. Lastly, clinical resistance, defined as the treatment failure in the patient, does not always correlate with in vitro resistance, measured as an increase in minimal inhibitory concentration of a drug. In this paper, only in vitro resistance incidence will be described.
2.3.1. 5-Fluorocytosine
5FC resistance is a very common phenomenon [9, 16, 84]. The development of resistance can be intrinsic, as is the case for C. tropicalis, or acquired through the selection of resistant mutants after antifungal exposure. Within the Candida genus, 7% to 8% of clinical isolates are resistant to 5FC, and this frequency increases to 22% when only nonalbicans Candida species are considered. One to two percent of Cryptococcus neoformans clinical isolates are resistant to 5FC [85]. Filamentous fungi such as Aspergillus and dermatophytes are not susceptible to 5FC.
2.3.2. Polyenes
Despite the reported increase of polyene resistance, it remains a relatively rare event in clinical isolates of fungal pathogens [86], probably in relation with their mode of action, and the absence of systematic and standardized determination of susceptibility of clinical isolates [87]. The incidence of strains resistant to polyenes may thus be largely underestimated. Most fungal species are considered as susceptible to polyene drugs. However, some of them are intrinsically poorly susceptible to these antifungals, such as C. glabrata, Scedosporium prolificans, or Aspergillus terreus [38]. Some species are more prone to acquire polyene resistance. Among yeasts, one may cite C. lusitaniae [88, 89], C. guilliermondii [88], C. krusei [38], and Trichosporon beigelii and among filamentous fungi Scedosporium apiospermum and Sporothrix schenckii [90, 91].
2.3.3. Azoles
The early 1990s was the start of a drastic increase in resistance among fungal clinical isolates. However, the improvement of antifungal therapeutic strategies throughout the last several years has helped to stabilize resistance frequencies. Increase in azoles use selected less susceptible species as well as those able to develop resistance. This led to a shift in the pathogenic fungal species encountered in clinic.
2.3.4. Echinocandins
Echinocandins resistance is a rare event [92]. For example, it is estimated that more than 97% of clinical isolates belonging to the Candida genus are susceptible to these drugs [93, 94]. Contrary to acquired resistance in other fungi, intrinsic echinocandin resistance in Cryptococcus neoformans is not linked with a FKS1 or FKS2 mutation. Indeed, C. neoformans β(1–3)-glucan synthase is inhibited by echinocandins, but this yeast is able to grow in the presence of high concentrations of these drugs. C. neoformans resistance to echinocandins seems to be due to a particular cell-wall polysaccharides composition in this species [95].
2.3.5. Incidence of In Vitro Resistance on Patient Care
As antifungal in vitro resistance poorly correlates with clinical outcome, better attention was needed to define parameters that produced reproducible and reliable intra- and interlaboratory results. For this purpose, two standardized methods for the testing of yeast and mould isolates (CLSI and Eucast) are recognized as the gold standards for drug susceptibility testing [96–98]. These standardized approaches produce susceptibility results comparable between laboratories, which may help to establish breakpoints for antifungal agents (see [96–98] for details). These breakpoints, defined as susceptibility ranges, together with pharmacokinetic and pharmacodynamic analyses and identification of resistance mechanisms, help to assess the in vivo activity of antifungal agents in invasive disease and therefore clinical outcome [99, 100].
3. Drug Resistance Molecular Mechanisms
Microorganisms develop mechanisms to counteract the fungicidal or fungistatic effects of all antifungals classes that are based on three major mechanisms, namely, (i) reducing the accumulation of the drug within the fungal cell, (ii) decreasing the affinity of the drug for its target, and (iii) modifications of metabolism to counterbalance the drug effect (Table 2). The molecular mechanisms leading to azole resistance have been most studied in yeast, and taking them as an example, such mechanisms are divided into four categories (Figure 8) [101]: (i) decrease in azole affinity for their target, (ii) increase in azole target copy number, (iii) alteration of ergosterol biosynthetic pathway after azoles action, and (iv) decrease in intracellular azole accumulation. In some highly resistant clinical isolates, sampled from long-term treated patients, several mechanisms of resistance are often combined [102, 103]. This increase in resistance along antifungal treatment is due to the sequential acquisition of different mechanisms [104–106]. In the following section, the molecular basis of the resistance mechanisms to antifungals will be described.
Table 2.
Nature, target, mode of action, and fungal resistance mechanisms of the major antifungal drugs used in human therapy.
| Antifungal agent | Mode of action and cellular target | Mechanism of resistance |
|---|---|---|
| polyenes | binding to ergosterol | absence of ergosterol (loss of function mutation in ERG3 or ERG6) |
| decrease of ergosterol content in cells | ||
|
| ||
| azoles | inhibition of cytochrome p450 function: 14α-lanosterol demethylase (ERG11) sterol Δ22 desaturase (ERG5) | efflux mediated by multidrug transporters |
| decrease of affinity in Erg11p by mutations | ||
| upregulation of ERG11 | ||
| alterations in the ergosterol biosynthetic pathway | ||
|
| ||
| allylamines | inhibition of squalene epoxidase (ERG1) | unknown |
|
| ||
| morpholines | inhibition of sterol Δ14 reductase (ERG24) and the Δ7–8 isomerase (ERG2) | unknown |
|
| ||
| 5-fluorocytosine | inhibition of nucleic acids synthesis | defect in cytosine permease |
| deficiency or lack of enzymes implicated in the metabolism of 5-FC | ||
| deregulation of the pyrimidine biosynthetic pathway | ||
|
| ||
| echinocandins | inhibition of β-1,3 glucan synthase (FKS1&2) | alteration of affinity of echinocandins for β(1,3)-glucan synthase |
Figure 8.
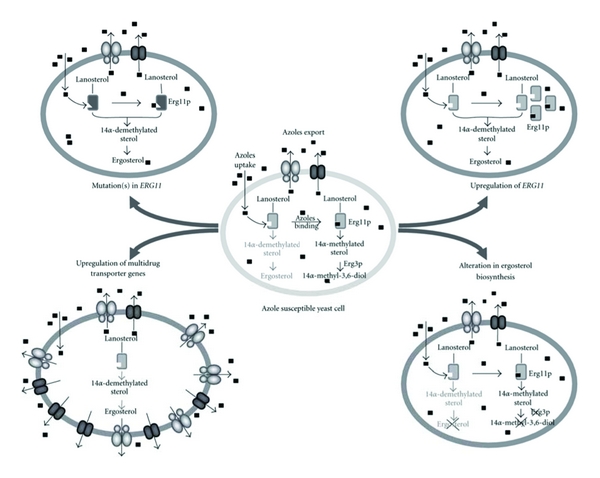
Mechanisms of resistance to azole compounds in C. albicans.
3.1. Increase of Drug Efflux
3.1.1. ABC Transporters
CDR1 and CDR2 (Candida drug resistance 1 and 2) from C. albicans are the two major ABC transporters involved in azole resistance in this species. CDR1 and CDR2 can be coordinately upregulated in some azole-resistant strains or by exposure to a wide variety of chemically unrelated inducers such as terbinafine, amorolfine, fluphenazine, or steroid hormones. Several cis-acting regulatory elements responsible for the regulation of these two genes were identified by several investigators [107–111]. Promoter deletion studies have revealed 5 different regulatory elements in the CDR1 promoter including a BEE (basal expression element), a DRE (drug responsive element), two SREs (steroid responsive element), and a NRE (negative regulatory element) (see Table 3 for details). Internal deletions of the BEE and DRE motifs in the CDR1 promoter affect basal CDR1 expression and drug-induced expression, respectively [107]. SRE1 and SRE2 were reported to be involved in the response to steroid hormones: with SRE1 responding only to progesterone and SRE2 to both progesterone and β-oestradiol [108]. Finally, the deletion of the NRE motif leads to an increase in the basal expression of CDR1 [110, 111]. In contrast to CDR1, the CDR2 promoter contains only a DRE motif (Table 3) [107]. Among these different cis-acting elements, DRE was the only element involved in constitutive high expression and in transient upregulation of both CDR1 and CDR2. This DRE sequence was functionally analyzed by systematic mutation each base of the initially described DRE sequence [107, 112]. The data obtained from systematic mutational studies are in agreement with ChIP-Chip assays performed with the trans-acting factor binding to the DRE [113]. In other Candida species, functional homologues to CDR1 and CDR2 were described as involved in drug resistance. In C. glabrata, CgCDR1 and CgCDR2 (formerly denoted PDH1) as well as SNQ2 (another ABC transporter coregulated with CgCDR1 and CgCDR2) are upregulated in azole-resistant clinical isolates and participate in azole resistance [114–118]. All the three genes contain cis-acting elements in their promoters, so-called PDRE. These elements are similar to those described in S. cerevisiae for PDR5, an ABC transporter involved in drug resistance of S. cerevisiae [119, 120]. Disruptions of CgCDR1 and CgCDR2 lead to hypersusceptibility to fluconazole, cycloheximide, and chloramphenicol [115, 117]. In both C. albicans and C. glabrata, CDR1 was shown to be the main contributor in azole-resistance among the ABC-transporters [121–123]. Other ABC-transporters from C. dubliniensis (CdCDR1 and CdCDR2) [124, 125], C. krusei (ABC1 and 2) [126, 127], C. tropicalis (CDR1-homologue), and C. neoformans (CnAFR1, AntiFungal Resistance 1) were reportedly upregulated in azole-resistant isolates. In A. fumigatus, atrF, and AfuMDR4 are upregulated in itraconazole-resistant strains [128–130]. The cis-acting regulatory elements of these genes are still awaiting in-depth dissection analysis. The overexpression of ABC-transporters have also been identified as a resistance mechanism to azole in Aspergillus nidulans [131, 132].
Table 3.
Cis-acting elements involved in drug resistance.
| Organism | Gene | Regulatory element | Position respectively to the ATG | Trans-acting factor | ||
|---|---|---|---|---|---|---|
| Name | Sequence | |||||
| ABC transporters | BEE | — | −960 to −710 | ? | ||
| DRE | ACGGATATCGGATATTTTTTT | −460 to −439 | Tac1 | |||
| CDR1 | NRE | CTGATTGA | −335 to −328 | ? | ||
| C. albicans | SRE1 | GGAGTAGCAAGTGTGTCAAGAACCTGAATTC | −740 to −711 | ? | ||
| SRE2 | TTATCCGAAACGCTTTACTCCTCTATTATT | −691 to −661 | ? | |||
| CDR2 | DRE | ACGGAAATCGGATATTTTTTT | −221 to −201 | Tac1 | ||
| C. glabrata | CgCDR1 | PDRE | TTCCGTGGAA | −1201 to −1192 | CgPdr1 | |
| CgCDR2 | PDRE | TTCCGTGGAA | −560 to −551 | CgPdr1 | ||
|
| ||||||
| MFS transporters | HRE/YRE | — | −561 to −520 | Cap1/? | ||
| BRE/ MDRE |
ACGGTAAAATCCTAATTGGGAAAAATACCGAGAATGA | −296 to −260 | Mcm1/Mrr1 | |||
| C. albicans | CaMDR1 | AR1 | — | −397 to –301 | ? | |
| AR2 | — | −588 to –500 | ? | |||
| AR3 | — | −287 to −209 | ? | |||
| C. glabrata | CgFLR1 | YRE3 | TTAGTAA | −372 to −366 | CgAp1 | |
|
| ||||||
| ERG11 | C. albicans | ERG11 | ARE | AATATCGTACCCGATTATGTCGTATATT | −224 to −251 | Upc2 |
| C. glabrata | ERG11 | SRE | Upc2A | |||
The identification of trans-acting factors regulating ABC-transporters in pathogenic fungi relied first on the well-described S. cerevisiae PDR network as a model [138–142]. Since the Zn2-Cys6 transcription factors PDR1/PDR3 are master regulators of this network in S. cerevisiae, an in silico search for PDR1/PDR3 homologues in fungal genomes was performed. Data so far available found only one functional homologue in C. glabrata [120]. CgPdr1p has 40% and 35% identity with Pdr1p and Pdr3p, respectively [143], and was able to complement a pdr1Δ S. cerevisiae mutant strain. Likewise, PDR1 deletion in C. glabrata leads to a loss of CgCDR1 and CgCDR2 regulation and to a sharp decrease in azole MICs. [144]. Three studies have identified separate gain-of-function mutations in CgPDR1 alleles of azole-resistant strains which are responsible for constitutive high expression of CgCDR1, CgCDR2, SNQ2, and CgPDR1 itself (Figure 9) [120, 145, 146].
Figure 9.
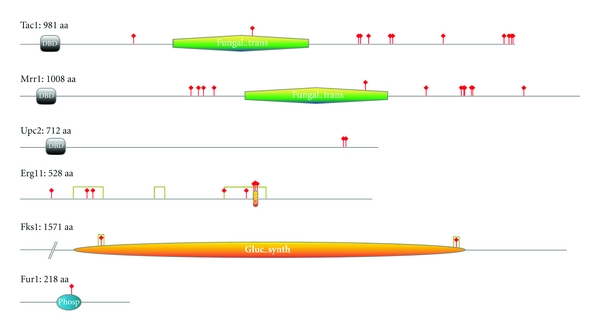
Point mutations affecting antifungal susceptibility in clinical isolates of C. albicans. Indicated functional domains were determined using either Prosite or Pfam tools. Only mutations which involvement in antifungal resistance was experimentally demonstrated are indicated by a red stick. Hot spot mutations in Erg11 and Fks1 are delimitated by gray boxes (Point mutation localization references: Tac1 [112], Mrr1 [133], Upc2 [134], Erg11 [135], Fks1 [136], Fur1 [137]). Drawings of the proteins were made with Prosite My Domain-Image Creator tool.
Attempts to identify C. albicans PDR1/3 functional homologues were undertaken to complement the absence of PDR1/PDR3 in S. cerevisiae by genetic screens. Several genes were identified including FCR1 and FCR3 (FluConazol Resistance) [147–149] and SHY1-3 (Suppressor of Hypersusceptibility) [150] (formerly, resp., named CTA4, ASG1 and ATF1). FCR1, CTA4, ASG1, and ATF1 encode Zn2-Cys6 transcription factors, while FCR3 encodes a bZip transcription factor. Even though FCR1 was able to restore PDR5 expression in a pdr1Δ/pdr3Δ S. cerevisiae mutant strain, its disruption in C. albicans resulted in decreased susceptibility to fluconazole, suggesting that FCR1 acts as a negative regulator of fluconazole susceptibility [147]. Nevertheless, the target genes of FCR1 in C. albicans are not yet known. Up to now, the relevance of FCR3 in azole resistance has not been addressed in C. albicans. CTA4, ASG1, and ATF1 expression in S. cerevisiae could restore PDR1/PDR3 functions in S. cerevisiae; however, their disruption in C. albicans did not affect azole susceptibility and expression of CDR1 and CDR2 [150]. An additional regulator of CDR1 was identified by a genetic screen in S. cerevisiae with a LacZ reporter system under the control of the CDR1 promoter. A C. albicans gene was subsequently identified that encodes for a protein CaNdt80p similar to the S. cerevisiae meiosis specific transcription factor Ndt80p. Disruption of CaNDT80 in C. albicans was shown to affect basal expression levels of CDR1 in C. albicans and reduce the ability of this gene to be upregulated in the presence of miconazole [151, 152]. More recently, Ndt80p was shown to have a global effect on azole-resistance through is regulon which includes many genes involved in ergosterol metabolism [153].
The release of the entire data from the C. albicans genome sequence has encouraged other approaches for identifying trans-regulators of CDR1 and CDR2. Since the DRE motifs present in the promoter of CDR genes contains two CGG triplets that are potentially recognized by Zn2-Cys6 transcription factors (TF) [154–157], it was likely that one of the 78 ORFs encoding proteins with Zn2-Cys6 signatures could be involved in the regulation of CDR1 and CDR2. Interestingly, genome data revealed that three of these ORF (the so-called “zinc cluster”) were located in tandem close to the mating type locus (MTL) at a distance of 14 kb [158]. Homozygosity at the MTL locus is associated with the development of azole resistance in C. albicans [159], thus indicating that one the genes of the zinc cluster could control CDR1 and CDR2 expression. As a matter of fact, deletion of one of these Zn2-Cys6 TF-encoding genes in an azole-susceptible strain led to increased drug susceptibility and loss of transient CDR1 and CDR2 upregulation in the presence of inducers. This gene was named TAC1 for transcriptional activator of CDR genes [158]. Consistent with the mutant phenotype, Tac1p can bind in vitro and in vivo to the DRE [112, 158]. However, TAC1 is not involved is the basal expression of CDR1 controlled at least by the BEE [158]. Hyperactive alleles that confer constitutive high CDR1 and CDR2 expression, and therefore drug resistance to a tac1Δ/Δ mutant strain of TAC1, were isolated from azole-resistant strains. Wild-type and hyperactive alleles differed by point mutations defined as gain-of-function mutations (GOF). Up to now, at least 15 GOF were described in TAC1 at 12 different positions [112, 158, 160, 161] (Figure 9). Wild-type and hyperactive alleles are co-dominant for the expression of their phenotypes [112, 158, 160, 161], and because of this property, high drug resistance levels correlate with homozygosity of hyperactive alleles. Interestingly, the TAC1 locus and the associated MTL are rendered homozygous in the development of azole resistance. Such events are accomplished by rearrangements on chromosome 5 including mitotic recombinations on one chromosome 5 arm or the loss of one chromosome 5 homologue followed by duplication [160]. Increase of resistance can still be obtained by isochromosome formation with the left arm of the chromosome 5. This allows for the increase of drug resistance genes present on this chromosome (among which TAC1 and ERG11) and thus can contribute to drug resistance increase [160–164]. Up to this date, regulation of Tac1p activity remains unknown.
3.1.2. Major Facilitator Superfamily (MFS) Transporters
In C. albicans, MDR1 (MultiDrug Resistance 1, previously named BENrfor Benomyl resistance) is a transporter currently shown to be the only MFS transporter involved in azole resistance of clinical isolates [165, 166]. MDR1 is not usually expressed at detectable levels in fluconazole-susceptible isolates, but is constitutively upregulated in some fluconazole-resistant isolates. As for CDR1 and CDR2, MDR1 can be specifically transiently upregulated by drugs such as benomyl, cycloheximide, methotrexate, and several oxidizing agents [165]. MFS transporters are known to be involved in azole resistance of other fungal species. Homologues of MDR1 in C. dubliniensis and C. tropicalis, named CdMDR1 and CtMDR1, respectively, are upregulated in azole-resistant strains [167–170]. In A. fumigatus, in vitro-generated itraconazole-resistant isolates show constitutive high expression level of the MFS transporter, AfuMDR3 [128]. The role of cis-acting regulatory elements in resistance was investigated in the C. albicans MDR1 gene by separate studies. Two of the studies undertaken by Rognon et al. and Riggle and Kumamoto identified a similar region, called BRE (benomyl response element) or MDRE (MDR1 drug resistance element) respectively. This region is responsible for the constitutive high expression of MDR1 in fluconazole-resistant isolates [171, 172] and was also shown to be responsible for the response to benomyl [172]. A second regulatory element involved in the response of MDR1 to oxidative stress is designated HRE (H2O2 response Element). This region contains two YRE (YAP1 response element) motifs [173], one perfectly conserved (-532 TTAGTAA-526) and the other with two mismatches (-549 TAACTAT-543). Interestingly, the HRE is not required for constitutive upregulation of MDR1 in azole-resistant isolates. A separate study undertaken by Hiller et al. described three distinct cis-activating regions (1, 2, and 3) in MDR1. Region 2, which overlaps with encompassing the HRE, was implicated in benomyl-dependent MDR1 response [174]. Region 1 and 3, close to the BRE/MDRE region, were required for a constitutive high expression of MDR1 in an azole-resistant isolate.
MDR1 expression was shown to be regulated by at least four trans-acting factors: Cap1p [175–177], Mrr1p [178, 179], Upc2p [180, 181], and Mcm1p [182]. Nucleotide sequence data of cis-acting elements has provided some clues to their identification. When comparing the MDR1 cis-acting elements with existing transcription factor binding site databases, several putative trans-acting elements were identified. As mentioned above, the HRE of MDR1 contains Yap1p binding sites. The bZip transcription factor Cap1 was shown to directly interact with the cis-acting domains [177] and to be involved in drug resistance [175]. The BRE/MDRE motif contains a perfect match for the Mads-box transcription factor Mcm1p in its sequence. Mogavero et al. showed that Mcm1p acts as a coregulator for Cap1 and Mrr1p and is not required for MDR1 upregulation by H2O2 but is required for full MDR1 induction by benomyl [182]. Genome-wide transcriptional analyses of clinical isolates that exhibit MDR1upregulation permitted the identification of a Zn2-Cys6 transcription factor that is coregulated with MDR1 [179]. Deletion of MRR1 in azole-resistant strains abolishes the constitutive overexpression of MDR1, therefore identifying Mrr1p as a central regulator of MDR1. Like for TAC1, two types of alleles were distinguished for MRR1. Wild-type alleles are needed for a transient upregulation of MDR1 by drug exposure. In contrast, hyperactive alleles confer constitutive overexpression of MDR1 and therefore also confer increased resistance to fluconazole [179]. Wild-type and hyperactive alleles differ by GOF mutations and until now, 14 GOF mutations at 13 positions were described in hyperactive Mrr1 [180] (Figure 9). Interestingly, hyperactive Mrr1 proteins were also shown to be able to confer Mdr1p-independent drug resistance probably through the regulation of oxydoreductases implicated in the detoxification of yeast cells after fluconazole exposure [183]. A blast analysis in C. dubliniensis allowed for the identification of a gene encoding for a protein that shares 91% of identity with Mrr1 of C. albicans [133] and able to complement a CaMrr1Δ mutant strain. The properties of CdMrr1 are similar to those of CaMrr1 and two types of alleles can also be distinguished. Until now 5 GOF mutations were identified and analyzed in hyperactive CdMrr1 proteins [133].
3.2. Target Alteration
3.2.1. Target Mutation
Another mechanism by which fungal pathogens are able to develop resistance is a decrease in antifungal affinity for their respective targets, without a major decrease in target activity. Such is the case for azole drugs, in which a decreased affinity between azole and a mutated lanosterol 14α-demethylase, can lead to resistance. A point mutation in the ERG11 gene that codes for lanosterol 14α-demethylase leads to the complete inhibition of the binding capacity of the azole drug to its target [184, 185]. Numerous of these point mutations identified in ERG11 were previously described, and their involvement in azole resistance was experimentally demonstrated for fungi such as Cryptococcus neoformans [186], C. albicans [187], (see also Figure 9), and C. tropicalis [167]. In Aspergillus fumigates, CYP51A and CYP51B encode two distinct forms of 14α-demethylase and mutations in the first of these two genes seem to be the most frequent mechanism responsible for azole resistance in clinical isolates. In this species, it was demonstrated that the nature of the nucleotide mutation, and therefore, the nature of the amino acid substitution, influences the development of resistance to different azole agents [188–192]. Interestingly, it was demonstrated that some clinical isolates share common mutations in Cyp51A with environmental azole-resistant strains, suggesting that some clinical azole resistant isolates might originate from the environment [193–195].
While target site alteration is far from being the most significant mechanism of resistance to azole drugs, it is the only known mechanism by which fungal pathogens are able to develop resistance to echinocandin drugs. This was demonstrated for S. cerevisiae and C. albicans. Echinocandin resistance is systematically associated with point mutations in either FKS1 or FKS2 [196, 197]. Analysis of the location of these mutations within the FKS genes led to the characterization of two regions, the so-called “hot spots”, integrity of which seems to be essential for enzyme activity [136]. In contrast to azoles and ERG11, mutations in FKS1 did not alter the β-glucane synthase affinity for its target but decreased only the enzyme processivity [198]. Hot-spot mutations have also been identified in other species, such as C. glabrata [196, 199], C. krusei [200], Scedosporium apiospermum [201], and A. fumigatus [202, 203] (Figure 9).
Numerous enzymes of the pyrimidine salvage pathway are involved in 5FC mode of action and thus numerous molecular mechanisms could lead to resistance to this drug [16, 204]. The most frequently found mechanism in clinical isolates of pathogenic fungi is a point mutation in the FUR1 gene that encodes the enzyme responsible for the conversion of 5FU into metabolites able to enter the cytosine metabolism (Figure 9) [14]. FUR1 mutation leads to complete resistance to both 5FC and 5FU in fungi. A second, frequently reported mutation leads only to resistance of 5FC. This second mutation is a point mutation in the FCY1 gene that codes for cytosine deaminase, the enzyme responsible for the conversion from 5FC into 5FU. Several such point mutations that lead to decreased activity of the cytosine deaminase were identified, essentially in Candida yeast species such as C. glabrata [205, 206] and in S. cerevisiae [207].
3.2.2. Target Expression Deregulation
A third mechanism of drug resistance is the deregulation of the drug target expression. For drugs targeting, the biosynthesis of ergosterol, such as azoles, terbinafine, or fenpropimorph, even relative short exposures of two to three hours lead to the transient upregulation of the ERG gene family in C. albicans, glabrata, tropicalis, and krusei [208]. These data suggest a common regulation of the ergosterol biosynthetic pathway in the presence of inhibitors. Longer azoles in vitro exposures (minimum 24 h) lead to constitutive upregulation of ERG genes, including ERG11 [209], and decrease drug susceptibility. In clinical isolates of C. albicans and C. dubliniensis resistant to azoles isolated from HIV patients, upregulation of ERG11 was described as a minor mechanism often combined with other more major mechanisms of resistance such as pump overexpression or ERG11 mutations [103, 169, 210]. The overexpression of ERG11 originates either by gene dosage effect through duplication of the gene or by upregulation of the gene by a trans-acting factors, both hypotheses were verified. In C. albicans and C. glabrata, it was shown that increased azole resistance due to ERG11 upregulation was in fact due to genome rearrangement via formation of an isochromosome in C. albicans and duplication of a chromosome in C. glabrata, and therefore amplification of ERG11 [163, 211]. In Cryptococcus neoformans, the well-known SRE1 gene was shown to regulate the ergosterol biosynthesis pathway and also to be involved with virulence of the fungus [212]. In S. cerevisiae, the ERG gene family was shown to be regulated by two zinc cluster transcription factors encoded by ScUPC2 and ScECM22. Two homologues of ScUPC2 were found in the genome of C. glabrata: CgUPC2A and CgUPC2B. It appears that while both transcription factors regulate sterol biosynthesis and exogenous uptake, only CgUpc2A is responsible for the regulation of the transcription of the ERG gene family in response to sterol inhibitors [213]. In C. albicans, only one gene homologue of ScUPC2/ScECM22 was found and named CaUPC2 [214, 215]. It was shown that CaUpc2 is necessary for the upregulation of ERG genes in the presence of ergosterol synthesis inhibitors. Moreover, the upc2 Δ/Δ mutant shows increased susceptibility to most drugs and a decrease in sterol uptake as compared to the wild-type strain [214, 215]. Further studies demonstrated the ability of CaUpc2 to bind to the ARE motif in the promoter of C. albicans ERG11 (Table 3) [215, 216]. Genome-wide location analysis of CaUpc2 confirmed the SRE motif as the DNA binding site, and also confirmed the ERG gene family as a CaUpc2 target as well as CDR1, MDR1, and UPC2 itself as new target genes. Analysis of clinical strains resistant to fluconazole with upregulated ERG11 expression, demonstrated the existence of a hyperactive allele of CaUPC2 that confers intrinsic upregulation of ERG genes. Currently, two GOF mutations were described for CaUpc2 (Figure 9) [134, 180].
3.3. Metabolism Modification
3.3.1. Echinocandins Paradoxical Effect
Some yeasts and filamentous fungi are able to grow in elevated echinocandin concentrations much higher than the MICs [136, 217]. This phenomenon, called paradoxical effect, is due to the metabolic adaptation of microorganism and is mediated by the cell wall integrity signalization pathway. This response is the direct consequence of the β(1-3)-glucans synthesis inhibition and the subsequent cell wall composition modifications, upon echinocandin administration [218, 219]. Several studies suggest that the magnitude of the paradoxical effect is variable depending on the microorganism itself as well as on the echinocandin nature. Therefore, the paradoxical effect would be more pronounced in the presence of caspofungin as compared to anidulafungin or micafungin [220]. However, the clinical significance of paradoxical effect has never been studied nor has it ever been observed in echinocandin-treated patients [86].
3.3.2. De Novo Synthesis of Pyrimidines
It is possible that 5FC resistance could be the consequence of an overall induction of the de novo pyrimidine biosynthetic pathway. In this case, the antifungal drug competes with the regular pyrimidine intermediate metabolites for incorporation into nucleic acids [16]. This increase in activity of the de novo pyrimidine biosynthetic pathway is reflected by an increased expression of the CDC21 gene, whose product is inhibited by 5FC [205]. FUR1 mutations could lead to 5FC resistance. However, its downregulation has also been demonstrated to be involved in 5FC decreased susceptibility. A 4-fold decreased expression of this gene of high importance in 5FC mode of action is sufficient to lead to a total resistance to this pyrimidine fluorinated analog in C. glabrata [84].
3.3.3. Ergosterol Biosynthesis Pathway Alteration
Modifications of main metabolic pathways could also lead to azole drugs resistance. For example, alteration of the late steps of the ergosterol biosynthetic pathway through inactivation of the ERG3 gene gives rise to cross-resistance to all azole drugs [101]. Indeed, the antifungal activity of azole drugs relies on the synthesis of toxic 14α methylated sterols by the late enzymes of this pathway. A point mutation that occurs in the ERG3 gene can lead to the total inactivation of C5 sterol desaturase. In this case, toxic 14α methylated sterols are no longer synthesized and even in the presence of azole drugs sterols species able to replace ergosterol are generated. While very uncommon, this mechanism was identified in several clinical isolates of C. albicans [221–223].
3.3.4. Plasma Membrane Composition Variation
Polyene drugs do not require internalization into fungal cells in order to exert their antifungal activity, since they incorporate into the plasma membrane from the external side. Thus, they escape metabolizing enzymes and efflux systems, and the only possibility for fungi to develop resistance to polyene is to modify their target, ergosterol. However, ergosterol is responsible for the integrity and fluidity of the plasma membrane, and therefore, possibilities to compensate for its absence are very limited. Although rarely described, resistance mechanisms responsible for acquired or innate resistance to polyene drugs were studied in several fungal species. In each case, resistance to polyenes results from a decrease or total absence of ergosterol in the plasma membrane through mutations in nonessential genes of the ergosterol biosynthetic pathway [224]. Molecular polyene resistance mechanisms were described in laboratory mutants of yeasts belonging to the Candida genus and in S. cerevisiae. Thus, both ERG11 deletion in C. albicans [225] and ERG3 deletion in S. cerevisiae [226] lead to mutants with cross-resistance to azole and polyene drugs. Likewise, ERG6 inactivation in C. lusitaniae [227] and S. cerevisiae results in polyene resistance [228]. Regarding clinical isolates, very few data is available. Only a few studies have associated polyene resistance to an ERG3 mutation in clinical isolates of C. albicans [221, 229] and to an ERG6 mutation in C. glabrata [230, 231].
3.3.5. Biofilms
“United we stand, divided we fall”. This statement is certainly true concerning the fight between fungi and antifungals. It is well characterized that microbial communities engulfed in a polysaccharides-rich extracellular matrix, also known as biofilm, are by far more resistant to antifungal drugs than isolated cells. Fortunately, few pathogenic species within the fungal kingdom are able to form biofilms. The mostly known and widely studied of those species able to form biofilms are the species of the Candida genus [232]. Another yeast frequently responsible for biofilm-associated infections is Cryptococcus neoformans [233, 234]. Some clinical cases have also reported the involvement of other yeast species, such as Pichia fabianii [235] or Trichosporon asahii [236]. Additionally, it is now accepted that filamentous fungi, and particularly those of the Aspergillus genus, can grow as biofilms in humans [237–239]. Fungal biofilms are frequently polymicrobial biofilms, meaning that bacterial species frequently associate with one or several fungi [240, 241]. In medical mycology, biofilms constitute a real concern in the fields of invasive and dental medicine. They constitute a nonnegligible source of nosocomial fungal infections, essentially through the use medical devices. Moreover fungal biofilms are resistant to almost all the currently used antifungals, with the exceptions of echinocandins and lipid formulations of AmB [242]. The molecular mechanisms underlying the persistence of the fungal biofilms despite antifungal treatment remain unclear. It is likely that biofilm resistance is the result of a combination a multiple factors, among them an increased expression of efflux pumps, a modification of plasma membrane composition, and the biofilm-produced extracellular matrix itself [232, 243].
4. Development of New Antifungal Strategies
Current antifungal treatments are limited in their capacity to treat infections, especially systemic infections and no considerable advancements in antifungal therapies were developed recently. New therapies are therefore needed against pathogenic fungi. Several approaches were developed during the last several years in order to find new solutions. Researchers aim to discover new antifungal drugs either by testing already existing medical compounds, compounds from natural sources such as plants, sea, microorganisms or by systematic screens of chemical compound libraries. Researchers also strive to elucidate the underlying biology of fungal microorganism both in vitro and in vivo. Host-fungal interactions play a critical role for all fungal pathogens. Targeting this interaction provides novel therapies, which could be used alone or in combination with existing antifungal drugs. Such a combination may also determine the development of antifungal drug resistance.
4.1. Development of New Antifungal Active Compounds
Much effort has gone towards analyzing the antifungal properties of what is called natural compounds (NP) or natural bioactive compounds isolated from plants, other microorganisms, or marine organisms [244–246]. Some such compounds are investigated because their known triggering mechanisms important for fungi, while other compounds are tested blindly for their antifungal properties. Currently, none of these studies have produced a compound suitable for the clinical trial stage although interesting results were obtained.
Other studies focused on in vitro screens of several drugs currently used in clinical practice for their potentiation of the antifungal effect of the fungistatic agent fluconazole (FLC) on Candida albicans. This facilitated the discovery of several compounds, such as inhibitors of the calcineurin [247, 248] or Tor pathways [249–251], efflux pump inhibitors (derived compounds of milbemycin) [252–254], and more recently, antibodies against heat-shock 90 protein (HSP90) [255]. In particular, inhibitors of the calcineurin pathway were shown to be fully active in vivo in the potentiation of fluconazole, and they also led to a dramatic decrease in fungi virulence [256–260].
Systematic screening of chemical compounds libraries was also undertaken, essentially by industrial laboratories as an attempt to discover new antifungal compounds. High throughput screening of the legacy Schering-Plough compound collection has recently lead to the discovery of a new glucan synthase inhibitor effective again C. albicans and C. glabrata [261–263].
Some analysis used reverse genetic assay in which, C. albicans heterozygous deletion or transposon disruption mutants collection were screened for growth under treatment with collections of chemical compounds [264, 265]. This approach allowed identification of both antifungal drugs and the genes related to the mechanism of action of the related compounds.
Another type of high-throughput screens of chemical libraries was achieved measuring the viability of drug-treated Caenorhabditis elegans infected with C. albicans [266]. Compounds can be simultaneously screened for antifungal efficacy and host toxicity, which overcomes one of the main obstacles in current antimicrobial discovery. A pilot screen for antifungal compounds using this novel C. elegans system identified 15 compounds that prolonged survival of nematodes infected with the medically important human pathogen C. albicans. One of these compounds, caffeic acid phenethyl ester (CAPE), had effective antifungal activity in a murine model of systemic candidiasis and had in vitro activity against several other fungal species [266]. In addition, this whole-animal system may enable the identification of compounds that modulate immune responses and/or affect fungal virulence factors that are only expressed during infection.
4.2. Genome-Wide Studies to Detect Potential New Antifungal Targets
The improvement of already existing antifungal drugs and the limitation of drugs resistance apparition has helped to elucidate the basic biology of the fungal pathogen. For this purpose, several groups made efforts to develop collection of systematic mutants essentially for C. albicans. An important difficulty for antifungal therapy is to develop drugs that exploit factors unique to fungi, which can be challenging considering that organism are eukaryotic and share many conserved biological pathways. Genes that are essential to fungal survival are possible targets for drug development.
Using the GRACE (gene replacement and conditional expression) or CPR (conditional promoter replacement) technologies, some research groups have assessed the essentiality of C. albicans and Aspergillus fumigatus genes [267, 268]. One study identified 567 essential genes in C. albicans [267]. And another study screened 54 genes of A. fumigatus based on ortholog functions and essentiality in C. albicans and S. cerevisiae [268], of which 35 were defined as essential in A. fumigatus. Authors were able to show that while the ERG11 gene family (CYP51A and ERG11B) is essential in A. fumigatus, the individual genes themselves are not. These analyses provide interesting and fully informative data for antifungal drug design and improve upon previous in silico analyses that when using S. cerevisiae data were only able to identify 61% of homologous genes reported in the genes found in the Roemer et al. analysis [267].
The diploid state of the genome presents a major problem to the development of a mutant collection. Therefore, some collections consist of heterozygous deletion [265] or transposon disruption mutants [264, 265]. Other collections contain homozygous transposon disruption mutants based on the random insertion thanks to the Tn7 transposon to a UAU cassette [269, 270]. These collection were first restricted to the transcription factors of C. albicans [269, 271, 272] but continue to be enlarged for the entire genome [270, 273]. Other collections consist of deletion mutants constructed with PCR generated deletion cassettes, with two different markers for each allele in the case of C. albicans [274, 275]. Such collections are now being constructed for C. glabrata.
Three kinds of analyses detailed below were performed with these collections. They aimed a better understanding of the modifications occurring in the fungi submitted to antifungal treatments or of the relationship developed between the fungus and its host all along the infectious process. Such knowledge might improve the actual therapy to avoid resistance development or might allow playing on the host-fungus equilibrium to improve recovery of patients.
First of all, treating strains with already known antifungal drugs and analyzing for example, growth modification and later transcriptional rewiring, some authors try to better understand drugs mechanisms of action and/or to find synergistic effect between them [272, 274]. Gene encoding the transcription factor Cas5 was found to be involved in the response to caspofungin [272]. Other studies showed that AGE3, which encodes an ADP-ribosylation factor GTPase activating effector protein, if deleted, abrogates fluconazole tolerance in C. albicans. Interestingly, Brefeldin A, an inhibitor of ADP-ribosylation factor, resulted in a synergistic effect with other drugs for C. albicans as well as for Aspergillus [273]. Finally, Homann et al. screened a collection of 143 transcription factor mutants under 55 distinct conditions among which exposition to fluconazole and 5FC, and they conclude in their analysis that nearly a quarter of the knockout strains affected sensitivity to commonly used antifungal drugs [274].
Other studies were geared better understand the biology of fungal species. For this purpose, mutant collections were subjected to a wide range of environmental conditions, modifying elements such as pH, salt concentration, carbon sources, oxidative conditions, temperature, and availability of essential elements such as metals (iron, copper, zinc, etc.) [274, 276].
Understanding the relationship between fungus and host during infection may provide further information useful for the improvement of antifungal treatment. In order to analyze the cross-talk occurring between fungus and host during the infectious process, researchers screened the colonization properties of mutants directly in hosts. One study that was performed with 1201 gene knockout mutants of Cryptococcus neoformans analyzed their in vivo proliferation profile in the murine lung, and they were able to identify 40 infectivity mutants [277]. Gene deletions in these mutants were previously uncharacterized and did not show any defect in traits known to be linked to virulence (polysaccharide capsule formation, melanization, and growth at body temperature). At least, four other similar studies were performed with C. albicans mutants. Two of them were done in invertebrate host models such as C. elegans or D. melanogaster [266, 278]. Interestingly, the Cas5 Δ/Δ mutant, which has already been shown to be important for caspofungin response, was shown to be less virulent in both invertebrate models of infection [278, 279]. Finally, this transcription factor was demonstrated as crucial for cell wall integrity, and its importance in virulence was confirmed in the mice intravenous model of infection [278]. Two other studies screened collections of C. albicans mutants directly in mice by pools of mutants that were previously tagged [280, 281]. One collection was restricted to Zn2-Cys6 transcription factors (TF) mutants and the other was composed of mutants affecting about 11% of the entire C. albicans genome with no respect to a gene class. In both cases, mutants were also screened for traits known to be linked to virulence, such as the ability to filament and proliferate as well as the ability to grow at 42°C, at high and low pH, and in oxidative conditions. Noble et al. identified 115 mutants among the 674 screened with attenuated infectivity, but normal morphological switching and proliferation [281]. More precisely, they identified glycolipid and glucosylceramide as the first small molecules synthesized by C. albicans that are specifically required for virulence. Vandeputte et al. identified two Zn2-Cys6 TF mutants within 77 tested. These mutants displayed attenuated infectivity in their pool test, which was also confirmed in independent single strains infection of mice ZCF13 and ZCF18 [280]. Both genes were previously uncharacterized. ZCF18 showed a slight growth defect in contrast to ZCF13 which grew normally at body temperature, but slightly less at 42°C. ZCF13 mutant displayed an abnormal morphology, producing strongly filamentous colonies on YPD medium at 35°C and displaying high invasion ability. ZCF18 deletion also led to a slight enhancement of colony wrinkling. Both genes are currently under further analysis.
Unfortunately, whenever promising, up to now, no new compound and/or new target have been selected for further development from these approaches.
5. Conclusion
These last years were very rich in better knowledge of molecular basis of antifungal resistance and more generally of the metabolism of pathogenic fungi. Antifungal drug resistance appears to essentially be due to point mutations in either drug targets or transcription factors regulating actors of the resistance. In the near future, high throughput diagnostic tools could be used in the course of treatment of fungal infections in order to detect resistance and adjust therapeutic strategies accordingly before any clinical evidence and therefore allow a rapid adjustment of the antifungal treatment.
One of the challenges of finding new antifungal targets in C. albicans was the lack of sophisticated screening technologies often employed with other fungal species such as Saccharomyces cerevisiae. The recent application of genome-wide studies to pathogenic fungi for both host-pathogen interactions and the biological study will hopefully encourage and facilitate the development of new effective therapeutic strategies. Such improvements in antifungal treatment may lead to a better clinical outcome.
Acknowledgment
The authors would like to thank Shawna McCallin for proofreading of the paper.
References
- 1.Morishita N, Sei Y. Microreview of Pityriasis versicolor and Malassezia species. Mycopathologia. 2006;162(6):373–376. doi: 10.1007/s11046-006-0081-2. [DOI] [PubMed] [Google Scholar]
- 2.Thomas J, Jacobson GA, Narkowicz CK, Peterson GM, Burnet H, Sharpe C. Toenail onychomycosis: an important global disease burden. Journal of Clinical Pharmacy and Therapeutics. 2010;35(5):497–519. doi: 10.1111/j.1365-2710.2009.01107.x. [DOI] [PubMed] [Google Scholar]
- 3.Diamond RD. The growing problem of mycoses in patients infected with the human immunodeficiency virus. Reviews of Infectious Diseases. 1991;13(3):480–486. doi: 10.1093/clinids/13.3.480. [DOI] [PubMed] [Google Scholar]
- 4.Arendrup MC. Epidemiology of invasive candidiasis. Current Opinion in Critical Care. 2010;16(5):445–452. doi: 10.1097/MCC.0b013e32833e84d2. [DOI] [PubMed] [Google Scholar]
- 5.Walsh TJ, Dixon DM. Spectrum of mycoses. In: Baron S, editor. Medical Microbiology. Galveston, Tex, USA: 1996. [PubMed] [Google Scholar]
- 6.Eucker J, Sezer O, Graf B, Possinger K. Mucormycoses. Mycoses. 2001;44(7-8):253–260. [PubMed] [Google Scholar]
- 7.Duschinsky R, Pleven E, Oberhansli W. Synthesis of 5-fluoropyrimidine metabolites. Acta—Unio Internationalis Contra Cancrum. 1960;16:599–604. [PubMed] [Google Scholar]
- 8.Grunberg E, Titsworth E, Bennett M. Chemotherapeutic activity of 5-fluorocytosine. Antimicrobial Agents and Chemotherapy. 1963;161:566–568. [PubMed] [Google Scholar]
- 9.Tassel D, Madoff MA. Treatment of Candida sepsis and Cryptococcus meningitis with 5-fluorocytosine. A new antifungal agent. Journal of the American Medical Association. 1968;206(4):830–832. [PubMed] [Google Scholar]
- 10.Stiller RL, Bennett JE, Scholer HJ. Correlation of in vitro susceptibility test results with in vivo response: flucytosine therapy in a systemic candidiasis model. The Journal of Infectious Diseases. 1983;147(6):1070–1077. doi: 10.1093/infdis/147.6.1070. [DOI] [PubMed] [Google Scholar]
- 11.Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. The Journal of Antimicrobial Chemotherapy. 2000;46(2):171–179. doi: 10.1093/jac/46.2.171. [DOI] [PubMed] [Google Scholar]
- 12.Daneshmend TK, Warnock DW. Clinical pharmacokinetics of systemic antifungal drugs. Clinical Pharmacokinetics. 1983;8(1):17–42. doi: 10.2165/00003088-198308010-00002. [DOI] [PubMed] [Google Scholar]
- 13.Stamm AM, Diasio RB, Dismukes WE, et al. Toxicity of amphotericin B plus flucytosine in 194 patients with cryptococcal meningitis. American Journal of Medicine. 1987;83(2):236–242. doi: 10.1016/0002-9343(87)90691-7. [DOI] [PubMed] [Google Scholar]
- 14.Francis P, Walsh TJ. Evolving role of flucytosine in immunocompromised patients: new insights into safety, pharmacokinetics, and antifungal therapy. Clinical Infectious Diseases. 1992;15(6):1003–1018. doi: 10.1093/clind/15.6.1003. [DOI] [PubMed] [Google Scholar]
- 15.Kauffman CA, Frame PT. Bone marrow toxicity associated with 5 fluorocytosine therapy. Antimicrobial Agents and Chemotherapy. 1977;11(2):244–247. doi: 10.1128/aac.11.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polak A, Scholer HJ. Mode of action of 5 fluorocytosine and mechanisms of resistance. Chemotherapy. 1975;21(3-4):113–130. doi: 10.1159/000221854. [DOI] [PubMed] [Google Scholar]
- 17.Diasio R, Lakings D, Bennett J. Evidence for conversion of 5-fluorocytosine to 5-fluorouracil in humans: possible factor in 5-fluorocytosine clinical toxicity. Antimicrobial Agents and Chemotherapy. 1978;14(6):903–908. doi: 10.1128/aac.14.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams KM, Duffield AM, Christopher RK, Finlayson PJ. Identification of minor metabolites of 5-fluorocytosine in man by chemical ionization gas chromatography mass spectrometry. Biomedical Mass Spectrometry. 1981;8(4):179–182. doi: 10.1002/bms.1200080410. [DOI] [PubMed] [Google Scholar]
- 19.Harris BE, Manning BW, Federle TW, Diasio RB. Conversion of 5-fluorocytosine to 5-fluorouracil by human intestinal microflora. Antimicrobial Agents and Chemotherapy. 1986;29(1):44–48. doi: 10.1128/aac.29.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benson JM, Nahata MC. Clinical use of systemic antifungal agents. Clinical Pharmacy. 1988;7(6):424–438. [PubMed] [Google Scholar]
- 21.Bennett JE, Dismukes WE, Duma RJ. A comparison of amphotericin B alone and combined with flucytosine in the treatment of cryptococcal meningitis. New England Journal of Medicine. 1979;301(3):126–131. doi: 10.1056/NEJM197907193010303. [DOI] [PubMed] [Google Scholar]
- 22.Patel R. Antifungal agents. Part I. Amphotericin B preparations and flucytosine. Mayo Clinic Proceedings. 1998;73(12):1205–1225. doi: 10.4065/73.12.1205. [DOI] [PubMed] [Google Scholar]
- 23.Hospenthal DR, Bennett JE. Flucytosine monotherapy for cryptococcosis. Clinical Infectious Diseases. 1998;27(2):260–264. doi: 10.1086/514669. [DOI] [PubMed] [Google Scholar]
- 24.Polak A, Grenson M. Evidence for a common transport system for cytosine, adenine and hypoxanthine in Saccharomyces cerevisiae and Candida albicans . European Journal of Biochemistry. 1973;32(2):276–282. doi: 10.1111/j.1432-1033.1973.tb02608.x. [DOI] [PubMed] [Google Scholar]
- 25.Waldorf AR, Polak A. Mechanisms of action of 5-fluorocytosine. Antimicrobial Agents and Chemotherapy. 1983;23(1):79–85. doi: 10.1128/aac.23.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett JE. Antifungal agents. In: Hardman JG, et al., editors. Goodman and Gilman's The Pharmacological Basis of Therapeurics. New York, NY, USA: McGraw-Hill; 1996. pp. 1175–1190. [Google Scholar]
- 27.Caffrey P, Lynch S, Flood E, Finnan S, Oliynyk M. Amphotericin biosynthesis in Streptomyces nodosus: deductions from analysis of polyketide synthase and late genes. Chemistry and Biology. 2001;8(7):713–723. doi: 10.1016/s1074-5521(01)00046-1. [DOI] [PubMed] [Google Scholar]
- 28.Lemke A, Kiderlen AF, Kayser O. Amphotericin B. Applied Microbiology and Biotechnology. 2005;68(2):151–162. doi: 10.1007/s00253-005-1955-9. [DOI] [PubMed] [Google Scholar]
- 29.Baginski M, Resat H, McCammon JA. Molecular properties of amphotericin B membrane channel: a molecular dynamics simulation. Molecular Pharmacology. 1997;52(4):560–570. doi: 10.1124/mol.52.4.560. [DOI] [PubMed] [Google Scholar]
- 30.Milhaud J, Ponsinet V, Takashi M, Michels B. Interactions of the drug amphotericin B with phospholipid membranes containing or not ergosterol: new insight into the role of ergosterol. Biochimica et Biophysica Acta. 2002;1558(2):95–108. doi: 10.1016/s0005-2736(01)00416-3. [DOI] [PubMed] [Google Scholar]
- 31.Paquet MJ, Fournier I, Barwicz J, Tancrède P, Auger M. The effects of amphotericin B on pure and ergosterol- or cholesterol-containing dipalmitoylphosphatidylcholine bilayers as viewed by 2H NMR. Chemistry and Physics of Lipids. 2002;119(1-2):1–11. doi: 10.1016/s0009-3084(02)00071-3. [DOI] [PubMed] [Google Scholar]
- 32.Ghannoum MA, Rice LB. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clinical Microbiology Reviews. 1999;12(4):501–517. doi: 10.1128/cmr.12.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akins RA. An update on antifungal targets and mechanisms of resistance in Candida albicans . Medical Mycology. 2005;43(4):285–318. doi: 10.1080/13693780500138971. [DOI] [PubMed] [Google Scholar]
- 34.Warn PA, Sharp A, Guinea J, Denning DW. Effect of hypoxic conditions on in vitro susceptibility testing of amphotericin B, itraconazole and micafungin against Asperigillus and Candida . The Journal of Antimicrobial Chemotherapy. 2004;53(5):743–749. doi: 10.1093/jac/dkh153. [DOI] [PubMed] [Google Scholar]
- 35.Zotchev SB. Polyne macrolide antibiotics and their applications in human therapy. Current Medicinal Chemistry. 2003;10(3):211–223. doi: 10.2174/0929867033368448. [DOI] [PubMed] [Google Scholar]
- 36.Cevher E, Sensoy D, Taha MAM, Araman A. Effect of thiolated polymers to textural and mucoadhesive properties of vaginal gel Formulations prepared with polycarbophil and chitosan. AAPS PharmSciTech. 2008;9(3):953–965. doi: 10.1208/s12249-008-9132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett JP, Vardulaki KA, Conlon C, et al. A systematic review of the antifungal effectiveness and tolerability of amphotericin B formulations. Clinical Therapeutics. 2003;25(5):1295–1320. doi: 10.1016/s0149-2918(03)80125-x. [DOI] [PubMed] [Google Scholar]
- 38.Ellis D. Amphotericin B: spectrum and resistance. The Journal of Antimicrobial Chemotherapy. 2002;49(supplement 1):7–10. doi: 10.1093/jac/49.suppl_1.7. [DOI] [PubMed] [Google Scholar]
- 39.Maertens JA. History of the development of azole derivatives. Clinical Microbiology and Infection. 2004;10(supplement 1):1–10. doi: 10.1111/j.1470-9465.2004.00841.x. [DOI] [PubMed] [Google Scholar]
- 40.Carrillo-Muñoz AJ, Giusiano G, Ezkurra PA, Quindós G. Antifungal agents: mode of action in yeast cells. Revista Espanola de Quimioterapia. 2006;19(2):130–139. [PubMed] [Google Scholar]
- 41.Woolley DW. Some new aspects of the relationship of chemical structure to biological activity. Science. 1944;100(2609):579–583. doi: 10.1126/science.100.2609.579. [DOI] [PubMed] [Google Scholar]
- 42.Fromtling RA. Overview of medically important antifungal azole derivatives. Clinical Microbiology Reviews. 1988;1(2):187–217. doi: 10.1128/cmr.1.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgess MA, Bodey GP. Clotrimazole (Bay b 5097): in vitro and clinical pharmacological studies. Antimicrobial Agents and Chemotherapy. 1972;2(6):423–426. doi: 10.1128/aac.2.6.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettenborn D. Toxicity of clotrimazole. Postgraduate Medical Journal. 1974;50(supplement 1):17–20. [PubMed] [Google Scholar]
- 45.Heel RC, Brogden RN, Pakes GE. Miconazole: a preliminary review of its therapeutic efficacy in systemic fungal infections. Drugs. 1980;19(1):7–30. doi: 10.2165/00003495-198019010-00002. [DOI] [PubMed] [Google Scholar]
- 46.Heeres J, Backx LJJ, Mostmans JH, Van Cutsem J. Antimycotic imidazoles. Part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. Journal of Medicinal Chemistry. 1979;22(8):1003–1005. doi: 10.1021/jm00194a023. [DOI] [PubMed] [Google Scholar]
- 47.Van Der Meer JW, Keuning JJ, Scheijgrond HW. The influence of gastric acidity on the bio-availability of ketoconazole. The Journal of Antimicrobial Chemotherapy. 1980;6(4):552–554. doi: 10.1093/jac/6.4.552. [DOI] [PubMed] [Google Scholar]
- 48.Brass C, Galgiani JN, Blaschke TF. Disposition of ketoconazole, an oral antifungal, in humans. Antimicrobial Agents and Chemotherapy. 1982;21(1):151–158. doi: 10.1128/aac.21.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perfect JR, Durack DT, Hamilton JD, Gallis HA. Failure of ketoconazole in cryptococcal meningitis. Journal of the American Medical Association. 1982;247(24):3349–3351. [PubMed] [Google Scholar]
- 50.Pont A, Williams PL, Loose DS. Ketoconazole blocks adrenal steroid synthesis. Annals of Internal Medicine. 1982;97(3):370–372. doi: 10.7326/0003-4819-97-3-370. [DOI] [PubMed] [Google Scholar]
- 51.Lewis JH, Zimmerman HJ, Benson GD, Ishak KG. Hepatic injury associated with ketoconazole therapy. Analysis of 33 cases. Gastroenterology. 1984;86(3):503–513. [PubMed] [Google Scholar]
- 52.Dismukes WE, Cloud G, Bowles C. Treatment of blastomycosis and histoplasmosis with ketoconazole: results of a prospective randomized clinical trial. Annals of Internal Medicine. 1985;103(6, part 1):861–872. [PubMed] [Google Scholar]
- 53.Arndt CA, Walsh TJ, McCully CL, Balis FM, Pizzo PA, Poplack DG. Fluconazole penetration into cerebrospinal fluid: implications for treating fungal infections of the central nervous system. The Journal of Infectious Diseases. 1988;157(1):178–180. doi: 10.1093/infdis/157.1.178. [DOI] [PubMed] [Google Scholar]
- 54.Brammer KW, Farrow PR, Faulkner JK. Pharmacokinetics and tissue penetration of fluconazole in humans. Reviews of Infectious Diseases. 1990;12(supplement 3):S318–S326. doi: 10.1093/clinids/12.supplement_3.s318. [DOI] [PubMed] [Google Scholar]
- 55.Espinel-Ingroff A, Shadomy S, Gebhart RJ. In vitro studies with R 51,211 (itraconazole) Antimicrobial Agents and Chemotherapy. 1984;26(1):5–9. doi: 10.1128/aac.26.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terrell CL. Antifimgal agents. part II. the azoles. Mayo Clinic Proceedings. 1999;74(1):78–100. doi: 10.4065/74.1.78. [DOI] [PubMed] [Google Scholar]
- 57.Barone JA, Moskovitz BL, Guarnieri J, et al. Enhanced bioavailability of itraconazole in hydroxypropyl-β- cyclodextrin solution versus capsules in healthy volunteers. Antimicrobial Agents and Chemotherapy. 1998;42(7):1862–1865. doi: 10.1128/aac.42.7.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boogaerts MA, Maertens J, Van der Geest R, et al. Pharmacokinetics and safety of a 7-day administration of intravenous itraconazole followed by a 14-day administration of itraconazole oral solution in patients with hematologic malignancy. Antimicrobial Agents and Chemotherapy. 2001;45(3):981–985. doi: 10.1128/AAC.45.3.981-985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Albengres E, Le Louët H, Tillement J-P. Systemic antifungal agents. Drug interactions of clinical significance. Drug Safety. 1998;18(2):83–97. doi: 10.2165/00002018-199818020-00001. [DOI] [PubMed] [Google Scholar]
- 60.Denning DW, Venkateswarlu K, Oakley KL, et al. Itraconazole resistance in Aspergillus fumigatus . Antimicrobial Agents and Chemotherapy. 1997;41(6):1364–1368. doi: 10.1128/aac.41.6.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sabo JA, Abdel-Rahman SM. Voriconazole: a new triazole antifungal. Annals of Pharmacotherapy. 2000;34(9):1032–1043. doi: 10.1345/aph.19237. [DOI] [PubMed] [Google Scholar]
- 62.Chiou C, Groll A, Walsh T. New drugs and novel targets for treatment of invasive fungal infections in patients with cancer. Oncologist. 2000;5(2):120–135. doi: 10.1634/theoncologist.5-2-120. [DOI] [PubMed] [Google Scholar]
- 63.Potoski BA, Brown J. The safety of voriconazole. Clinical Infectious Diseases. 2002;35(10):1273–1275. doi: 10.1086/343746. [DOI] [PubMed] [Google Scholar]
- 64.Denning DW. Echinocandins: a new class of antifungal. The Journal of Antimicrobial Chemotherapy. 2002;49(6):889–891. doi: 10.1093/jac/dkf045. [DOI] [PubMed] [Google Scholar]
- 65.Kurtz M, Douglas C. Lipopeptide inhibitors of fungal glucan synthase. Journal of Medical and Veterinary Mycology. 1997;35(2):79–86. doi: 10.1080/02681219780000961. [DOI] [PubMed] [Google Scholar]
- 66.Marco F, Pfaller MA, Messer SA, Jones RN. Antifungal activity of a new triazole, voriconazole (UK-109,496), compared with three other antifungal agents tested against clinical isolates of filamentous fungi. Medical Mycology. 1998;36(6):433–436. [PubMed] [Google Scholar]
- 67.Mazur P, Morin N, Baginsky W, et al. Differential expression and function of two homologous subunits of yeast 1,3-β-D-glucan synthase. Molecular and Cellular Biology. 1995;15(10):5671–5681. doi: 10.1128/mcb.15.10.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Georgopapadakou NH, Tkacz JS. The fungal cell wall as a drug target. Trends in Microbiology. 1995;3(3):98–104. doi: 10.1016/s0966-842x(00)88890-3. [DOI] [PubMed] [Google Scholar]
- 69.Stone EA, Fung HB, Kirschenbaum HL. Caspofungin: an echinocandin antifungal agent. Clinical Therapeutics. 2002;24(3):351–377. doi: 10.1016/s0149-2918(02)85039-1. [DOI] [PubMed] [Google Scholar]
- 70.Franzot SP, Casadevall A. Pneumocandin L-743,872 enhances the activities of amphotericin B and fluconazole against Cryptococcus neoformans in vitro. Antimicrobial Agents and Chemotherapy. 1997;41(2):331–336. doi: 10.1128/aac.41.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pfaller MA, Messer SA, Gee S, et al. In vitro susceptibilities of Candida dubliniensis isolates tested against the new triazole and echinocandin antifungal agents. Journal of Clinical Microbiology. 1999;37(3):870–872. doi: 10.1128/jcm.37.3.870-872.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Espinel-Ingroff A. Comparison of in vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. Journal of Clinical Microbiology. 1998;36(10):2950–2956. doi: 10.1128/jcm.36.10.2950-2956.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oakley KL, Moore CB, Denning DW. In vitro activity of the echinocandin antifungal agent LY303,366 in comparison with itraconazole and amphotericin B against Aspergillus spp. Antimicrobial Agents and Chemotherapy. 1998;42(10):2726–2730. doi: 10.1128/aac.42.10.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pound MW, Townsend ML, Drew RH. Echinocandin pharmacodynamics: review and clinical implications. The Journal of Antimicrobial Chemotherapy. 2010;65(6):1108–1118. doi: 10.1093/jac/dkq081. Article ID dkq081. [DOI] [PubMed] [Google Scholar]
- 75.Feldmesser M, Kress Y, Mednick A, Casadevall A. The effect of the echinocandin analogue caspofungin on cell wall glucan synthesis by Cryptococcus neoformans . The Journal of Infectious Diseases. 2000;182(6):1791–1795. doi: 10.1086/317614. [DOI] [PubMed] [Google Scholar]
- 76.Cappelletty D, Eiselstein-McKitrick K. The echinocandins. Pharmacotherapy. 2007;27(3):369–388. doi: 10.1592/phco.27.3.369. [DOI] [PubMed] [Google Scholar]
- 77.Hector RF. Compounds active against cell walls of medically important fungi. Clinical Microbiology Reviews. 1993;6(1):1–21. doi: 10.1128/cmr.6.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Niewerth M, Kunze D, Seibold M, Schaller M, Korting HC, Hube B. Ciclopirox olamine treatment affects the expression pattern of Candida albicans genes encoding virulence factors, iron metabolism proteins, and drug resistance factors. Antimicrobial Agents and Chemotherapy. 2003;47(6):1805–1817. doi: 10.1128/AAC.47.6.1805-1817.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leem SH, Park JE, Kim IS, Chae JY, Sugino A, Sunwoo Y. The possible mechanism of action of ciclopirox olamine in the yeast Saccharomyces cerevisiae . Molecules and Cells. 2003;15(1):55–61. [PubMed] [Google Scholar]
- 80.Borgers M. Mechanism of action of antifungal drugs, with special reference to the imidazole derivatives. Reviews of Infectious Diseases. 1980;2(4):520–534. doi: 10.1093/clinids/2.4.520. [DOI] [PubMed] [Google Scholar]
- 81.Pfaller M, Moet GJ, Messer SA, Jones RN, Castanheira M. Geographic variations in species distribution and echinocandin and azole antifungal resistance rates among Candida bloodstream infection isolates: Report from the SENTRY Antimicrobial Surveillance Program (2008 to 2009) Journal of Clinical Microbiology. 2011;49(1):396–399. doi: 10.1128/JCM.01398-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008-2009. Antimicrobial Agents and Chemotherapy. 2011;55(2):561–566. doi: 10.1128/AAC.01079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pfaller MA, Messer SA, Moet GJ, Jones RN, Castanheira M. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008-2009) International Journal of Antimicrobial Agents. 2011;38(1):65–69. doi: 10.1016/j.ijantimicag.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 84.Normark S, Schönebeck J. In vitro studies of 5-fluorocytosine resistance in Candida albicans and Torulopsis glabrata . Antimicrobial Agents and Chemotherapy. 1972;2(3):114–121. doi: 10.1128/aac.2.3.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medoff G, Kobayashi GS. Strategies in the treatment of systemic fungal infections. New England Journal of Medicine. 1980;302(3):145–155. doi: 10.1056/NEJM198001173020304. [DOI] [PubMed] [Google Scholar]
- 86.Kanafani ZA, Perfect JR. Antimicrobial resistance: resistance to antifungal agents: mechanisms and clinical impact. Clinical Infectious Diseases. 2008;46(1):120–128. doi: 10.1086/524071. [DOI] [PubMed] [Google Scholar]
- 87.Pfaller MA, Yu WL. Antifungal susceptibility testing: new technology and clinical applications. Infectious Disease Clinics of North America. 2001;15(4):1227–1261. doi: 10.1016/s0891-5520(05)70192-6. [DOI] [PubMed] [Google Scholar]
- 88.Powderly W, Kobayashi GS, Herzig GP, Medoff G. Amphotericin B-resistant yeast infection in severely immunocompromised patients. American Journal of Medicine. 1988;84(5):826–832. doi: 10.1016/0002-9343(88)90059-9. [DOI] [PubMed] [Google Scholar]
- 89.Anaissie E, Karyotakis NC, Hachem R, Dignani MC, Rex JH, Paetznick V. Correlation between in vitro and in vivo activity of antifungal agents against Candida species. The Journal of Infectious Diseases. 1994;170(2):384–389. doi: 10.1093/infdis/170.2.384. [DOI] [PubMed] [Google Scholar]
- 90.White T, Marr K, Bowden R. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clinical Microbiology Reviews. 1998;11(2):382–402. doi: 10.1128/cmr.11.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perea S, Patterson T. Antifungal resistance in pathogenic fungi. Clinical Infectious Diseases. 2002;35(9):1073–1080. doi: 10.1086/344058. [DOI] [PubMed] [Google Scholar]
- 92.Espinel-Ingroff A. Mechanisms of resistance to antifungal agents: yeasts and filamentous fungi. Revista Iberoamericana de Micologia. 2008;25(2):101–106. doi: 10.1016/s1130-1406(08)70027-5. [DOI] [PubMed] [Google Scholar]
- 93.Espinel-Ingroff A. In vitro antifungal activities of anidulafungin and micafungin, licensed agents and the investigational triazole posaconazole as determined by NCCLS methods for 12,052 fungal isolates: review of the literature. Revista Iberoamericana de Micologia. 2003;20(4):121–136. [PubMed] [Google Scholar]
- 94.Pfaller M, Boyken L, Hollis RJ, et al. In vitro susceptibility of invasive isolates of Candida spp. to anidulafungin, caspofungin, and micafungin: six years of global surveillance. Journal of Clinical Microbiology. 2008;46(1):150–156. doi: 10.1128/JCM.01901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maligie M, Selitrennikoff C. Cryptococcus neoformans resistance to echinocandins: (1,3)β-glucan synthase activity is sensitive to echinocandins. Antimicrobial Agents and Chemotherapy. 2005;49(7):2851–2856. doi: 10.1128/AAC.49.7.2851-2856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.CLSI, C.a.L.S.I. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard—Third Edition—Document M27-A3. Wayne, Pa, USA: CLSI; 2008. [Google Scholar]
- 97.CLSI, C.a.L.S.I. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous fungi; Approved Standard—Second Edition—Document M38-A2. Wayne, Pa, USA: CLSI; 2008. [Google Scholar]
- 98.Rodríguez-Tudela JL, Arendrup MC, Cuenca-Estrella M, Donnelly JP, Lass-Flörl C. EUCAST breakpoints for antifungals. Drug News and Perspectives. 2010;23(2):93–97. doi: 10.1358/dnp.2010.23.2.1400855. [DOI] [PubMed] [Google Scholar]
- 99.Pfaller MA, Diekema DJ, Andes D, et al. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resistance Updates. 2011;14(3):164–176. doi: 10.1016/j.drup.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 100.Pfaller MA, Castanheira M, Messer SA, Moet GJ, Jones RN. Echinocandin and triazole antifungal susceptibility profiles for Candida spp., Cryptococcus neoformans, and Aspergillus fumigatus: application of new CLSI clinical breakpoints and epidemiologic cutoff values to characterize resistance in the SENTRY Antimicrobial Surveillance Program (2009) Diagnostic Microbiology and Infectious Disease. 2011;69(1):45–50. doi: 10.1016/j.diagmicrobio.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 101.Sanglard D. Clinical relevance of mechanisms of antifungal drug resistance in yeasts. Enfermedades Infecciosas y Microbiologia Clinica. 2002;20(9):462–479. doi: 10.1016/s0213-005x(02)72842-5. [DOI] [PubMed] [Google Scholar]
- 102.Sanglard D, Ischer F, Calabrese D, Micheli MD, Bille J. Multiple resistance mechanisms to azole antifungals in yeast clinical isolates. Drug Resistance Updates. 1998;1(4):255–265. doi: 10.1016/s1368-7646(98)80006-x. [DOI] [PubMed] [Google Scholar]
- 103.Perea S, López-Ribot JL, Kirkpatrick WR, et al. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrobial Agents and Chemotherapy. 2001;45(10):2676–2684. doi: 10.1128/AAC.45.10.2676-2684.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrobial Agents and Chemotherapy. 2010;54(4):1476–1483. doi: 10.1128/AAC.01645-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Franz R, Kelly SL, Lamb DC, Kelly DE, Ruhnke M, Morschhäuser J. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrobial Agents and Chemotherapy. 1998;42(12):3065–3072. doi: 10.1128/aac.42.12.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marr KA, Lyons CN, Rustad T, Bowden RA, White TC. Rapid, transient fluconazole resistance in Candida albicans is associated with increased mRNA levels of CDR. Antimicrobial Agents and Chemotherapy. 1998;42(10):2584–2589. doi: 10.1128/aac.42.10.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.de Micheli M, Bille J, Schueller C, Sanglard D. A common drug-responsive element mediates the upregulation of the Candida albicans ABC transporters CDR1 and CDR2, two genes involved in antifungal drug resistance. Molecular Microbiology. 2002;43(5):1197–1214. doi: 10.1046/j.1365-2958.2002.02814.x. [DOI] [PubMed] [Google Scholar]
- 108.Kamani N, Akhtar Gaur N, Jha S, et al. SRE1 and SRE2 are two specific steroid-responsive modules of Candida drug resistance gene I (CDRI) promoter. Yeast. 2004;21(3):219–239. doi: 10.1002/yea.1067. [DOI] [PubMed] [Google Scholar]
- 109.Puri N, Krishnamurthy S, Habib S, Hasnain SE, Goswami SK, Prasad R. CDR1, a multidrug resistance gene from Candida albicans, contains multiple regulatory domains in its promoter and the distal AP-1 element mediates its induction by miconazole. FEMS Microbiology Letters. 1999;180(2):213–219. doi: 10.1111/j.1574-6968.1999.tb08798.x. [DOI] [PubMed] [Google Scholar]
- 110.Gaur N, Manoharlal R, Saini P, et al. Expression of the CDR1 efflux pump in clinical Candida albicans isolates is controlled by a negative regulatory element. Biochemical and Biophysical Research Communications. 2005;332(1):206–214. doi: 10.1016/j.bbrc.2005.04.113. [DOI] [PubMed] [Google Scholar]
- 111.Gaur NA, Puri N, Karnani N, Mukhopadhyay G, Goswami SK, Prasad R. Identification of a negative regulatory element which regulates basal transcription of a multidrug resistance gene CDR1 of Candida albicans . FEMS Yeast Research. 2004;4(4-5):389–399. doi: 10.1016/S1567-1356(03)00204-6. [DOI] [PubMed] [Google Scholar]
- 112.Coste AT, Crittin J, Bauser C, Rohde B, Sanglard D. Functional analysis of cis-and trans-acting elements of the Candida albicans CDR2 promoter with a novel promoter reporter system. Eukaryotic Cell. 2009;8(8):1250–1267. doi: 10.1128/EC.00069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu TT, Znaidi S, Barker KS, et al. Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryotic Cell. 2007;6(11):2122–2138. doi: 10.1128/EC.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bennett JE, Izumikawa K, Marr KA. Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrobial Agents and Chemotherapy. 2004;48(5):1773–1777. doi: 10.1128/AAC.48.5.1773-1777.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Izumikawa K, Kakeya H, Tsai HF, Grimberg B, Bennett JE. Function of Candida glabrata ABC transporter gene, PDH1. Yeast. 2003;20(3):249–261. doi: 10.1002/yea.962. [DOI] [PubMed] [Google Scholar]
- 116.Sanglard D, Ischer F, Bille J. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata . Antimicrobial Agents and Chemotherapy. 2001;45(4):1174–1183. doi: 10.1128/AAC.45.4.1174-1183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrobial Agents and Chemotherapy. 1999;43(11):2753–2765. doi: 10.1128/aac.43.11.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Torelli R, Posteraro B, Ferrari S, et al. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata . Molecular Microbiology. 2008;68(1):186–201. doi: 10.1111/j.1365-2958.2008.06143.x. [DOI] [PubMed] [Google Scholar]
- 119.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Molecular Microbiology. 2006;61(3):704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 120.Vermitsky J-P, Edlind TD. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrobial Agents and Chemotherapy. 2004;48(10):3773–3781. doi: 10.1128/AAC.48.10.3773-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsao S, Rahkhoodaee F, Raymond M. Relative contributions of the Candida albicans ABC transporters Cdrlp and Cdr2p to clinical azole resistance. Antimicrobial Agents and Chemotherapy. 2009;53(4):1344–1352. doi: 10.1128/AAC.00926-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Holmes A, Lin Y-H, Niimi K, et al. ABC transporter Cdr1p contributes more than Cdr2p does to fluconazole efflux in fluconazole-resistant Candida albicans clinical isolates. Antimicrobial Agents and Chemotherapy. 2008;52(11):3851–3862. doi: 10.1128/AAC.00463-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata . PLoS One. 2011;6(3) doi: 10.1371/journal.pone.0017589. Article ID e17589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moran G, Sullivan D, Morschhäuser J, Coleman D. The Candida dubliniensis CdCDR1 gene is not essential for fluconazole resistance. Antimicrobial Agents and Chemotherapy. 2002;46(9):2829–2841. doi: 10.1128/AAC.46.9.2829-2841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moran GP, Sanglard D, Donnelly SM, Shanley DB, Sullivan DJ, Coleman DC. Identification and expression of multidrug transporters responsible for fluconazole resistance in Candida dubliniensis . Antimicrobial Agents and Chemotherapy. 1998;42(7):1819–1830. doi: 10.1128/aac.42.7.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lamping E, Ranchod A, Nakamura K, et al. Abc1p is a multidrug efflux transporter that tips the balance in favor of innate azole resistance in Candida krusei . Antimicrobial Agents and Chemotherapy. 2009;53(2):354–369. doi: 10.1128/AAC.01095-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Katiyar S, Edlind T. Identification and expression of multidrug resistance-related ABC transporter genes in Candida krusei . Medical Mycology. 2001;39(1):109–116. doi: 10.1080/mmy.39.1.109.116. [DOI] [PubMed] [Google Scholar]
- 128.Nascimento AM, Goldman GH, Park S, et al. Multiple resistance mechanisms among Aspergillus fumigatus mutants with high-level resistance to itraconazole. Antimicrobial Agents and Chemotherapy. 2003;47(5):1719–1726. doi: 10.1128/AAC.47.5.1719-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Slaven J, Anderson M, Sanglard D, et al. Increased expression of a novel Aspergillus fumigatus ABC transporter gene, atrF, in the presence of itraconazole in an itraconazole resistant clinical isolate. Fungal Genetics and Biology. 2002;36(3):199–206. doi: 10.1016/s1087-1845(02)00016-6. [DOI] [PubMed] [Google Scholar]
- 130.Slaven JW, et al. Induced expression of a novel Aspergillus fumigatus ABC transporter gene, atrF, in response to itraconazole. Fungal Genetics and Biology. 2002;36:199–206. doi: 10.1016/s1087-1845(02)00016-6. [DOI] [PubMed] [Google Scholar]
- 131.Semighini CP, Marins M, Goldman MHS, Goldman GH. Quantitative analysis of the relative transcript levels of ABC transporter Atr genes in Aspergillus nidulans by real-time reverse transcription-PCR assay. Applied and Environmental Microbiology. 2002;68(3):1351–1357. doi: 10.1128/AEM.68.3.1351-1357.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.do Nascimento AM, Goldman MH, Goldman GH. Molecular characterization of ABC transporter-encoding genes in Aspergillus nidulans . Genetics and Molecular Research. 2002;1(4):337–349. [PubMed] [Google Scholar]
- 133.Schubert S, Rogers P, Morschhäuser J. Gain-of-function mutations in the transcription factor MRR1 are responsible for overexpression of the MDR1 efflux pump in fluconazole-resistant Candida dubliniensis strains. Antimicrobial Agents and Chemotherapy. 2008;52(12):4274–4280. doi: 10.1128/AAC.00740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Heilmann C, Schneider S, Barker KS, Rogers PD, Morschhäuser J. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans . Antimicrobial Agents and Chemotherapy. 2010;54(1):353–359. doi: 10.1128/AAC.01102-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morio F, Loge C, Besse B, Hennequin C, Le Pape P. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagnostic Microbiology and Infectious Disease. 2010;66(4):373–384. doi: 10.1016/j.diagmicrobio.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 136.Perlin DS. Resistance to echinocandin-class antifungal drugs. Drug Resistance Updates. 2007;10(3):121–130. doi: 10.1016/j.drup.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hope WW, Tabernero L, Denning DW, Anderson MJ. Molecular mechanisms of primary resistance to flucytosine in Candida albicans . Antimicrobial Agents and Chemotherapy. 2004;48(11):4377–4386. doi: 10.1128/AAC.48.11.4377-4386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fardeau V, Lelandais G, Oldfield A, et al. The central role of PDR1 in the foundation of yeast drug resistance. Journal of Biological Chemistry. 2007;282(7):5063–5074. doi: 10.1074/jbc.M610197200. [DOI] [PubMed] [Google Scholar]
- 139.Le Crom S, Devaux F, Marc P, Zhang X, Moye-Rowley WS, Jacq C. New insights into the pleiotropic drug resistance network from genome-wide characterization of the YRR1 transcription factor regulation system. Molecular and Cellular Biology. 2002;22(8):2642–2649. doi: 10.1128/MCB.22.8.2642-2649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Delahodde A, Delaveau T, Jacq C. Positive autoregulation of the yeast transcription factor Pdr3p, which is involved in control of drug resistance. Molecular and Cellular Biology. 1995;15(8):4043–4051. doi: 10.1128/mcb.15.8.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Katzmann D, Burnett PE, Golin J, Mahe Y, Moye-Rowley WS. Transcriptional control of the yeast PDR5 gene by the PDR3 gene product. Molecular and Cellular Biology. 1994;14(7):4653–4661. doi: 10.1128/mcb.14.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mamnun Y, Pandjaitan R, Mahé Y, Delahodde A, Kuchler K. The yeast zinc finger regulators Pdr1p and Pdr3p control pleiotropic drug resistance (PDR) as homo- and heterodimers in vivo. Molecular Microbiology. 2002;46(5):1429–1440. doi: 10.1046/j.1365-2958.2002.03262.x. [DOI] [PubMed] [Google Scholar]
- 143.Tsai H, Krol AA, Sarti KE, Bennett JE. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrobial Agents and Chemotherapy. 2006;50(4):1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Molecular Microbiology. 2006;61(3):704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 145.Ferrari S, Ischer F, Calabrese D, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathogens. 2009;5(1) doi: 10.1371/journal.ppat.1000268. Article ID e1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tsai H-F, Krol AA, Sarti KE, Bennett JE. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrobial Agents and Chemotherapy. 2006;50(4):1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shen H, An MM, Wang DJ, et al. Fcr1p inhibits development of fluconazole resistance in Candida albicans by abolishing CDR1 induction. Biological and Pharmaceutical Bulletin. 2007;30(1):68–73. doi: 10.1248/bpb.30.68. [DOI] [PubMed] [Google Scholar]
- 148.Talibi D, Raymond M. Isolation of a putative Candida albicans transcriptional regulator involved in pleiotropic drug resistance by functional complementation of a pdr1 pdr3 mutation in Saccharomyces cerevisiae . Journal of Bacteriology. 1999;181(1):231–240. doi: 10.1128/jb.181.1.231-240.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang X, Talibi D, Weber S, Poisson G, Raymond M. Functional isolation of the Candida albicans FCR3 gene encoding a bZip transcription factor homologous to Saccharomyces cerevisiae Yap3p. Yeast. 2001;18(13):1217–1225. doi: 10.1002/yea.770. [DOI] [PubMed] [Google Scholar]
- 150.Coste AT, Ramsdale M, Ischer F, Sanglard D. Divergent functions of three Candida albicans zinc-cluster transcription factors (CTA4, ASG1 and CTF1) complementing pleiotropic drug resistance in Saccharomyces cerevisiae . Microbiology. 2008;154(5):1491–1501. doi: 10.1099/mic.0.2007/016063-0. [DOI] [PubMed] [Google Scholar]
- 151.Chen C-G, Yang Y-L, Shih H-I, Su C-L, Lo H-J. CaNdt80 is involved in drug resistance in Candida albicans by regulating CDR1. Antimicrobial Agents and Chemotherapy. 2004;48(12):4505–4512. doi: 10.1128/AAC.48.12.4505-4512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang J, Yang YL, Wu CJ, et al. The DNA-binding domain of CaNdt80p is required to activate CDR1 involved in drug resistance in Candida albicans . Journal of Medical Microbiology. 2006;55(10):1403–1411. doi: 10.1099/jmm.0.46650-0. [DOI] [PubMed] [Google Scholar]
- 153.Sellam A, Tebbji F, Nantel A. Role of Ndt80p in sterol metabolism regulation and azole resistance in Candida albicans . Eukaryotic Cell. 2009;8(8):1174–1183. doi: 10.1128/EC.00074-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hikkel I, Lucau-Danila A, Delaveau T, Marc P, Devaux F, Jacq C. A general strategy to uncover transcription factor properties identifies a new regulator of drug resistance in yeast. Journal of Biological Chemistry. 2003;278(13):11427–11432. doi: 10.1074/jbc.M208549200. [DOI] [PubMed] [Google Scholar]
- 155.Kren A, Mamnun YM, Bauer BE, et al. War1p, a novel transcription factor controlling weak acid stress response in yeast. Molecular and Cellular Biology. 2003;23(5):1775–1785. doi: 10.1128/MCB.23.5.1775-1785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mendizabal I, Rios G, Mulet JM, Serrano R, De Larrinoa IF. Yeast putative transcription factors involved in salt tolerance. FEBS Letters. 1998;425(2):323–328. doi: 10.1016/s0014-5793(98)00249-x. [DOI] [PubMed] [Google Scholar]
- 157.Schjerling P, Holmberg S. Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Research. 1996;24(23):4599–4607. doi: 10.1093/nar/24.23.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryotic Cell. 2004;3(6):1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rustad T, Stevens DA, Pfaller MA, White TC. Homozygosity at the Candida albicans MTL locus associated with azole resistance. Microbiology. 2002;148(4):1061–1072. doi: 10.1099/00221287-148-4-1061. [DOI] [PubMed] [Google Scholar]
- 160.Coste A, Selmecki A, Forche A, et al. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryotic Cell. 2007;6(10):1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Coste A, Turner V, Ischer F, et al. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans . Genetics. 2006;172(4):2139–2156. doi: 10.1534/genetics.105.054767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans . Science. 2006;313(5785):367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Molecular Microbiology. 2008;68(3):624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 164.Wu W, Pujol C, Lockhart SR, Soll DR. Chromosome loss followed by duplication is the major mechanism of spontaneous mating-type locus homozygosis in Candida albicans . Genetics. 2005;169(3):1311–1327. doi: 10.1534/genetics.104.033167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fling M, Kopf J, Tamarkin A, Gorman JA, Smith HA, Koltin Y. Analysis of a Candida albicans gene that encodes a novel mechanism for resistance to benomyl and methotrexate. Molecular and General Genetics. 1991;227(2):318–329. doi: 10.1007/BF00259685. [DOI] [PubMed] [Google Scholar]
- 166.Ben-Yaacov R, Knoller S, Caldwell GA, Becker JM, Koltin Y. Candida albicans gene encoding resistance to benomyl and methotrexate is a multidrug resistance gene. Antimicrobial Agents and Chemotherapy. 1994;38(4):648–652. doi: 10.1128/aac.38.4.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vandeputte P, Larcher G, Bergès T, Renier G, Chabasse D, Bouchara J-P. Mechanisms of azole resistance in a clinical isolate of Candida tropicalis . Antimicrobial Agents and Chemotherapy. 2005;49(11):4608–4615. doi: 10.1128/AAC.49.11.4608-4615.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pinjon E, Jackson CJ, Kelly SL, et al. Reduced azole susceptibility in genotype 3 Candida dubliniensis isolates associated with increased CaCDR1 and CdCDR2 expression. Antimicrobial Agents and Chemotherapy. 2005;49(4):1312–1318. doi: 10.1128/AAC.49.4.1312-1318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pinjon E, Moran GP, Jackson CJ, et al. Molecular mechanisms of itraconazole resistance in Candida dubliniensis . Antimicrobial Agents and Chemotherapy. 2003;47(8):2424–2437. doi: 10.1128/AAC.47.8.2424-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wirsching S, Moran GP, Sullivan DJ, Coleman DC, Morschhäuser J. MDR1-mediated drug resistance in Candida dubliniensis . Antimicrobial Agents and Chemotherapy. 2001;45(12):3416–3421. doi: 10.1128/AAC.45.12.3416-3421.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Riggle P, Kumamoto C. Transcriptional regulation of MDR1, encoding a drug efflux determinant, in fluconazole-resistant Candida albicans strains through an Mcm1p binding site. Eukaryotic Cell. 2006;5(12):1957–1968. doi: 10.1128/EC.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rognon B, Kozovska Z, Coste AT, Pardini G, Sanglard D. Identification of promoter elements responsible for the regulation of MDR1 from Candida albicans, a major facilitator transporter involved in azole resistance. Microbiology. 2006;152(12):3701–3722. doi: 10.1099/mic.0.29277-0. [DOI] [PubMed] [Google Scholar]
- 173.Nguyên D, Alarco A, Raymond M. Multiple Yap1p-binding sites mediate induction of the yeast major facilitator FLR1 gene in response to drugs, oxidants, and alkylating agents. Journal of Biological Chemistry. 2001;276(2):1138–1145. doi: 10.1074/jbc.M008377200. [DOI] [PubMed] [Google Scholar]
- 174.Hiller D, Stahl S, Morschhäuser J. Multiple cis-acting sequences mediate upregulation of the MDR1 efflux pump in a fluconazole-resistant clinical Candida albicans isolate. Antimicrobial Agents and Chemotherapy. 2006;50(7):2300–2308. doi: 10.1128/AAC.00196-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Alarco AM, Raymond M. The bZip transcription factor Cap1p is involved in multidrug resistance and oxidative stress response in Candida albicans . Journal of Bacteriology. 1999;181(3):700–708. doi: 10.1128/jb.181.3.700-708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang X, De Micheli M, Coleman ST, Sanglard D, Moye-Rowley WS. Analysis of the oxidative stress regulation of the Candida albicans transcription factor, Cap1p. Molecular Microbiology. 2000;36(3):618–629. doi: 10.1046/j.1365-2958.2000.01877.x. [DOI] [PubMed] [Google Scholar]
- 177.Znaidi S, Barker KS, Weber S, et al. Identification of the Candida albicans Cap1p regulon. Eukaryotic Cell. 2009;8(6):806–820. doi: 10.1128/EC.00002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dunkel N, Blaß J, Rogers PD, Morschhäuser J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Molecular Microbiology. 2008;69(4):827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Morschhäuser J, Barker KS, Liu TT, BlaB-Warmuth J, Homayouni R, Rogers PD. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans . PLoS Pathogens. 2007;3(11) doi: 10.1371/journal.ppat.0030164. Article ID e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, Rogers PD. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryotic Cell. 2008;7(7):1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Znaidi S, Weber S, Al-Abdin OZ, et al. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryotic Cell. 2008;7(5):836–847. doi: 10.1128/EC.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mogavero S, Tavanti A, Senesi S, Rogers PD, Morschhäuser J. Differential requirement of the transcription factor Mcm1 for activation of the Candida albicans multidrug efflux pump MDR1 by its regulators Mrr1 and Cap1. Antimicrobial Agents and Chemotherapy. 2011;55(5):2061–2066. doi: 10.1128/AAC.01467-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schubert S, Barker KS, Znaidi S, et al. Regulation of efflux pump expression and drug resistance by the transcription factors Mrr1, Upc2, and Cap1 in Candida albicans . Antimicrobial Agents and Chemotherapy. 2011;55(5):2212–2223. doi: 10.1128/AAC.01343-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vanden Bossche H, Marichal P, Gorrens J, Bellens D, Moereels H, Janssen PAJ. Mutation in cytochrome P-450-dependent 14α-demethylase results in decreased affinity for azole antifungals. Biochemical Society Transactions. 1990;18(1):56–59. doi: 10.1042/bst0180056. [DOI] [PubMed] [Google Scholar]
- 185.Marichal P, Koymans L, Willemsens S, et al. Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans . Microbiology. 1999;145(10):2701–2713. doi: 10.1099/00221287-145-10-2701. [DOI] [PubMed] [Google Scholar]
- 186.Lamb DC, Corran A, Baldwin BC, Kwon-Chung J, Kelly SL. Resistant P45051A1 activity in azole antifungal tolerant Cryptococcus neoformans from AIDS patients. FEBS Letters. 1995;368(2):326–330. doi: 10.1016/0014-5793(95)00684-2. [DOI] [PubMed] [Google Scholar]
- 187.Sanglard D, Ischer F, Koymans L, Bille J. Amino acid substitutions in the cytochrome P-450 lanosterol 14α- demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrobial Agents and Chemotherapy. 1998;42(2):241–253. doi: 10.1128/aac.42.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Cuenca-Estrella M, Rodriguez-Tudela JL. Substitutions at methionine 220 in the 14α-sterol demethylase (cyp51A) of Aspergillus fumigatus are responsible for resistance in vitro to azole antifungal drugs. Antimicrobial Agents and Chemotherapy. 2004;48(7):2747–2750. doi: 10.1128/AAC.48.7.2747-2750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen J, Li H, Li R, Bu D, Wan Z. Mutations in the cyp51A gene and susceptibility to itraconazole in Aspergillus fumigatus serially isolated from a patient with lung aspergilloma. The Journal of Antimicrobial Chemotherapy. 2005;55(1):31–37. doi: 10.1093/jac/dkh507. [DOI] [PubMed] [Google Scholar]
- 190.Diaz-Guerra TM, Mellado E, Cuenca-Estrella M, Rodriguez-Tudela JL. A point mutation in the 14α-sterol demethylase gene cyp51a contributes to itraconazole resistance in Aspergillus fumigatus . Antimicrobial Agents and Chemotherapy. 2003;47(3):1120–1224. doi: 10.1128/AAC.47.3.1120-1124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mann PA, Parmegiani RM, Wei SQ, et al. Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14α-demethylase. Antimicrobial Agents and Chemotherapy. 2003;47(2):577–581. doi: 10.1128/AAC.47.2.577-581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mellado E, Garcia-Effron G, Alcázar-Fuoli L, et al. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrobial Agents and Chemotherapy. 2007;51(6):1897–1904. doi: 10.1128/AAC.01092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Verweij PE, Snelders E, Kema GH, Mellado E, Melchers WJ. Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use? The Lancet Infectious Diseases. 2009;9(12):789–795. doi: 10.1016/S1473-3099(09)70265-8. [DOI] [PubMed] [Google Scholar]
- 194.Snelders E, Huis In’t Veld RAG, Rijs AJMM, Kema GHJ, Melchers WJG, Verweij PE. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Applied and Environmental Microbiology. 2009;75(12):4053–4057. doi: 10.1128/AEM.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mortensen KL, Mellado E, Lass-Flörl C, Rodriguez-Tudela JL, Johansen HK, Arendrup MC. Environmental study of azole-resistant Aspergillus fumigatus and other aspergilli in Austria, Denmark, and Spain. Antimicrobial Agents and Chemotherapy. 2010;54(11):4545–4549. doi: 10.1128/AAC.00692-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Park S, Kelly R, Kahn JN, et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrobial Agents and Chemotherapy. 2005;49(8):3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Balashov SV, Park S, Perlin DS. Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FKS1 . Antimicrobial Agents and Chemotherapy. 2006;50(6):2058–2063. doi: 10.1128/AAC.01653-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Garcia-Effron G, Park S, Perlin DS. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrobial Agents and Chemotherapy. 2009;53(1):112–122. doi: 10.1128/AAC.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Katiyar S, Pfaller M, Edlind T. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrobial Agents and Chemotherapy. 2006;50(8):2892–2894. doi: 10.1128/AAC.00349-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kahn J, Garcia-Effron G, Hsu M-J, Park S, Marr KA, Perlin DS. Acquired echinocandin resistance in a Candida krusei isolate due to modification of glucan synthase. Antimicrobial Agents and Chemotherapy. 2007;51(5):1876–1878. doi: 10.1128/AAC.00067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Johnson ME, Katiyar SK, Edlind TD. New Fks hot spot for acquired echinocandin resistance in Saccharomyces cerevisiae and its contribution to intrinsic resistance of Scedosporium species. Antimicrobial Agents and Chemotherapy. 2011;55(8):3774–3781. doi: 10.1128/AAC.01811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gardiner RE, Souteropoulos P, Park S, Perlin DS. Characterization of Aspergillus fumigatus mutants with reduced susceptibility to caspofungin. Medical Mycology. 2005;43(supplement 1):S299–S305. doi: 10.1080/13693780400029023. [DOI] [PubMed] [Google Scholar]
- 203.Rocha E, Garcia-Effron G, Park S, Perlin DS. A Ser678Pro substitution in Fks1p confers resistance to echinocandin drugs in Aspergillus fumigatus . Antimicrobial Agents and Chemotherapy. 2007;51(11):4174–4176. doi: 10.1128/AAC.00917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kerridge D, Fasoli M, Wayman FJ. Drug resistance in Candida albicans and Candida glabrata . Annals of the New York Academy of Sciences. 1988;544:245–259. doi: 10.1111/j.1749-6632.1988.tb40410.x. [DOI] [PubMed] [Google Scholar]
- 205.Vandeputte P, Pineau L, Larcher G, et al. Molecular mechanisms of resistance to 5-fluorocytosine in laboratory mutants of Candida glabrata . Mycopathologia. 2011;171(1):11–21. doi: 10.1007/s11046-010-9342-1. [DOI] [PubMed] [Google Scholar]
- 206.Edlind TD, Katiyar SK. Mutational analysis of flucytosine resistance in Candida glabrata . Antimicrobial Agents and Chemotherapy. 2010;54(11):4733–4738. doi: 10.1128/AAC.00605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ko TP, Lin JJ, Hu CY, Hsu YH, Wang AHJ, Liaw SH. Crystal structure of yeast cytosine deaminase: insights into enzyme mechanism and evolution. Journal of Biological Chemistry. 2003;278(21):19111–19117. doi: 10.1074/jbc.M300874200. [DOI] [PubMed] [Google Scholar]
- 208.Henry K, Nickels J, Edlind T. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrobial Agents and Chemotherapy. 2000;44(10):2693–2700. doi: 10.1128/aac.44.10.2693-2700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ribeiro MA, Paula CR. Up-regulation of ERG11 gene among fluconazole-resistant Candida albicans generated in vitro: is there any clinical implication? Diagnostic Microbiology and Infectious Disease. 2007;57(1):71–75. doi: 10.1016/j.diagmicrobio.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 210.Perea S, López-Ribot JL, Wickes BL, et al. Molecular mechanisms of fluconazole resistance in Candida dubliniensis isolates from human immunodeficiency virus-infected patients with oropharyngeal candidiasis. Antimicrobial Agents and Chemotherapy. 2002;46(6):1695–1703. doi: 10.1128/AAC.46.6.1695-1703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Marichal P, Vanden Bossche H, Odds FC, et al. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrobial Agents and Chemotherapy. 1997;41(10):2229–2237. doi: 10.1128/aac.41.10.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chang YC, Bien CM, Lee H, Espenshade PJ, Kwon-Chung KJ. Sre1p, a regulator of oxygen sensing and sterol homeostasis, is required for virulence in Cryptococcus neoformans . Molecular Microbiology. 2007;64(3):614–629. doi: 10.1111/j.1365-2958.2007.05676.x. [DOI] [PubMed] [Google Scholar]
- 213.Nagi M, Nakayama H, Tanabe K, et al. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata . Genes to Cells. 2011;16(1):80–89. doi: 10.1111/j.1365-2443.2010.01470.x. [DOI] [PubMed] [Google Scholar]
- 214.Silver PM, Oliver BG, White TC. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryotic Cell. 2004;3(6):1391–1397. doi: 10.1128/EC.3.6.1391-1397.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrobial Agents and Chemotherapy. 2005;49(5):1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Oliver B, Song JL, Choiniere JH, White TC. cis-acting elements within the Candida albicans ERG11 promoter mediate the azole response through transcription factor Upc2p. Eukaryotic Cell. 2007;6(12):2231–2239. doi: 10.1128/EC.00331-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stevens DA, White TC, Perlin DS, Selitrennikoff CP. Studies of the paradoxical effect of caspofungin at high drug concentrations. Diagnostic Microbiology and Infectious Disease. 2005;51(3):173–178. doi: 10.1016/j.diagmicrobio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 218.Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H. Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for β-1,6-glucan synthesis inhibition by caspofungin. Antimicrobial Agents and Chemotherapy. 2006;50(9):3160–3161. doi: 10.1128/AAC.00563-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cota JM, Grabinski JL, Talbert RL, et al. Increases in SLT2 expression and chitin content are associated with incomplete killing of Candida glabrata by caspofungin. Antimicrobial Agents and Chemotherapy. 2008;52(3):1144–1146. doi: 10.1128/AAC.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chamilos G, Lewis RE, Albert N, Kontoyiannis DP. Paradoxical effect of echinocandins across Candida species in vitro: evidence for echinocandin-specific and Candida species-related differences. Antimicrobial Agents and Chemotherapy. 2007;51(6):2257–2259. doi: 10.1128/AAC.00095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nolte FS, Parkinson T, Falconer DJ, et al. Isolation and characterization of fluconazole- and amphotericin B- resistant Candida albicans from blood of two patients with leukemia. Antimicrobial Agents and Chemotherapy. 1997;41(1):196–199. doi: 10.1128/aac.41.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Miyazaki Y, Geber A, Miyazaki H, et al. Cloning, sequencing, expression and allelic sequence diversity of ERG3 (C-5 sterol desaturase gene) in Candida albicans . Gene. 1999;236(1):43–51. doi: 10.1016/s0378-1119(99)00263-2. [DOI] [PubMed] [Google Scholar]
- 223.Martel CM, Parker JE, Bader O, et al. Identification and characterization of four azole-resistant erg3 mutants of Candida albicans . Antimicrobial Agents and Chemotherapy. 2010;54(11):4527–4533. doi: 10.1128/AAC.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Vanden Bossche H, Marichal P, Odds FC. Molecular mechanisms of drug resistance in fungi. Trends in Microbiology. 1994;2(10):393–400. doi: 10.1016/0966-842x(94)90618-1. [DOI] [PubMed] [Google Scholar]
- 225.Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrobial Agents and Chemotherapy. 2003;47(8):2404–2412. doi: 10.1128/AAC.47.8.2404-2412.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Watson PF, Rose ME, Kelly SL. Isolation and analysis of ketoconazole resistant mutants of Saccharomyces cerevisiae . Journal of Medical and Veterinary Mycology. 1988;26(3):153–162. doi: 10.1080/02681218880000231. [DOI] [PubMed] [Google Scholar]
- 227.Young L, Hull C, Heitman J. Disruption of ergosterol biosynthesis confers resistance to amphotericin B in Candida lusitaniae . Antimicrobial Agents and Chemotherapy. 2003;47(9):2717–2724. doi: 10.1128/AAC.47.9.2717-2724.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jensen-Pergakes KL, Kennedy MA, Lees ND, Barbuch R, Koegel C, Bard M. Sequencing, disruption, and characterization of the Candida albicans sterol methyltransferase (ERG6) gene: drug susceptibility studies in erg6 mutants. Antimicrobial Agents and Chemotherapy. 1998;42(5):1160–1167. doi: 10.1128/aac.42.5.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kelly S, Lamb DC, Kelly DE, et al. Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol Δ5,6-desaturation. FEBS Letters. 1997;400(1):80–82. doi: 10.1016/s0014-5793(96)01360-9. [DOI] [PubMed] [Google Scholar]
- 230.Vandeputte P, Tronchin G, Bergès T, Hennequin C, Chabasse D, Bouchara J-P. Reduced susceptibility to polyenes associated with a missense mutation in the ERG6 gene in a clinical isolate of Candida glabrata with pseudohyphal growth. Antimicrobial Agents and Chemotherapy. 2007;51(3):982–990. doi: 10.1128/AAC.01510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Vandeputte P, Tronchin G, Larcher G, et al. A nonsense mutation in the ERG6 gene leads to reduced susceptibility to polyenes in a clinical isolate of Candida glabrata . Antimicrobial Agents and Chemotherapy. 2008;52(10):3701–3709. doi: 10.1128/AAC.00423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mukherjee PK, Chandra J. Candida biofilm resistance. Drug Resistance Updates. 2004;7(4-5):301–309. doi: 10.1016/j.drup.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 233.Martinez LR, Mihu MR, Han G, et al. The use of chitosan to damage Cryptococcus neoformans biofilms. Biomaterials. 2010;31(4):669–679. doi: 10.1016/j.biomaterials.2009.09.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pettit RK, Repp KK, Hazen KC. Temperature affects the susceptibility of Cryptococcus neoformans biofilms to antifungal agents. Medical Mycology. 2010;48(2):421–426. doi: 10.1080/13693780903136879. [DOI] [PubMed] [Google Scholar]
- 235.Hamal P, Ostransky J, Dendis M, et al. A case of endocarditis caused by the yeast Pichia fabianii with biofilm production and developed in vitro resistance to azoles in the course of antifungal treatment. Medical Mycology. 2008;46(6):601–605. doi: 10.1080/13693780802078180. [DOI] [PubMed] [Google Scholar]
- 236.Di Bonaventura G, Pompilio A, Picciani C, Iezzi M, D’Antonio D, Piccolomini R. Biofilm formation by the emerging fungal pathogen Trichosporon asahii: development, architecture, and antifungal resistance. Antimicrobial Agents and Chemotherapy. 2006;50(10):3269–3276. doi: 10.1128/AAC.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ueno A, Hamano T, Fujii A, et al. The effects of voriconazole and vascular lesions in invasion of aspergillosis into the central nerve system. Clinical Neurology. 2009;49(8):468–473. doi: 10.5692/clinicalneurol.49.468. [DOI] [PubMed] [Google Scholar]
- 238.Seidler MJ, Salvenmoser S, Müller FMC. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrobial Agents and Chemotherapy. 2008;52(11):4130–4136. doi: 10.1128/AAC.00234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chandrasekar PH, Manavathu EK. Do Aspergillus species produce biofilm? Future Microbiology. 2008;3(1):19–21. doi: 10.2217/17460913.3.1.19. [DOI] [PubMed] [Google Scholar]
- 240.Harriott MM, Noverr MC. Candida albicans and Staphylococcus aureus form polymicrobial biofilms: effects on antimicrobial resistance. Antimicrobial Agents and Chemotherapy. 2009;53(9):3914–3922. doi: 10.1128/AAC.00657-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Pierce GE. Pseudomonas aeruginosa, Candida albicans, and device-related nosocomial infections: implications, trends, and potential approaches for control. Journal of Industrial Microbiology and Biotechnology. 2005;32(7):309–318. doi: 10.1007/s10295-005-0225-2. [DOI] [PubMed] [Google Scholar]
- 242.Chandra J, Zhou G, Ghannoum MA. Fungal biofilms and antimycotics. Current Drug Targets. 2005;6(8):887–894. doi: 10.2174/138945005774912762. [DOI] [PubMed] [Google Scholar]
- 243.Jabra-Rizk M, Falkler WA, Meiller TF. Fungal biofilms and drug resistance. Emerging Infectious Diseases. 2004;10(1):14–19. doi: 10.3201/eid1001.030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Di Santo R. Natural products as antifungal agents against clinically relevant pathogens. Natural Product Reports. 2010;27(7):1084–1098. doi: 10.1039/b914961a. [DOI] [PubMed] [Google Scholar]
- 245.Arif T, Bhosale JD, Kumar N, et al. Natural products - Antifungal agents derived from plants. Journal of Asian Natural Products Research. 2009;11(7):621–638. doi: 10.1080/10286020902942350. [DOI] [PubMed] [Google Scholar]
- 246.Mayer AM, Rodríguez AD, Berlinck RGS, Fusetani N. Marine pharmacology in 2007-8: marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comparative Biochemistry and Physiology. 2011;153(2):191–222. doi: 10.1016/j.cbpc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Marchetti O, Entenza JM, Sanglard D, Bille J, Glauser MP, Moreillon P. Fluconazole plus cyclosporine: a fungicidal combination effective against experimental due to Candida albicans . Antimicrobial Agents and Chemotherapy. 2000;44(11):2932–2938. doi: 10.1128/aac.44.11.2932-2938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Marchetti O, Moreillon P, Glauser MP, Bille J, Sanglard D. Potent synergism of the combination of fluconazole and cyclosporine in Candida albicans . Antimicrobial Agents and Chemotherapy. 2000;44(9):2373–2381. doi: 10.1128/aac.44.9.2373-2381.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Cruz MC, Cavallo LM, Görlach JM, et al. Rapamycin antifungal action is mediated via conserved complexes with FKBP12 and TOR kinase homologs in Cryptococcus neoformans . Molecular and Cellular Biology. 1999;19(6):4101–4112. doi: 10.1128/mcb.19.6.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Cruz MC, Del Poeta M, Wang P, et al. Immunosuppressive and nonimmunosuppressive cyclosporine analogs are toxic to the opportunistic fungal pathogen Cryptococcus neoformans via cyclophilin-dependent inhibition of calcineurin. Antimicrobial Agents and Chemotherapy. 2000;44(1):143–149. doi: 10.1128/aac.44.1.143-149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cruz MC, Goldstein AL, Blankenship J, et al. Rapamycin and less immunosuppressive analogs are toxic to Candida albicans and Cryptococcus neoformans via FKBP12-dependent inhibition of TOR. Antimicrobial Agents and Chemotherapy. 2001;45(11):3162–3170. doi: 10.1128/AAC.45.11.3162-3170.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cannon RD, Lamping E, Holmes AR, et al. Efflux-mediated antifungal drug resistance. Clinical Microbiology Reviews. 2009;22(2):291–321. doi: 10.1128/CMR.00051-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Dryden M, Payne P. Preventing parasites in cats. Veterinary Therapeutics. 2005;6(3):260–267. [PubMed] [Google Scholar]
- 254.Sharma M, Manoharlal R, Shukla S, et al. Curcumin modulates efflux mediated by yeast ABC multidrug transporters and is synergistic with antifungals. Antimicrobial Agents and Chemotherapy. 2009;53(8):3256–3265. doi: 10.1128/AAC.01497-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Pachl J, Svododa P, Jacobs F, et al. A randomized, blinded, multicenter trial of lipid-associated amphotericin B alone versus in combination with an antibody-based inhibitor of heat shock protein 90 in patients with invasive candidiasis. Clinical Infectious Diseases. 2006;42(10):1404–1413. doi: 10.1086/503428. [DOI] [PubMed] [Google Scholar]
- 256.Bader T, Schröppel K, Bentink S, Agabian N, Köhler G, Morschhäuser J. Role of calcineurin in stress resistance, morphogenesis, and virulence of a Candida albicans wild-type strain. Infection and Immunity. 2006;74(7):4366–4369. doi: 10.1128/IAI.00142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Blankenship J, Wormley FL, Boyce MK, et al. Calcineurin is essential for Candida albicans survival in serum and virulence. Eukaryotic Cell. 2003;2(3):422–430. doi: 10.1128/EC.2.3.422-430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Odom A, Muir S, Lim E, Toffaletti DL, Perfect J, Heitman J. Calcineurin is required for virulence of Cryptococcos neoformans . The EMBO Journal. 1997;16(10):2576–2589. doi: 10.1093/emboj/16.10.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sanglard D, Ischer F, Marchetti O, Entenza J, Bille J. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Molecular Microbiology. 2003;48(4):959–976. doi: 10.1046/j.1365-2958.2003.03495.x. [DOI] [PubMed] [Google Scholar]
- 260.Steinbach WJ, Cramer RA, Perfect BZ, et al. Calcineurin controls growth, morphology, and pathogenicity in Aspergillus fumigatus . Eukaryotic Cell. 2006;5(7):1091–1103. doi: 10.1128/EC.00139-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ting PC, Kuang R, Wu H, et al. The synthesis and structure-activity relationship of pyridazinones as glucan synthase inhibitors. Bioorganic and Medicinal Chemistry Letters. 2011;21(6):1819–1822. doi: 10.1016/j.bmcl.2011.01.058. [DOI] [PubMed] [Google Scholar]
- 262.Zhou G, Ting PC, Aslanian R, et al. SAR studies of pyridazinone derivatives as novel glucan synthase inhibitors. Bioorganic and Medicinal Chemistry Letters. 2011;21(10):2890–2893. doi: 10.1016/j.bmcl.2011.03.083. [DOI] [PubMed] [Google Scholar]
- 263.Walker SS, Xu Y, Triantafyllou I, et al. Discovery of a novel class of orally active antifungal {beta}-1,3-D-glucan synthase inhibitors. Antimicrob Agents Chemother. 2011;55(11):5099–5106. doi: 10.1128/AAC.00432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Oh J, Fung E, Schlecht U, et al. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathogens. 2010;6(10) doi: 10.1371/journal.ppat.1001140. Article ID e1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Xu D, Jiang B, Ketela T, et al. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans . PLoS pathogens. 2007;3(6):p. e92. doi: 10.1371/journal.ppat.0030092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Pukkila-Worley R, Holson E, Wagner F, Mylonakis E. Antifungal drug discovery through the study of invertebrate model hosts. Current Medicinal Chemistry. 2009;16(13):1588–1595. doi: 10.2174/092986709788186237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Roemer T, Jiang B, Davison J, et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Molecular Microbiology. 2003;50(1):167–181. doi: 10.1046/j.1365-2958.2003.03697.x. [DOI] [PubMed] [Google Scholar]
- 268.Hu W, Sillaots S, Lemieux S, et al. Essential gene identification and drug target prioritization in Aspergillus fumigatus . PLoS Pathogens. 2007;3(3):p. e24. doi: 10.1371/journal.ppat.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bruno V, Mitchell A. Large-scale gene function analysis in Candida albicans . Trends in Microbiology. 2004;12(4):157–161. doi: 10.1016/j.tim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 270.Nobile C, Mitchell A. Large-scale gene disruption using the UAU1 cassette. Methods in Molecular Biology. 2009;499:175–194. doi: 10.1007/978-1-60327-151-6_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Nobile C, Mitchell A. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Current Biology. 2005;15(12):1150–1155. doi: 10.1016/j.cub.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 272.Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, Mitchell AP. Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathogens. 2006;2(3):0204–0210. doi: 10.1371/journal.ppat.0020021. Article ID e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Epp E, Vanier G, Harcus D, et al. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLoS Pathogens. 2010;6(2) doi: 10.1371/journal.ppat.1000753. Article ID e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Homann OR, Dea J, Noble SM, Johnson AD. A phenotypic profile of the Candida albicans regulatory network. PLoS Genetics. 2009;5(12) doi: 10.1371/journal.pgen.1000783. Article ID e1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Noble S, Johnson A. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans . Eukaryotic Cell. 2005;4(2):298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Epp E, Walther A, Lépine G, et al. Forward genetics in Candida albicans that reveals the Arp2/3 complex is required for hyphal formation, but not endocytosis. Molecular Microbiology. 2010;75(5):1182–1198. doi: 10.1111/j.1365-2958.2009.07038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans . Cell. 2008;135(1):174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Chamilos G, Nobile CJ, Bruno VM, Lewis RE, Mitchell AP, Kontoyiannis DP. Candida albicans Cas5, a regulator of cell wall integrity, is required for virulence in murine and toll mutant fly models. The Journal of Infectious Diseases. 2009;200(1):152–157. doi: 10.1086/599363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Pukkila-Worley R, Peleg AY, Tampakakis E, Mylonakis E. Candida albicans hyphal formation and virulence assessed using a caenorhabditis elegans infection model. Eukaryotic Cell. 2009;8(11):1750–1758. doi: 10.1128/EC.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Vandeputte P, Ischer F, Sanglard D, Coste AT. In vivo systematic analysis of Candida albicans Zn2-Cys6 transcription factors mutants for mice organ colonization. Plos One. 2011;6(10):p. e26962. doi: 10.1371/journal.pone.0026962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Noble SM, French S, Kohn LA, Chen V, Johnson AD. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nature Genetics. 2010;42(7):590–598. doi: 10.1038/ng.605. [DOI] [PMC free article] [PubMed] [Google Scholar]