

Abstract
The threat of West Nile virus (WNV) epidemics necessitates the development of a technology platform that can produce reagents to support detection and diagnosis rapidly and inexpensively. A plant expression system is attractive for protein production due to its low-cost and high-scalability nature and its ability to make appropriate posttranslational modifications. Here, we investigated the feasibility of using plants to produce two WNV detection and diagnostic reagents to address the current cost and scalability issues. We demonstrated that WNV DIII antigen and E16 monoclonal antibody are rapidly produced at high levels in two plant species and are easily purified. Furthermore, they are effective in identifying WNV and in detecting human IgM response to WNV infection. E16 mAb does not cross-react with other flaviviruses, therefore, is valuable for improving diagnostic accuracy. This study provides a proof of principle for using plants as a robust and economical system to produce diagnostic reagents for arboviruses.
1. Introduction
West Nile virus (WNV) is an arbovirus that belongs to the Flavivirus genus of the Flaviviridae family. It is a neurotropic, enveloped virus with a single-stranded, positive polarity, 11 kilobase RNA genome. The transmission cycles of WNV involve mosquitoes of the genus Culex and birds, while humans and other mammals are incidental hosts. Until 1999, WNV was found in the Eastern Hemisphere, with wide distribution in Africa, Asia, the Middle East, and Europe [1]. In 1999, WNV entered the Western Hemisphere through New York City. Since then, more than 30,000 humans with severe WNV infection in the continental United States have been diagnosed, with many more cases of infection remaining undiagnosed. Advanced age is one of the greatest risk factors for severe neurological disease, long-term morbidity, and death [2], although a genetic basis of susceptibility has also been recently identified [3].
Several methods have been developed for the detection of WNV and WNV infections. Specific work in mosquito vectors and infected avian or mammalian hosts has been successful and is protein or nucleic acid based [4–6]. Human WNV infection can be diagnosed by detecting host immune responses such as WNV-specific IgM or IgG production with enzyme-linked immunosorbent assays (ELISAs) or by directly identifying WNV from cell cultures of serum, cerebrospinal fluid, or tissues with WNV-specific monoclonal antibodies (mAbs) [4, 7, 8]. Reverse-transcriptase polymerase-chain-reaction- (RT-PCR-) based assays have also been developed to identify the presence of the RNA genome of WNV [9, 10]. Both protein and nucleic acid-based assays are being evaluated for identifying WNV or its genome in other mammals, mosquito pools, and avian specimens. However, due to the short viremic phase and low viral count of WNV in human blood and cerebrospinal fluid, protein-based assays such as ELISA have increasingly become the standard methods of detection and diagnosis [7]. Currently, WNV-specific mAbs, recombinant WNV antigens, and other protein-based detecting reagents are produced in mouse hybridoma, mammalian, insect, or bacterial cell cultures [11–13]. The high production costs and limited scalability associated with the bioreactor-based cell culture system may limit their application. The expanding epidemics of WNV demand the development of a technology platform that can rapidly produce reagents to accommodate the detection and diagnostic needs at a low cost.
Plants have been proposed as bioreactors for protein production because of their capacity to generate large volumes of proteins at low cost and their ability to make appropriate posttranslational modification of recombinant proteins [14, 15]. Traditionally, proteins are produced in transgenic plants that require an extensive time period to generate transgenic lines [16]. In contrast, transient expression of a target gene in plant material can produce the target protein within 1 to 2 weeks [17]. The recently developed transient expression systems based on plant viral vectors promote high-level accumulation of foreign protein due to their efficient replication, which results in high copy numbers of transgenes and their mRNA transcripts [18, 19]. An example of a viral-based vector uses tobacco mosaic virus (TMV) and potato virus X (PVX) genomes under the control of plant promoters (the MagnICON system) [20]. Another example is the geminivirus-based expression system: a DNA replicon system derived from the bean yellow dwarf virus (BeYDV) that allows rapid high-yield production of proteins in plants [21]. These plant-based transient expression systems provide the speed and flexibility of bacterial gene expression system coupled with the posttranslational protein modification capability and yield of mammalian cell cultures.
In this study, we demonstrate the feasibility of using plant transient expression systems to produce two groups of protein reagents that are required for the detection and diagnosis of WNV infection: a recombinant antigen derived from the domain III (DIII) of WNV envelope (E) protein and a mAb (E16) that specifically recognizes WNV DIII. Our plant transient expression systems permit high-level expression of WNV DIII antigen and E16 mAb in both Nicotiana benthamiana and lettuce plants. These detection and diagnostic reagents can be easily purified to greater than 95% purity. In addition, our results demonstrate their functional activity and utility in identifying WNV and in detecting human IgM response to WNV infections. Therefore, this study provides proof of principle for using plants as a robust, rapid, and flexible production system for protein reagents for the detection and diagnosis of WNV and can be broadly applied to other arboviruses.
2. Materials and Methods
2.1. Experimental Materials
The DIII protein of Dengue virus serotype 2 (DENV-2) was purchased from Genway Biotech (San Diego, Calif, USA). The E. coli-produced DIII protein of WNV was a gift from Dr. M. Diamond (Washington University School of Medicine, St. Louis, Mo, USA). The West Nile IgM capture ELISA kit was obtained from Diagnostic Automation Inc. (Calabasas, Calif, USA).
2.2. Construction of Plant Expression Vectors
The coding sequence of WNV E protein DIII 13.5 kDa ectodomain (amino acids 296–415 of WNV E protein of the New York 1999 strain, Genbank Accession number AF196835) was optimized in silico with N. benthamiana-optimized codons using an algorithm as described in Villalobos et al. [22]. An 18-bp sequence coding for a hexahistidine (His6) purification tag was added to the C-terminus of the DIII gene. Optimized DIII-His6 sequence was synthesized (DNA 2.0, Menlo Park, Calif, USA) and cloned into the 5′ modules of plant expression vector pICH11599 of the MagnICON system as described previously [20]. The geminiviral vector pBY-HL(hE16-no-KDEL).R for expressing E16, a mAb against WNV E protein [13, 23], was constructed by replacing the E16 heavy chain (HC)-KDEL sequence in vector pBY-HL(hE16).R [24] with the KDEL-less E16 HC sequence. The expression of the resulting pBY-HL(hE16-no-KDEL).R vector produces E16 mAb molecules in which the C-termini of the HC are not attached by the ER-retention signal KDEL.
2.3. Agroinfiltration of N. benthamiana and Lettuce
Plant expression vectors were individually transformed into Agrobacterium tumefaciens GV3101 by electroporation as previously described [25]. Wild-type N. benthamiana and lettuce (L. sativa) plants were grown in a greenhouse with 16/8 hr light/dark cycle at 25°C for 6 weeks. For DIII expression N. benthamiana leaves were co-agroinfiltrated with GV3101 strains containing the DIII 5′ module along with the TMV 3′ module, and an integrase construct, with a syringe as described previously [26]. For E16 expression in N. benthamiana, the syringe-agroinfiltration method was also used to deliver the geminiviral vector pBY-HL(hE16-no-KDEL).R into leaves. Lettuce heads were infiltrated with the GV3101 strain containing the geminiviral vector pBY-HL(hE16-no-KDEL).R for 2 × 5 min under vacuum (100 mbar) as described previously [27].
2.4. Extraction of Total Protein from Plant Leaves
Agroinfiltrated N. benthamiana or lettuce leaves were harvested 3–8 days following infiltration (dpi) for evaluating the temporal pattern of DIII and E16 mAb expression. For other protein analysis, plant leaves were harvested 4 dpi. Total leaf protein was extracted by homogenization in extraction buffer I (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM phenylmethylsufonyl flouride) for DIII antigen or extraction buffer II (PBS, pH 7.4, 1 mM EDTA, 10 mg/mL sodium ascorbate, 10 μg/mL leupeptin, 0.3 mg/mL phenylmethylsufonyl flouride) for E16 mAb using a FastPrep machine (Qbiogene, Carlsbad, Calif, USA) following manufacturers instructions. Extraction buffers I and II were specifically developed based on the biochemical properties of DIII antigen and E16 mAb for the maximal extraction and stability of each target protein, while minimizing the coextraction of plant host proteins. The crude plant extract was clarified by centrifugation at 18,000 ×g for 30 min at 4°C. The concentration of leaf total soluble protein (TSP) was measured by using the Protein Bradford Assay (Hercules, Calif, USA).
2.5. SDS-PAGE and Western Blot Analysis
SDS-PAGE and western blotting were performed as described previously [28]. Protein samples were subjected to 12% or 4–20% gradient SDS-PAGE under reducing (5% v/v β-mercaptoethanol) or nonreducing conditions. Gels were then either stained with Coomassie blue or used to transfer proteins onto PVDF membranes. Horseradish-peroxidase- (HRP-) conjugated antibodies against human-kappa light chain (LC) or gamma heavy chain (HC) (Southern Biotech, Birmingham, Ala, USA) were used for western blot analysis of E16 mAb. For DIII antigen, a rabbit anti-WNV DIII polyclonal antibody and an HRP-conjugated goat anti-rabbit IgG (Southern Biotech, Birmingham, Ala, USA) were used for the analysis.
2.6. Purification of DIII Antigen and E16 mAbs from Plant Tissue
N. benthamiana or lettuce leaves were harvested on 4 dpi and extracted with a blender in extraction buffer I (for DIII) or extraction buffer II (for E16), and the extract was clarified by filtering through Miracloth, followed by centrifugation at 17,700 ×g for 30 min at 4°C. For DIII purification, the pH of the clarified extract was adjusted to pH 5.0 and followed by centrifugation at 17,700 × g for 30 min at 4°C to precipitate the most abundant plant protein, the photosynthetic enzyme RuBisCo, and other host proteins. The supernatant was adjusted to pH 8.0 and respun to remove residue precipitates. The supernatant was then applied to a Ni-NTA His-Bind column and purified by metal chelation chromatography according to a protocol supplied by the manufacturer (Novagen, Madison, Wis, USA). The column eluate containing the purified DIII antigen was buffer-exchanged to PBS. For E16 purification, the clarified leaf extract was filtered through a 0.2-micron filter and loaded directly to a MAbSelect Protein A column (GE Healthcare, Piscataway, NJ, USA). The column was eluted with 50 mM sodium citrate, pH 2.5. Tris-base (1 M) was added immediately to the eluate to attain a final pH of 7.0. The purity of DIII or E16 was determined by quantitating Coomassie blue—stained protein bands on SDS-PAGE using a densitometer as described previously [26].
2.7. ELISA
E16 quantitative ELISA was designed to detect the assembled form of E16 mAb (with both LC and HC) as described previously [23]. Plates were coated with a goat anti-human-gamma HC antibody (1 : 2,000 dilution, Southern Biotech, Birmingham, Ala, USA). After incubation with plant protein extract, an HRP-conjugated anti-human-kappa LC antibody (1 : 10,000 dilution) was used as the detection antibody. A mammalian cell-culture-derived E16 was used as a reference standard [13]. The plates were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (KPL Inc, Gaithersburg, Md, USA).
DIII expression analysis was performed by coating plates with a monoclonal mAb against WNV DIII (1 : 2,000 dilution) [13]. Clarified plant extract was then applied to the wells followed by detection with a rabbit anti-WNV DIII antibody (1 : 10,000 dilution) and an HRP-conjugated goat anti-rabbit IgG antibody (1 : 10,000 dilution).
Binding specificity of plant-derived E16 to DIII protein of various flaviviruses was performed using a previous published method [29]. An E. coli-derived WNV DIII [29] or DENV-2 DIII (100 uL, 2 ug/mL) was immobilized on microtiter plates. An HRP-conjugated anti-human-kappa LC antibody (1 : 10,000 dilution) was used as the detection antibody. A mammalian cell-culture-derived E16 and a generic human IgG (Southern Biotech, Birmingham, Ala, USA) were used as the positive and negative control, respectively.
The WNV IgM capture ELISA was performed according to the procedures provided by the manufacturer (Diagnostic Automation Inc. Calabasas, Calif, USA). Briefly, E16 mAbs derived from N. benthamiana and lettuce were conjugated to HRP with a commercial Lightning-Link HRP conjugation kit (Innova Biosciences, Cambridge, UK). Human WNV IgM positive control (PC) or negative control (NC) serum was incubated in microtiter wells coated with anti-human IgM antibodies. This was followed by incubation with a negative control antigen (NCA), a kit-supplied WNV recombinant antigen protein (WNRA), or plant-derived WNV DIII antigen (50 uL, 1 : 25 dilution). After washing, wells were incubated with either a kit-supplied WNRA-specific antibody (anti-WNRA) labeled with HRP or with the HRP-conjugated plant-derived E16 (50 uL, 1 : 100 dilution). High concentrations of plant-derived DIII and E16 mAb were used to saturate their respective target-binding sites, ensuring all WNV-specific IgM molecules captured on the plate are detected. After further washing, TMB substrate was used to develop the plate. The Immune Status Ratio (ISR) is calculated by dividing the average absorbance of the WNRA or plant-derived DIII antigen by the average absorbance of NCA (WNRA/NCA or Plant DIII/NCA). The quality control specifications for this ELISA are PC ISR > 5.66 and NC ISR < 4.47.
2.8. Flow Cytometry of Yeast Displaying WNV E Protein
Analysis of the detection of WNV E protein by plant-derived E16 was performed using a yeast strain that expresses WNV E protein on its surface. The staining of yeast cells and flow cytometry analysis were performed as described previously [13]. Briefly, yeast cells were grown to log phase, followed by an additional 24 h in tryptophan-free media containing 2% galactose to induce WNV E protein expression. The yeast cells were then incubated with E16 mAbs derived from N. benthamiana or lettuce. A generic human IgG was used as a negative control mAb. The yeast cells were stained with a goat anti-human secondary antibody conjugated to Alexa Fluor 488 (Invitrogen, Carlsbad, Calif, USA). Subsequently, the yeast cells were analyzed on a BD FACSCalibur flow cytometer (Franklin Lakes, NJ, USA).
3. Results
3.1. Expression of WNV DIII Recombinant Antigen with Transient Expression Vectors in N. benthamiana Plants
As an initial test of the feasibility of using plant to produce WNV diagnostic reagent, we examined the expression of DIII antigen in the plant host of N. benthamiana. This is a plant species that is related to the common tobacco plants and has been used extensively in producing recombinant proteins [30]. Western blot analysis showed that DIII antigen was detected in the plant sample infiltrated with DIII construct with the expected molecular weights of 13.5 kDa along with an E. coli-produced DIII positive control (Figure 1(a)). This band was not detected in negative control leaf samples that were infiltrated with infiltration buffer, confirming the specificity of the DIII band (Figure 1(a)). The slightly smaller molecular weight of plant-derived DIII reflects the fact that it lacks the extra N-terminal peptide tags (His-Tag and T7-Tag) in the E. coli-derived DIII. Western blot confirmed the stability of plant-derived DIII as only the expected band was observed. The production of DIII was confirmed by a sandwich ELISA using two WNV specific antibodies. ELISA results also indicated that DIII reached a high expression level within 4 days after infiltration, with an average accumulation of 1% TSP (Figure 2), which corresponds to ~0.1 mg/g fresh leaf weight (FLW). This level is comparable to the high range of accumulation of other antigens in plants [25] and is the highest expression level of any flavivirus antigen ever reported in plants. This convincingly demonstrates that plants can rapidly express DIII antigen at high levels. Furthermore, in contrast to E. coli-produced DIII which requires a cumbersome solubilization and refolding process [11], plant-derived DIII is produced as a soluble protein and can be easily purified to greater than 95% purity (data not shown) by a metal chelation chromatography.
Figure 1.
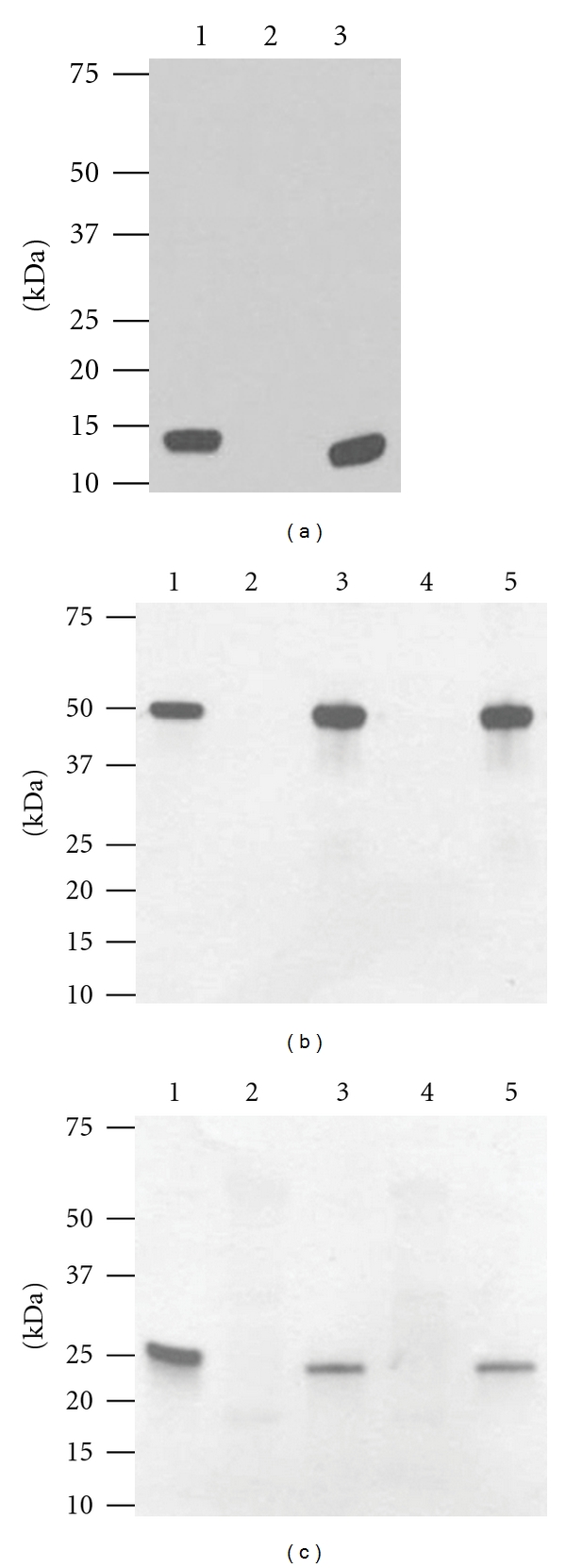
Western blot analysis of WNV DIII antigen and E16 mAb expression in N. benthamiana and lettuce. (a) WNV DIII. Leaf protein extracts were separated on a 4–20% SDS-PAGE gradient gel and transferred onto PVDF membranes. The membranes were probed with a rabbit anti-WNV DIII polyclonal antibody. Lane 1: E. coli-derived DIII standard; lane 2: protein extract from leaves infiltrated with buffer (negative control); lane 3: extract from DIII construct infiltrated N. benthamiana leaves. (b) and (c) Expression of E16. Wild-type laboratory-grown lettuce or N. benthamiana were infiltrated with dual-replicon geminiviral vector pBY-HL (hE16-no-KDEL). R and harvested on 4 dpi. Total leaf protein extracts were separated on 4–20% SDS-PAGE gradient gels under reducing conditions and transferred to PVDF membranes. The membranes were incubated with a goat anti-human-gamma chain antibody to detect HC (b) or a goat anti-human-kappa chain antibody to detect LC (c). Lane 1: human IgG reference standard; lane 2: extract from lettuce leaves infiltrated with buffer (lettuce negative control); lane 3: protein samples from lettuce infiltrated with geminiviral vector pBY-HL(hE16-no-KDEL). R; lane 4: extract from buffer-infiltrated N. benthamiana leaves (negative control); Lane 5: N. benthamiana leaf protein extract infiltrated with pBY-HL(hE16-no-KDEL). R vector.
Figure 2.
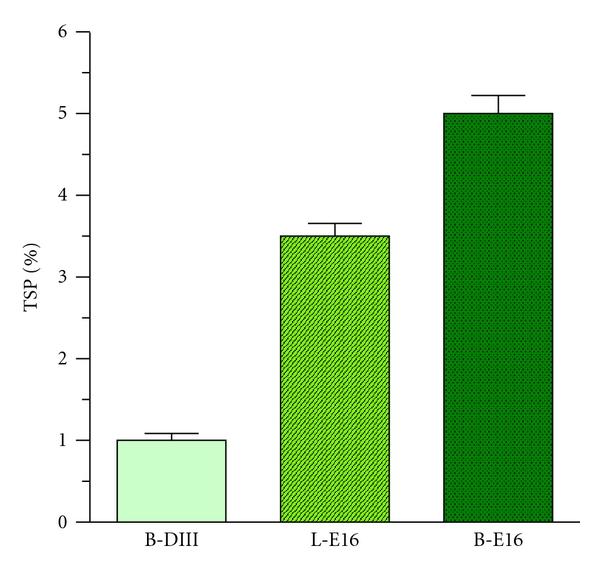
Accumulation of WNV DIII antigen and E16 mAbs in N. benthamiana and lettuce. Total proteins from plant leaves infiltrated with DIII or E16 expression vectors were extracted on 4 dpi and analyzed by ELISA with E. coli-produced WNV DIII or mammalian cell-produced E16 as the reference standard. For DIII ELISA, a monoclonal mAb against WNV DIII [13] was the capture antibody, and a rabbit anti-WNV DIII antibody was the detection antibody. For E16, goat anti-human gamma- and kappa-chain antibodies were used as capture and detection reagents, respectively, to detect the assembled form of E16 mAb. Mean ± SD of samples from three independent infiltrations are presented. B-DIII: WNV DIII produced in N. benthamiana; L-E16: lettuce-produced E16; B-E16: N. benthamiana-derived E16.
3.2. High-Level Expression and Assembly of a mAb against WNV in Wild-Type Plants
To further demonstrate the utility of plants as a platform for production of protein reagents, we examined the production of a mAb, a representative of another important group of relevant molecules used in the detection and diagnosis of WNV infection. To compare the expression of E16 mAb in two species of plant hosts (N. benthamiana and lettuce), we used the BeYDV-based geminiviral replicon vectors. In contrast to the MagnICON system, these vectors have been shown to direct transgenic protein expression in both tobacco and lettuce [24]. The Agrobacterium strain containing replicons of the LC- and HC-coding sequences of E16 were infiltrated into laboratory-grown lettuce and N. benthamiana leaves. Western blot analysis revealed that the LC and HC of E16 were expressed in both lettuce and N. benthamiana leaves with the expected molecular weights of 25 kDa and 50 kDa, respectively, (Figures 1(b) and 1(c)). The full tetrameric (2HC + 2LC) assembly of these mAbs was demonstrated by western blot (data not shown) and Coomassie blue staining analysis (Figure 3, Lanes 5–7) under nonreducing conditions. The assembly of pHu-E16 was corroborated by an ELISA that detects the assembled form of E16 (HC capture, LC probe, Figure 2). Since only the expected bands were observed on western blots, the results established that E16 is stable during biosynthesis in both plant hosts and proteolytic degradation of the LC or HC did not occur. Further ELISA analysis also showed that E16 was produced rapidly in both plant species and reached a high level of accumulation to 3.5% (in lettuce) or 5% TSP (in N. benthamiana) on 4 dpi (Figure 2). This expression level is comparable to the highest expression level of mAbs reported by geminiviral vectors in both lettuce and tobacco [24] and by MagnICON vectors in tobacco [26]. Overall, these results demonstrate that fully assembled E16 mAb can be produced rapidly at high levels in both lettuce and N. benthamiana plant hosts.
Figure 3.
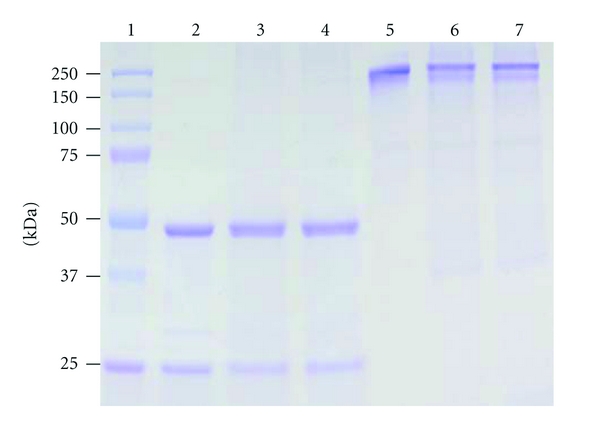
Purification of E16 mAbs produced in lettuce or N. benthamiana. Purified E16 mAbs were analyzed on a 12% SDS-PAGE gel using either reducing (Lanes 2–4) or nonreducing (Lanes 5–7) sample buffer. Lane 1: Molecular weight marker; lanes 2 and 5: mammalian cell-derived E16 as reference standard; lanes 3 and 6: E16 purified from lettuce leaves; lanes 4 and 7: purified E16 from N. benthamiana. One representative of several independent experiments is shown.
3.3. Purification of E16 mAbs from Plant Tissue
To examine whether plant-derived E16 mAbs can be recovered from plant tissue effectively, we extracted and purified the mAbs from infiltrated lettuce and N. benthamiana leaves using an affinity chromatography method based on Protein A resin. Coomassie blue staining analysis of SDS-PAGE showed that E16 mAbs produced in lettuce and N. benthamiana can be purified to >95% purity with intact HC and LC using this single-affinity chromatography step (Figure 3). A similar analysis under nonreducing conditions confirmed that mAbs produced in both plant hosts remained fully assembled after purification (Figure 3 lanes 5–7). These results demonstrated that E16 mAb not only can be rapidly produced at high levels in two host plant species but also can be isolated and purified to high purity using a facile purification method.
3.4. Plant-Derived DIII WNV Antigen and mAbs Are Functional in Detection and Diagnosis of WNV Infection
Functional characterizations were performed to determine if DIII antigen and E16 mAbs produced in plants retained their biological activities and can be used in detection and diagnosis assays for WNV infection. As shown in Figures 1(a) and 2, plant-derived DIII antigen retains the ability to bind antibodies that specifically recognize DIII of WNV E protein. This binding specificity was confirmed by the lack of binding of plant-derived DIII to other non-DIII-specific mAbs (data not shown).
One of the issues facing the current immunobased WNV diagnosis is the cross-reactivity of antibodies against WNV with other flaviviruses. The E16 mAb produced from mammalian cells binds to a specific region of WNV DIII and does not cross-react with DIII of other flaviviruses [13]. To examine the binding specificity of plant-derived E16, various concentrations of this mAb were incubated with either DIII of WNV or DIII of DENV-2 that was immobilized on an ELISA plate. As shown in Figure 4, the binding to WNV DIII increased with the concentration of lettuce or N. benthamiana-derived E16 in the reaction in a similar manner as the mammalian cell-derived E16 positive control. In contrast, none of the E16s showed specific binding to DIII of DENV-2 (Figure 4). The negative control IgG (a generic human IgG) showed no specific binding to either WNV or DENV-2 DIII (Figure 4). These results indicate that the specific avidity for WNV DIII is retained by the plant-derived E16s. This high specificity makes it a valuable reagent in obtaining unambiguous diagnostic results for detecting WNV and WNV infection.
Figure 4.
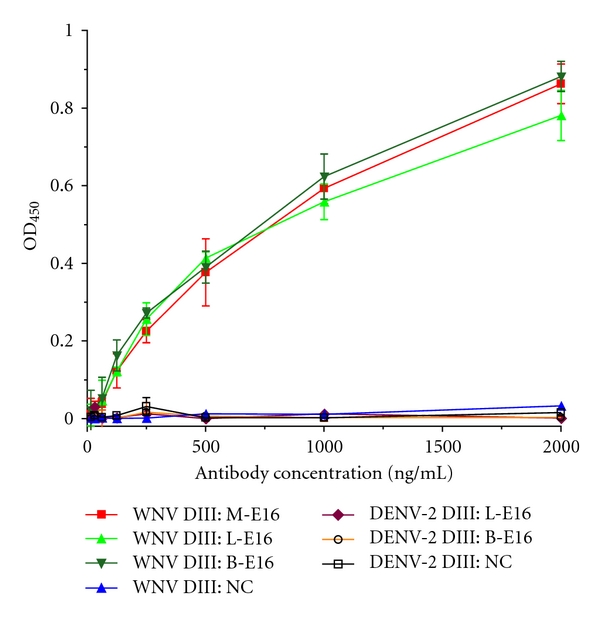
Highly specific binding of plant-derived E16 to WNV DIII antigen. WNV DIII or DENV-2 DIII antigen produced in E. coli was immobilized on an ELISA plate and incubated with increasing concentrations of lettuce or N. benthamiana-derived E16 mAb (L-E16 or B-E16), mammalian cell-derived E16 (M-E16, positive control), or a negative control generic human IgG (NC). A HRP-conjugated anti-human IgG was used to detect mAbs bound to DIII antigens. Mean ± SD of OD450 from three independent experiments are presented.
We first examined the application of plant-derived E16 mAbs in detecting the presence of WNV antigens by using a flow cytometry assay. In this assay, WNV E protein is displayed on the surface of yeast, and the ability of mAbs in detecting WMV E protein is measured by flow cytometry [13]. Our analysis showed that the percentage of yeast cells expressing WNV E protein and the mean fluorescence intensity of binding by lettuce or N. benthamiana-derived E16 were significantly higher compared to the negative control antibodies (Figure 5). This result indicates that plant-derived E16s are effective in specifically detecting WNV proteins. We further demonstrated the utility of plant-derived WNV DIII antigen and E16 mAb in a WNV IgM capture ELISA as another example of their potential application as diagnostic reagents. In the original commercial kit for diagnosis of WNV infection, controls and serum samples are incubated in microtiter wells which have been coated with anti-human IgM antibodies. The microtiter wells are then incubated with WNV recombinant antigen (WNRA) or a negative control antigen (NCA) and followed by incubation with WNRA-specific antibody labeled with HRP. The presence of IgMs against WNV is determined by whether the ratio of the absorbance of the WNRA and the NCA is above a preset threshold. In our modified ELISA, plant-derived E16 was conjugated to HRP first and then used to replace the WNRA-specific antibody in the original kit. Accordingly, WNRA was replaced by our plant-derived DIII antigen in the modified assay. Our results showed that the plant-derived DIII antigen and E16 mAb effectively differentiated the WNV human IgM-positive serum from the negative control serum in a similar manner as the reagents in the commercial kit (Figure 6). The ISRs of our modified assay for the negative control serum are 1.09 (lettuce-derived E16) and 1.24 (N. benthamiana-E16), which are similar to that of the original assay (1.61) and well within the required ISR range (<4.47). Similarly, ISR for the WNV IgM-positive serum is comparable between the modified and original assay (8.68 and 8.98 versus 13.48), which are also within the required specification range (>5.66). The positive IgM control serum showed a lower absorbance in the plant-derived DIII/E16 samples than that of WNRA/anti-WNRA (Figure 6). Since both WNRA and plant-produced DIII were added to the reaction in concentrations high enough to saturate all binding sites of WNV-specific IgMs that were captured on the plate by anti-IgM antibodies, it is unlikely that this is caused by the concentration difference of the two antigens. Instead, the potential difference in antigen-binding site and/or valency between the reference standard mAb and plant-derived E16 mAb is a more likely factor in causing the observed difference. It also remains possible that this result corroborates the previous observation that WMV DIII-specific IgM titers in the sera of infected mammals are generally lower than those of WNV E protein [29]. We are currently expressing a full-length WNV E protein in plants to address this issue. Nonetheless, this example clearly demonstrated the effectiveness of plant-derived antigen and mAb as reagents for detection and diagnosis of WNV infection and suggested their potential applications in other diagnostic assays.
Figure 5.
Identification of WNV E protein displayed on the cell surface of yeast by plant-derived E16 mAbs. Lettuce (L-E16) or N. benthamiana-produced E16 mAb (B-E16), mammalian cell-derived E16 (M-E16, positive control), or a generic human IgG (NC, negative control) was used to stain yeast cells displaying WNV E protein, which were then processed by flow cytometry. Representative data from three independent experiments are shown.
Figure 6.
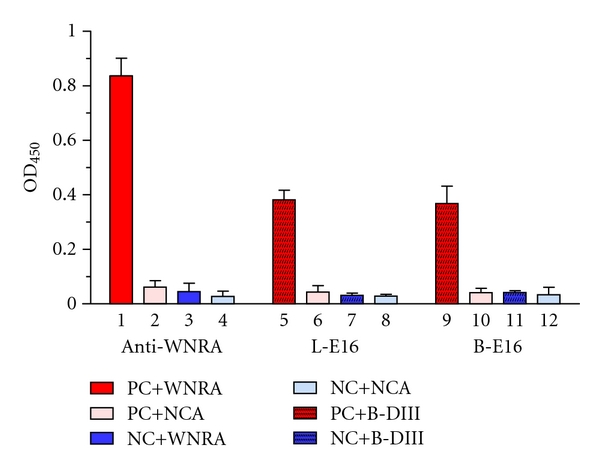
WNV IgM capture ELISA with plant-derived DIII antigen and E16 mAbs. Columns 1 and 2 (positive controls): WNV IgM-positive serum (PC) detected by kit-supplied positive control antigen WNRA (1) or negative control antigen NCA (2) with the kit-supplied WNRA-specific antibody (Anti-WNRA); Columns 3 and 4 (negative controls): WNV IgM negative serum (NC) detected by WNRA (3) or NCA (4) with Anti-WNRA; Columns 5 and 6: PC detected by N. benthamiana-derived DIII antigen (B-DIII) (5) or NCA (6) with lettuce-derived E16 (L-E16); Columns 7 and 8: NC detected by B-DIII (7) or NCA (8) with L-E16; Columns 9 and 10: PC detected by B-DIII (9) or NCA (10) with N. benthamiana-derived E16 (B-E16); Columns 11 and 12: NC detected by B-DIII (11) or NCA (12) with B-E16.
4. Discussion
The rapidity of the spread of WNV and the potential threat to bird, mammal, and human populations demand the development of a technology platform that can rapidly produce a variety of protein reagents, flexibly and at low cost, to support the need of detection and diagnosis sufficient to address the potential public health crisis. Here, we investigated the feasibility of using plants to produce a WNV antigen and a mAb against WNV to address the cost and scalability issues associated with mouse hybridoma mammalian, insect, or bacterial cell cultures. Our results demonstrated that WNV DIII antigen and E16 mAb can be rapidly produced at high levels in two host plant species. Furthermore, these proteins can be easily isolated to high purity with simple extraction and purification methods and they retain the functionality and specificity of their original counterparts.
Plants are suitable for antigen and mAb production because they can produce large volumes of protein at low cost and be rapidly scaled up for commercial production without the high-capital investment associated with bioreactor facilities for mammalian, insect, or bacterial cell cultures [14, 15, 31, 32]. The WNV E glycoprotein is essential for membrane fusion and mediates binding to cellular receptors, and is a major target of host antibody responses [33]. DIII of WNV E protein contains the cellular receptor-binding motifs and the majority of the neutralizing epitopes that induce strong host antibody responses and/or protective immunity [13]. As a result, E protein and DIII have been extensively explored as WNV vaccine candidates and as detection and diagnostic reagents [7]. The WNV DIII protein has been produced in insect cell and bacterial cultures [11, 12]. These culture systems are difficult to scale up for large-scale protein production. In addition, production of recombinant DIII in bacterial cultures requires a cumbersome solubilization and refolding process due to the formation of inclusion bodies [11]. In contrast, DIII antigen was expressed rapidly in N. benthamiana plant as a soluble protein and accumulated to 1% of TSP or 0.1 mg/g FLW, the highest expression level of any flavivirus antigen ever reported in plants. In addition, plant-derived DIII can be easily purified to >95% purity without the needs of solubilization and refolding.
We previously investigated the possibility of producing E16 as a post-exposure therapeutic mAb in N. benthamiana with the TMV and PVX-based MagnICON system. In that investigation, the “KDEL” tetrapeptide ER-retention signal was added to the C-terminus of the HC [23], and/or E16 was expressed in “humanized” plant lines that can add mammalian glycoforms to mAbs (Q. Chen, unpublished results). These extra procedures were performed to avoid the production of plant-specific glycans, which may trigger a potentially harmful immune response in human patients. Since diagnostic reagents are used in vitro, the concern for the potential adverse host immune response becomes irrelevant. Thus, in this study we examined the feasibility of using wild-type plants and HC without the extra-KDEL ER-retention peptide to produce E16. Furthermore, instead of using the MagnICON system of the previous study, we tested the ability of geminiviral replicon vectors for E16 production because it can direct recombinant protein expression in both tobacco and lettuce [24]. Our results showed that the KDEL-less E16 can be produced in wild-type plants with geminiviral vectors and accumulated to a level comparable to the highest expression level for mAbs in plants ever reported. The ease of using wild-type plants to produce E16 not only simplifies the experimental procedure, but also minimizes biosafety and regulatory concerns and costs associated with transgenic crops and genetically modified plants [15]. Furthermore, E16 mAbs produced from both wild-type N. benthamiana and lettuce can be easily purified to >95% purity and retain their highly-specific antigen binding avidity.
In addition to wild-type N. benthamiana, we also explored the possibility of using laboratory-grown lettuce to produce E16. Similar to N. benthamiana, lettuce is a robust-growing plant that produces large quantities of biomass rapidly. In contrast to tobacco, it produces negligible quantities of phenolics and alkaloids and, thus, can potentially simplify the protein purification process and reduce the overall cost of goods in commercial production. Our results demonstrated that lettuce-produced E16 mAb has equivalent structure and functionality as N. benthamiana-derived E16. Previously we showed that mAbs can be expressed in grocery store-bought lettuce [24]. This potentially would allow us to have access to unlimited quantities of inexpensive plant material for the large-scale commercial production of E16 as a detection and diagnostic reagent. Overall, the capability of performing posttranslational modifications by plant cells and the availability of the glycoengineered plant lines to produce recombinant proteins with mammalian glycoforms provide our plant system the advantage over bacterial cultures in producing mAbs and antigens that require posttranslational modification. Moreover, the rapid high-level production and assembly of E16 and a simple current good manufacture-practice- (cGMP-) compliant purification scheme—convincingly demonstrate the viability of this system for large-scale cost-effective production of mAbs compared to hybridoma and mammalian cell cultures.
We further demonstrated the utility of plant-derived antigen and mAbs in detection of WNV or host immune responses to WNV infection in a flow-cytometry-based and a WNV IgM capture ELISA assay. The results of these examples clearly indicated their value in the detection of WNV and diagnosis of WNV infection and suggest their potential application in other protein-based assays. One critical issue of immunobased WNV diagnosis is the cross-reactivity of antibodies among flaviviruses. The high specificity of plant-derived E16s may provide us a valuable tool in addressing such problems. For example, plant E16 can be used in a VecTest-like WNV antigen assay to rapidly detect WNV infection in wild bird and mosquito populations [34]. Since plant E16 does not recognize DIII protein from other flaviviruses, this high specificity will improve the accuracy of the current assay and reduce ambiguity. Sotelo and colleagues have recently developed a new epitope-blocking ELISA that has demonstrated utility in detecting WNV infection in a wide range of hosts, including humans, birds, and other mammals [35]. This ELISA utilizes a mouse hybridoma cell-derived neutralizing mAb that binds WNV E protein and requires very small volumes of sera, making it feasible to directly test small-size birds and mammals without harming their health. As a result, this new method promises to be helpful for both disease diagnosis and surveillance. Since plant-derived E16 is a neutralizing mAb that binds to WNV E protein, we speculate that its high specificity to WNV DIII will enhance the specificity of this assay. In addition, using plant-derived E16 for this assay will address the issue of production scalability of mouse hybridoma cells and greatly reduce the cost of mAb production.
The research of our laboratory and others has demonstrated the ability of plants in producing a variety of recombinant proteins at low cost [15, 31, 32, 36, 37]. This study extends the utility of plant expression system to the production of mAbs and antigens as detection and diagnostic reagents for arboviruses. One of the concerns of plant biotechnology has been whether the product yield can be high enough to allow large-scale production and advance the technology beyond the proof-of-principle stage. While this was a valid issue for traditional plant expression systems, the high-level product yield of new plant expression systems have suggested that such concern is no longer necessary. These new expression systems such as the MagnICON and geminiviral expression systems used in this study have allowed high and consistent level of target protein accumulation in plants sufficient for commercial-scale manufacturing [14, 15, 21]. For example, several of pharmaceutical proteins produced by these plant expression systems with similar product yield as WNV DIII and E16 mAb have not only been produced on a large scale under the United States Food and Drug Administration's (FDA) cGMP regulation but also have been tested in late-stage human clinical trials [14, 15, 38]. The high-level product yield of DIII antigen and E16 mAb and their simple cGMP-compliant purification schemes, demonstrate the viability of our plant production system for their large-scale cost-effective production and suggest a promising future beyond the proof-of-principle stage.
In addition to promoting high product yield, one of the other advantages of plant-transient expression systems is the rapid speed of target protein production [17]. With both MagnICON and geminiviral expression systems, our antigen and mAbs were produced within a week of vector infiltration. These rapid and high protein accumulation levels provide the flexibility and versatility of the plant production system that are superior over other production systems in accommodating the demands of new reagents by the fast-evolving diagnostic assays. For example, if a novel assay has been approved for viral detection and diagnosis, the plant-transient expression system can be rapidly adjusted to produce the new reagents within one to two months. This flexibility and versatility also give the plant production system an advantage in producing a virtually unlimited number of protein detection and diagnostic reagents for not only WNV but for many other arboviruses, such as dengue virus, Japanese encephalitis virus, St. Louis encephalitis virus, and yellow fever virus.
5. Conclusions
This research demonstrated the effective production of functional protein reagents in plants for the detection of WNV and diagnosis of WNV infection. The robustness, cost effectiveness, scalability, and flexibility of the plant system will make it an attractive platform for the production of detection and diagnostic reagents for a broad range of arboviruses.
Acknowledgments
The authors thank Dr. M. Diamond for the gift of E. coli-produced WNV DIII protein. The critical reading of this paper by F. Farmer is appreciated. This research was supported by Grant number U01AI075549 from NIAID of NIH to Q. Chen and a SSE grant from College of Technology and Innovation of Arizona State University to Q. Chen. J. He and H. Lai contributed equally to this publication.
References
- 1.Hubálek Z, Halouzka J. West Nile fever—a reemerging mosquito-borne viral disease in Europe. Emerging Infectious Diseases. 1999;5(5):643–650. doi: 10.3201/eid0505.990505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bode AV, Sejvar JJ, Pape WJ, Campbell GL, Marfin AA. West Nile Virus disease: A descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clinical Infectious Diseases. 2006;42(9):1234–1240. doi: 10.1086/503038. [DOI] [PubMed] [Google Scholar]
- 3.Diamond MS, Klein RS. A genetic basis for human susceptibility to West Nile virus. Trends in Microbiology. 2006;14(7):287–289. doi: 10.1016/j.tim.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Wu J, Li Y, Li F, Njoo H. Rapid and accurate in vitro assays for detection of West Nile virus in blood and tissues. Transfusion Medicine Reviews. 2009;23(2):146–154. doi: 10.1016/j.tmrv.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Linnen JM, Deras ML, Cline J, et al. Performance evaluation of the PROCLEIX® West Nile virus assay on semi-automated and automated systems. Journal of Medical Virology. 2007;79(9):1422–1430. doi: 10.1002/jmv.20930. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Anne Hapip C, Liu B, Fang CT. Highly sensitive TaqMan RT-PCR assay for detection and quantification of both lineages of West Nile virus RNA. Journal of Clinical Virology. 2006;36(3):177–182. doi: 10.1016/j.jcv.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Prince HE, Hogrefe WR. Assays for detecting West Nile Virus antibodies in human serum, plasma, and cerebrospinal fluid. Clinical and Applied Immunology Reviews. 2005;5(1):45–63. [Google Scholar]
- 8.Sambol AR, Hinrichs SH. Evaluation of a new West Nile virus lateral-flow rapid IgM assay. Journal of Virological Methods. 2009;157(2):223–226. doi: 10.1016/j.jviromet.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Parida M, Posadas G, Inoue S, Hasebe F, Morita K. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. Journal of Clinical Microbiology. 2004;42(1):257–263. doi: 10.1128/JCM.42.1.257-263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanciotti RS, Kerst AJ, Nasci RS, et al. Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. Journal of Clinical Microbiology. 2000;38(11):4066–4071. doi: 10.1128/jcm.38.11.4066-4071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu JJH, Rajamanonmani R, Li J, Bhuvananakantham R, Lescar J, Ng ML. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. Journal of General Virology. 2005;86(2):405–412. doi: 10.1099/vir.0.80411-0. [DOI] [PubMed] [Google Scholar]
- 12.Alonso-Padilla J, de Oya NJ, Blázquez A-B, Escribano-Romero E, Escribano JM, Saiz J-C. Recombinant West Nile virus envelope protein E and domain III expressed in insect larvae protects mice against West Nile disease. Vaccine. 2011;29(9):1830–1835. doi: 10.1016/j.vaccine.2010.12.081. [DOI] [PubMed] [Google Scholar]
- 13.Oliphant T, Engle M, Nybakken GE, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nature Medicine. 2005;11(5):522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faye L, Gomord V. Success stories in molecular farming—a brief overview. Plant Biotechnology Journal. 2010;8(5):525–528. doi: 10.1111/j.1467-7652.2010.00521.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen Q. Expression and manufacture of pharmaceutical proteins in genetically engineered horticultural plants. In: Mou B, Scorza R, editors. Transgenic Horticultural Crops: Challenges and Opportunities—Essays by Experts. Boca Raton, Fla, USA: Taylor & Francis; 2011. pp. 86–126. [Google Scholar]
- 16.Davies HM. Commercialization of whole-plant systems for biomanufacturing of protein products: evolution and prospects. Plant Biotechnology Journal. 2010;8(8):845–861. doi: 10.1111/j.1467-7652.2010.00550.x. [DOI] [PubMed] [Google Scholar]
- 17.Komarova TV, Baschieri S, Donini M, Marusic C, Benvenuto E, Dorokhov YL. Transient expression systems for plant-derived biopharmaceuticals. Expert Review of Vaccines. 2010;9(8):859–876. doi: 10.1586/erv.10.85. [DOI] [PubMed] [Google Scholar]
- 18.Cañizares MC, Nicholson L, Lomonossoff GP. Use of viral vectors for vaccine production in plants. Immunology and Cell Biology. 2005;83(3):263–270. doi: 10.1111/j.1440-1711.2005.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lico C, Chen Q, Santi L. Viral vectors for production of recombinant proteins in plants. Journal of Cellular Physiology. 2008;216(2):366–377. doi: 10.1002/jcp.21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giritch A, Marillonnet S, Engler C, et al. Rapid high-yield expression of full-size IgG antibodies in plants coinfected with noncompeting viral vectros. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(40):14701–14706. doi: 10.1073/pnas.0606631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q, He J, Phoolcharoen W, Mason HS. Geminiviral vectors based on bean yellow dwarf virus for production of vaccine antigens and monoclonal antibodies in plants. Human Vaccines. 2011;7(3):331–338. doi: 10.4161/hv.7.3.14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S. Gene Designer: a synthetic biology tool for constructuring artificial DNA segments. BMC Bioinformatics. 2006;7, article no. 285 doi: 10.1186/1471-2105-7-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai H, Engle M, Fuchs A, et al. Monoclonal antibody produced in plants efficiently treats West Nile virus infection in mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2419–2424. doi: 10.1073/pnas.0914503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai H, He J, Engle M, et al. Robust production of virus-like particles and monoclonal antibodies with geminiviral replicon vectors in lettuce. doi: 10.1111/j.1467-7652.2011.00649.x. Plant Biotechnology Journal. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Z, Chen Q, Hjelm B, Arntzen C, Mason H. A DNA replicon system for rapid high-level production of virus-like particles in plants. Biotechnology and Bioengineering. 2009;103(4):706–714. doi: 10.1002/bit.22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Z, Phoolcharoen W, Lai H, et al. High-level rapid production of full-size monoclonal antibodies in plants by a single-vector DNA replicon system. Biotechnology and Bioengineering. 2010;106(1):9–17. doi: 10.1002/bit.22652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Negrouk V, Eisner G, Lee HI, Han K, Taylor D, Wong HC. Highly efficient transient expression of functional recombinant antibodies in lettuce. Plant Science. 2005;169(2):433–438. [Google Scholar]
- 28.Santi L, Batchelor L, Huang Z, et al. An efficient plant viral expression system generating orally immunogenic Norwalk virus-like particles. Vaccine. 2008;26(15):1846–1854. doi: 10.1016/j.vaccine.2008.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliphant T, Nybakken GE, Austin SK, et al. Induction of epitope-specific neutralizing antibodies against West Nile virus. Journal of Virology. 2007;81(21):11828–11839. doi: 10.1128/JVI.00643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conley AJ, Zhu H, Le LC, et al. Recombinant protein production in a variety of Nicotiana hosts: a comparative analysis. Plant Biotechnology Journal. 2011;9(4):434–444. doi: 10.1111/j.1467-7652.2010.00563.x. [DOI] [PubMed] [Google Scholar]
- 31.Chen Q. Turning a new leaf. European Biopharmaceutical Review. 2011;2(56):64–68. [Google Scholar]
- 32.Chen Q. Expression and purification of pharmaceutical proteins in plants. Biological Engineering. 2008;1(4):291–321. [Google Scholar]
- 33.Crill WD, Chang GJJ. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. Journal of Virology. 2004;78(24):13975–13986. doi: 10.1128/JVI.78.24.13975-13986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panella NA, Burkhalter KL, Langevin SA, et al. Rapid West Nile virus antigen detection. Emerging Infectious Diseases. 2005;11(10):1633–1635. doi: 10.3201/eid1110.040394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sotelo E, Llorente F, Rebollo B, et al. Development and evaluation of a new epitope-blocking ELISA for universal detection of antibodies to West Nile virus. Journal of Virological Methods. 2011;174(1-2):35–41. doi: 10.1016/j.jviromet.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Chen Q, Tacket CO, Mason H, et al. Subunit vaccines produced using plant biotechnology. In: Levine MM, editor. New Generation Vaccines. 4th edition. New York, NY, USA: Informa Healthcare; 2009. pp. 306–315. [Google Scholar]
- 37.Phoolcharoen W, Bhoo SH, Lai H, et al. Expression of an immunogenic Ebola immune complex in Nicotiana benthamiana. Plant Biotechnology Journal. 2011;9(7):807–816. doi: 10.1111/j.1467-7652.2011.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai H, Chen Q. Bioprocessing of plant-derived virus-like particles of Norwalk virus capsid protein under current Good Manufacture Practice regulations. doi: 10.1007/s00299-011-1196-6. Plant Cell Reports. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]