

Abstract
Bacterial artificial chromosome (BAC) clones have proven invaluable for genetic manipulation of herpesvirus genomes. BAC cloning can also be useful for capturing representative genomes that comprise a viral stock or mixture. The Towne live attenuated cytomegalovirus vaccine was developed in the 1970s by serial passage in cultured fibroblasts. Although its safety, immunogenicity, and efficacy have been evaluated in nearly a thousand human subjects, the vaccine itself has been little studied. Instead, genetic composition and in vitro growth properties have been inferred from studies of laboratory stocks that may not always accurately represent the viruses that comprise the vaccine. Here we describe the use of BAC cloning to define the genotypic and phenotypic properties of viruses from the Towne vaccine. Given the extensive safety history of the Towne vaccine, these BACs provide a logical starting point for the development of next-generation rationally engineered cytomegalovirus vaccines.
1. Introduction
Cytomegalovirus (CMV) is a common cause of intrauterine infection, affecting 0.5–3% of all newborns worldwide. Each year in the United States, an estimated 8,000 babies are born with symptomatic CMV infections that result in serious complications including hearing loss, vision impairment, and varying degrees of mental retardation [1]. Another 6% to 23% of infants born infected but asymptomatic at birth will develop sensorineural hearing loss [2]. Overall, congenital CMV infections account for one fourth of all cases of sensorineural hearing loss in the United States [3]. This disease burden could be prevented by an effective vaccine [4].
The Towne vaccine is a live vaccine attenuated by extensive passage in human fibroblasts [5]. It has been used in nearly 1000 volunteers with no serious side effects [6]. In renal transplant patients, Towne reduced CMV-associated disease but did not prevent recipients from acquiring CMV infections [7]. When used at a very low dose, it also failed to protect immunocompetent mothers from acquiring CMV infections from their children [8]. These results suggest that Towne may be overattenuated and that by defining, characterizing, and selectively repairing attenuating mutations, modified next-generation Towne-based vaccines with enhanced efficacy could be engineered.
However, current Towne strain molecular clones, sequences, and phenotypic data do not derive from the vaccine, but from laboratory stocks that, due to serial distribution among research laboratories and attendant plaque purifications, may have diverged from the viruses that comprise the vaccine. The reference stock ATCC VR-977 is a laboratory stock that predates the C-107 vaccine lot (S. Plotkin, personal communication). VR-977 is comprised of a roughly 50/50 mixture of two genome variants: the Towne-varS (varS) genome contains large b/b′ sequence repeats and a 13 kb deletion from the UL/b′ region that removes 15 genes, whereas the Towne-varL (varL) genome retains the UL sequences that are deleted from varS and has short (59-bp) b/b′ repeats (Figure 1) [9, 11–13]. Existing sequence data and bacterial artificial chromosome (BAC) clones of Towne genomes also derive either from laboratory stocks of uncertain provenance or from VR-977. Moreover, viruses with varL genomes are incompletely characterized [9]. Indeed, only recently, by sequencing the VR-977 mixture, has it been possible to reconstruct an approximation of the varL genome; although within regions common to both genome types, 18 polymorphisms were detected that could not be assigned to particular variants [13].
Figure 1.
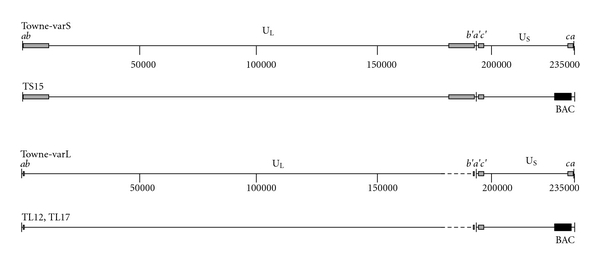
Genome maps of Towne-varS, Towne-varL, and BAC-cloned viruses. Inverted repeats b/b′ and c/c′ (grey boxes) flank unique long (UL) and unique short (US) regions to delineate long and short genome segments. One or more copies of the a sequence (black bars) can be found at long segment termini and inverted at the long and short segment junction (b′a′c′), while one or no copies are found at short segment termini [9, 10] (for simplicity single copies are shown). Dashed lines indicate UL sequences in varL that are missing from varS; black boxes represent the BAC ori cassette (BAC). Nucleotide positions are indicated; genome features are approximately to scale.
To identify potential attenuating mutations in viruses that comprise the vaccine and to establish defined molecular clones of known provenance for future development of modified Towne-based vaccines, BAC clones were derived from the vaccine with minimal passage in vitro. One BAC clone representing varL was fully sequenced and compared to the sequence obtained from VR-977, laboratory stocks of varS, and wild-type clinical isolates in order to establish a minimum set of attenuating mutations for the vaccine.
2. Materials and Methods
2.1. Cells and Virus
MRC-5 (ATCC CCL-171) and ARPE-19 (ATCC CRL-2302) cells and CMV strain BADrUL131-Y4 (a gift from Dai Wang and Thomas Shenk [14]) were propagated as described [15]. Infected cells expressing green fluorescent protein (GFP) were visualized and photographed using a Nikon Diaphot 300 inverted fluorescence microscope. Towne vaccine (lot C-107 [16]) was obtained from the Wistar Institute, Philadelphia, Pa, USA. It was manufactured in 1989 by Program Resources, McLean, Va, USA as lyophilized culture supernatant of MRC-5 cells infected with passage 131 seed stock. One vial was reconstituted with 1 ml sterile water and added to one 75 cm2 flask of confluent MRC-5 cells. After 1 week the culture medium was collected as stock Towne P2. Representative Towne varS and varL viruses were obtained from the vaccine by screening viral clones isolated by limiting dilution with PCR reactions specific for varS or varL genomes (not shown). Variant designations were subsequently confirmed by sequence and restriction pattern analyses as described in Section 3.1.
2.2. BAC Cloning
Plasmid pYD-C29 was a gift from Dong Yu. When restricted with PmlI, it forms a linear fragment containing LoxP-flanked BAC origin (ori) sequences and expression cassettes for GFP and puromycin N-acetyl transferase (puroR). This is flanked by ∼1-kb recombination sequences from the US28-US29 region of CMV strain AD169 [18] (see Figure 2(a)).
Figure 2.
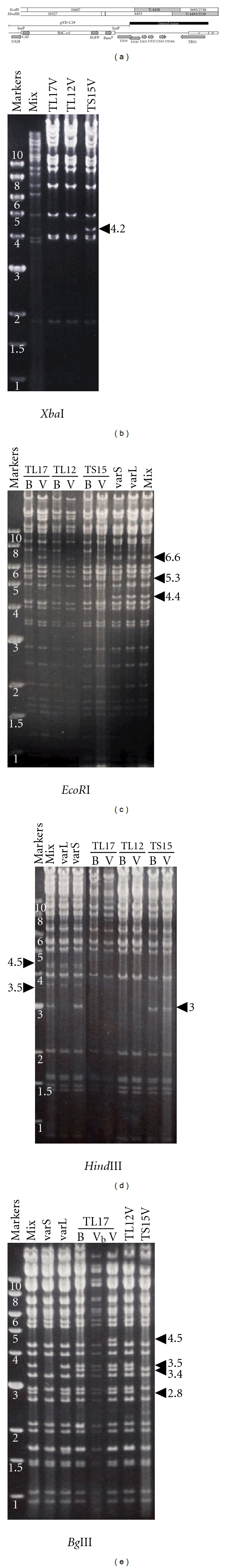
Restriction analysis of BACs and BAC-derived viral genomes. (a) EcoRI and HindIII restriction maps illustrate restriction fragments near the right end of BAC-cloned genomes that retain BAC ori. EcoRI T and HindIII V fragments (grey) are designated [17]. Genetic features of this region are illustrated, including the US28, BAC ori cassette, and US29 sequences in pYD-C29, CMV genes US29 to TRS1, and c repeat with single terminal a sequence. (b–e) Restriction digests of virion DNAs made from the vaccine virus mixture (mix) or varS and varL viruses isolated from the vaccine, virions from BAC-derived viruses (V), or circular BAC DNAs (B). Vb indicates TL17 virion DNA that retains the BAC ori; in all other BAC-derived viruses the BAC ori was excised. 4.2 kb XbaI, 5.3 kb EcoRI, and 3.0 kb HindIII fragments are unique to varS genomes, while 2.8 and 3.5 kb BglII fragments are unique to varL genomes. BACs lack 4.5 and 3.5 kb HindIII and 4.4 kb EcoRI fragments due to deletions and a 6.6 kb EcoRI terminal fragment because they are circular. All BACs contain an unexpected 3.4 kb BglII fragment due to a point mutation in the recombination vector. A 4.5 kb BglII fragment that may arise from a single copy of the a sequence is unique to TL17 virus with excised BAC ori.
BAC clones TS15 and TL12 were obtained by transfection/infection in which 4 μg PmeI-digested pYD-C29 DNA was transfected into MRC-5 cells using jetPEI reagent (Polyplus-transfection SA) followed two days later by infection (MOI = 1) with Towne P2. BAC clone TL17 was obtained by cotransfection of MRC-5 cells using jetPEI with 10 μg PmeI-digested pYD-C29 DNA and 17 μg virion DNA (purified, as previously described [19], from supernatants of MRC-5 cells infected by Towne P2 stock). When extensive CPE was reached, puromycin was added to a final concentration of 1–3 μg/mL. After 3-4 days, dead cells were removed by washing, 105-106 fresh cells were added, and the cultures were maintained without puromycin until extensive CPE was reached. This process was repeated 2-3 times until the majority of cells were GFP+. The supernatants were transferred to 50 cm2 dishes, and DNA was isolated 24 or 72 h post infection (p.i.) and used to transform electrocompetent E. coli as described previously [20].
2.3. Virus Reconstitution
Midiprep BAC DNA was prepared as described previously [20]. GFP+ viruses retaining the BAC ori were reconstituted by transfection of 10 μg BAC DNA into MRC-5 cells using jetPEI. BAC-ori-excised viruses were obtained in the same way except that BAC DNA was co-transfected with 1 μg pCre DNA (a gift from Wolfram Brune and Gabriele Hahn), and the resulting viruses were cloned by limiting dilution in 96-well plates and screened to identify wells containing GFP-negative viruses.
2.4. Viral Growth Curves
Confluent MRC-5 cells in 75 cm2 flasks were infected at MOIs of 0.1 or 0.05 pfu/cell. The cultures were washed 3 h p.i. Culture supernatants were titrated every 3 or 4 days for up to 14 days p.i. (d.p.i) using a 96-well method as described [20].
2.5. Sequencing
BAC TL12 was sequenced by the Genome Center at Washington University. Sheared fragments of TL12 BAC DNA were separated electrophoretically using 0.8% agarose. Fragments in the 4.0 to 6.5 kb range were extracted and subcloned into pSMART (Lucigen) and then sequenced using a 3730 DNA analyzer (Applied Biosystems). The data were assembled by Phrap (written by Phil Green). Coverage was approximately 8X. The annotated sequence has been deposited with GenBank (accession number GQ121041). Targeted Sanger dideoxy sequencing was performed using midiprep BAC or virion DNA templates. Sequencing primer UL130-3 (CAGACGCACCACAGAACGCAGAC) was used to determine the presence or absence of the TT mutation in the native UL130 locus.
3. Results
3.1. BAC Clones of VarS and VarL Derived from the Vaccine
Infectious BAC clones were isolated with limited passage of the vaccine in MRC-5 cells (see Section 2). As we did not plaque-purify recombinant viruses, BAC clones were anticipated to reflect the mixture of varL and varS genomes present in the vaccine. BAC clones were initially screened by restriction analysis to identify clones with apparently complete genomes and to differentiate varS and varL clones. Based on available sequence from a VR-977-derived varS genome (GenBank AY315197), a 4.2 kb XbaI fragment that spans the UL/b′ region of varS was predicted to be absent from varL genomes. Of 16 colonies resulting from transfection/infection, XbaI restriction patterns were identical, with the exception that 7 contained and 9 lacked the 4.2 kb XbaI fragment. Only 19 of 29 colonies resulting from cotransfection appeared to contain complete genomes, and of these, 3 contained and 16 lacked the 4.2 kb XbaI fragment (see data for representative clones, Figure 2(b)).
Selected BAC clones were tested for their ability to reconstitute infectious viruses upon transfection into MRC-5 cells. XbaI, HindIII, EcoRI, and BglII restriction patterns of BACs and BAC-derived viral genomes were then compared to patterns obtained from the vaccine-derived mixture and from varS and varL viruses isolated from the vaccine by limiting-dilution cloning (see Section 2). Figure 2 shows data for one representative varS (TS15) and two representative varL clones (TL12, TL17) that were selected based on ability to reconstitute infectious virus and minimal restriction pattern disparities with vaccine-derived viruses.
Restriction analyses revealed 5.3 kb EcoRI and 3.0 kb HindIII fragments specific for varS genomes (Figures 2(c) and 2(d)) and 2.8 and 3.5 kb BglII fragments specific for varL genomes (Figure 2(e)). As viral genomes are linear, they have terminal restriction fragments that are absent from circular BACs. Consequently, all BAC DNAs lacked a 6.6 kb terminal EcoRI fragment from the left end of the genome, but this fragment was restored in genomic DNA from BAC-derived viruses (Figure 2(c)). BACs and BAC-derived viral genomes also lacked the 4.4 kb EcoRI “T” fragment, the 4.5 kb terminal HindIII “V” fragment, and a 3.5 kb form of HindIII “V” that derives from genomes that lack a terminal a sequence [9, 10] (Figures 2(c) and 2(d)). Clustering of these missing fragments suggested a deletion distal of the BAC ori insertion site (Figure 2(a)). Targeted DNA sequencing confirmed that all BAC clones have ~7 kb deletions to the right of BAC ori that impact ORFs US29-TRS1 (Figure 2(a)).
A 3.4 kb BglII fragment was unique to all three BAC clones and BAC-derived viruses (Figure 2(e)). Sequencing of TL12 revealed that this fragment is due to a mutation from G to T at position 207132 in the VR-977 sequence [13] which creates a novel BglII site within US28. That pYD-C29 has a T at this position suggests that the mutation occurred during its construction and was incorporated into the BAC clones through recombination.
A 4.5 kb BglII fragment was unique to TL17 virus lacking the BAC ori (Figure 2(e)). Based on the TL12 sequence, excision of the BAC ori is predicted to produce a 5.7 kb BglII fragment that contains three reiterated a sequences. If, in TL17, this fragment contains only one a sequence copy (which predominates in strain AD169 [21]), a 4.5 kb BglII fragment is predicted. Thus, with the exception of the US29–TRS1 deletions, based on restriction digests with 4 enzymes, all three BACs appear to accurately represent varS and varL genomes present in the vaccine.
3.2. Complete Sequence of TL12
BAC TL12 was fully sequenced, annotated, and deposited in GenBank (GQ121041). When compared to VR-977-derived varS and varL partial sequences (AY315197 and AY446869), TL12 is missing 7657 bp comprising US29, US30, US31, US32, US34, US34A, and TRS1. In addition, TL12 was found to have mutations relative to wild-type CMV in RL13 (frame-shifted), UL1 (frameshifted), UL40 (partially deleted), UL130 (frame-shifted), US1 (prematurely terminated), US9 (frameshifted), and US28 (truncated). The mutation in US9 probably constitutes a natural variant, as it is shared with other CMV strains [22]. The US28 and US29 mutations are unique to TL12 because of the cloning procedure, whereas each of the others is present in previous sequences obtained from “Towne strain” (varS) laboratory stocks and the VR-977 mixture [13, 23–26]. The G to T change in US28 (position 207132, discussed above) is silent. However, sequences were also missing from the 3′ end of the US28 ORF, due to a flaw in pYD-C29 that resulted from an error in the strain AD169 sequence. This resulted in loss of 30 and addition of 4 novel amino acid residues at the C terminus of US28. Similarly, the US29-TRS1 deletion removed the 3′ end of US29, modifying the ORF to remove 78 residues from the C terminus and replacing them with 152 residues of novel sequence.
These data suggest that, during in vitro passage, varL evolved from the original wild-type Towne clinical isolate by acquiring mutations in RL13, UL1, UL40, UL130, and US1; then, varS arose from varL by deletion/duplication of UL sequences. Thus, only these five mutations appear to differentiate varL, as found in the vaccine, from the presumed wild-type CMV sequence.
3.3. Growth Properties of TS15, TL12, and TL17 in Fibroblasts
Growth curves for BAC-derived viruses were determined using MRC-5 fibroblast cells. At an MOI of 0.1, BAC-derived viruses exhibited modest growth delays on days 4 and 6, but by day 8 achieved titers similar to the Towne mixture (Figure 3(a)). The growth delay was slightly more pronounced at an MOI of 0.05, but by day 10 the BAC-derived viruses reached titers only slightly lower than those of the vaccine-derived mixture or vaccine-derived varS and varL viruses (Figure 3(b)). These growth impairments are most likely attributable to the lack of TRS1 in the BAC-derived viruses, as similar growth impairments at low MOI have been reported for TRS1-deletion mutants of strain AD169 [27].
Figure 3.
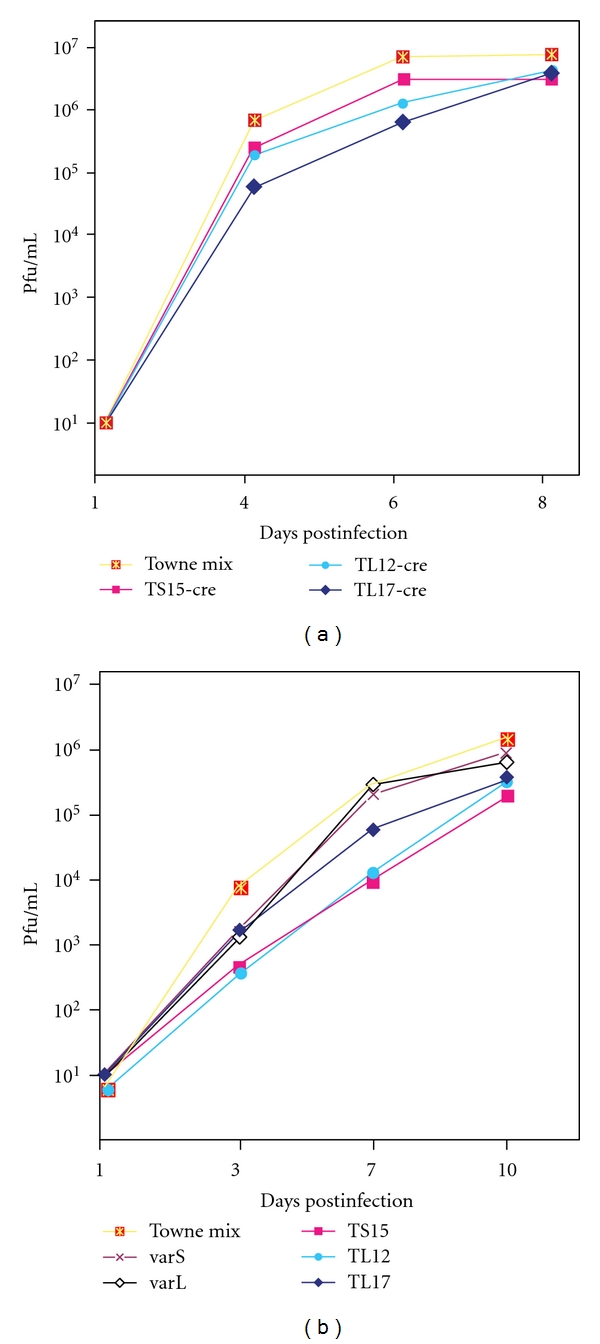
Growth curves of BAC-derived viruses and viruses derived from the vaccine. MRC-5 cultures were infected at MOIs of 0.1 (left) and 0.05 (right) with the vaccine virus mixture (Towne mix), varS, or varL viruses isolated from the vaccine and BAC-derived viruses lacking (-cre) or containing the BAC ori. Titers of each virus in the culture supernatants were determined on the days indicated.
3.4. Entry and Spread of TS15, TL12, and TL17 Is Impaired in Epithelial Cells
CMV entry into endothelial and epithelial cells utilizes an endocytic pathway that requires the virion-associated gH/gL/UL128-UL131 complex comprised of gH, gL, UL128, UL130, and UL131 (also known as UL131A [28, 29]). Laboratory varS stocks contain a TT insertion in UL130 that has been associated with inefficient entry and replication in endothelial cells [30, 31]. The TL12 sequence and targeted sequencing of TS15 and TL17 confirmed that all three vaccine-derived BACs retain the TT insertion in UL130. To determine whether the endocytic entry pathway is similarly impaired in these BAC-derived viruses, efficiencies of viral entry and spread were compared between fibroblasts and epithelial cells. MRC-5 and ARPE-19 cells were infected with replicate inocula of GFP+ viruses and monitored for green fluorescence on day 3 to assess entry and at later times to assess spread. Compared to entry into MRC-5s, TS15, TL12, and TL17 entered ARPE-19 epithelial cells with very poor efficiency; however, after entry TS15 and TL17 spread slowly in ARPE-19 cells, whereas TL12 did not (Figure 4). In contrast, control virus BADrUL131-Y4, a GFP-tagged virus known to replicate efficiently in epithelial cells [14], entered both cell types efficiently. Viral titers in the supernatants of TS15- or TL17-infected ARPE-19 cells did not exceed 4×102 pfu/mL, compared to 3×105 pfu/mL in MRC-5 cells, indicating a severe restriction to replication in epithelial cells that is consistent with the above observations of inefficient entry and spread.
Figure 4.
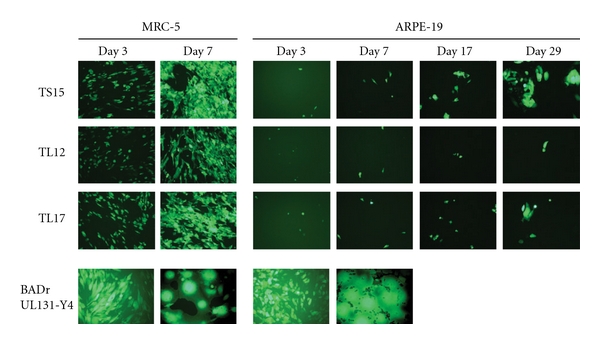
Entry and spread of BAC-derived viruses in MRC-5 or ARPE-19 cells. Confluent cell monolayers were infected with matched inocula (104 pfu) of the indicated GFP+ viruses. Representative micrographs (8X final magnification) were taken on the days indicated.
4. Discussion
The Towne vaccine has been well characterized in vivo through clinical trials that involved nearly 1000 human subjects. Remarkably, the viruses that comprise the vaccine have not been studied directly. Instead, in vitro data pertaining to the Towne strain has been obtained from laboratory stocks having uncertain lineage with respect to the vaccine. Presumptions that these data apply to the vaccine involve considerable risk. For example, because the first characterized laboratory stocks of strain Towne were exclusively varS, the possibility that the vaccine might contain less deleted forms of the CMV genome (i.e., varL) was for many years overlooked. As we now consider genetic modifications intended to improve vaccine immunogenicity and efficacy while maintaining safety, it is imperative to have an accurate description of the genotypic and phenotypic properties of the viruses that were evaluated in vivo. Moreover, next-generation Towne-based vaccines should be based not on laboratory stocks but on vaccine-derived viral genomes.
BAC cloning allowed the isolation, with minimal passage in vitro, of viral genomes representing the vaccine. The fact that among the BAC clones varS and varL genomes were present in roughly equal proportions suggests that, like VR-977, the vaccine is a mixture of the two variant genome types. This result is concordant with unpublished restriction and targeted sequence analyses suggesting the presence of varL genomes in vaccine lot C-107 (Greg Duke, personal communication). This finding confirms that the vaccine's safety history applies to varL as well as to varS genomes (a conclusion that could not be made if vaccine lots tested in humans had been found to contain only varS viruses). Therefore, varL should provide an acceptable starting point for future vaccine development. To this end, restriction analyses, sequencing, and growth properties in fibroblasts suggest that, with exception of mutations in US28 and US29 and deletion of US130-TRS1, the BAC clones are accurate representations of viruses comprising the vaccine. Moreover, as the BAC ori/GFP cassette is flanked by LoxP sites, the inserted foreign sequences can be easily excised with Cre recombinase, leaving only 35 bp (one LoxP site) of nonviral sequences.
The US130-TRS1 deletions that we observed were most likely compensatory in response to the increase in genome length caused by insertion of the 9.1-kb BAC ori/GFP cassette. Very similar deletions (also adjacent to BAC ori insertions) were previously observed in initial BAC clones of the guinea pig cytomegalovirus genome [20]. Further studies demonstrated that guinea pig cytomegalovirus does not tolerate genomes over length by 9 kb, presumably due to restrictions imposed by the packaging capacity of the capsid [32]. This problem may be particularly acute for cytomegaloviruses, as their genomes are the largest of the Herpesviridae. Currently the only solution is to repair the deletions using E. coli genetics, although in theory it may be possible to clone full-length herpesvirus genomes independent of packaging-imposed size restrictions by in vitro transposition of BAC ori into viral DNA, followed by transformation directly into E. coli, as has been suggested [33].
Sequence data from the BAC clones confirmed the presence of five potential attenuating mutations (RL13, UL1, UL40, UL130, and US1) in varL viruses comprising the vaccine. This information may provide the basis for future modifications designed to modify attenuation or enhance immunogenicity. For example, the UL130 mutation was shown to impair endothelial and epithelial but not fibroblast tropism of laboratory stocks by modifying the C-terminus of UL130 and impairing assembly and virion incorporation of gH/gL/UL128-UL131 complexes [30, 31]. That vaccine viruses contain the same mutation and exhibit similar restricted tropism in vitro suggests that in vivo the UL130 mutation contributes to safety of the vaccine by limiting the cells available for viral replication and dissemination to those of fibroblast/mesenchymal lineage. Conversely, suboptimal efficacy of the Towne vaccine may be linked to its failure to induce robust epithelial entry-specific neutralizing antibody responses [15]. As potent epithelial entry-specific neutralizing epitopes reside within gH/gL/UL128-UL131 complexes [34, 35], repair of the UL130 mutation (and consequent restoration of gH/gL/UL128-UL131 complex formation) may be essential for improving vaccine efficacy.
Thus, it may be necessary to accompany modifications designed to improve immunogenicity and efficacy with strategies to maintain or improve safety. For example, repair of UL130 could be accompanied by additional mutations that allow assembly of nonfunctional but antigenically native gH/gL/UL128-UL131 complexes, thus maintaining restricted tropism while expressing important neutralizing epitopes. In addition, mutations that partially impair viral replication in vitro (such as the US130-TRS1 deletions described here) may serve to enhance safety by limiting replication in vivo. Finally, viruses incapable of replication in vivo could be constructed by creation of conditional lethal mutations (which would require special in vitro culture conditions for propagation), or more simply by inactivation of infectious virus in vitro prior to immunization.
Towards the development of such rationally engineered whole-virus-based CMV vaccines, the BAC clones described here provide defined reagents of known provenance that can be readily manipulated with existing genetic technologies.
Acknowledgments
The authors are grateful to Dai Wang and Thomas Shenk for providing the BADrUL131-Y4 BAC, Dong Yu for plasmid pYD-C29, Wolfram Brune and Gabriele Hahn for plasmid pCre. This work was supported in part by NIH/NIAID Grants R21AI073615 and R01AI088750 (to M. A. McVoy).
References
- 1.Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. The New England Journal of Medicine. 1992;326(10):663–667. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- 2.Fowler KB, Boppana SB. Congenital cytomegalovirus (CMV) infection and hearing deficit. Journal of Clinical Virology. 2006;35(2):226–231. doi: 10.1016/j.jcv.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 3.Morton CC, Nance WE. Newborn hearing screening—a silent revolution. The New England Journal of Medicine. 2006;354(20):2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 4.Stratton KR, Durch SJ, Lawrence RS, editors. Vaccines for the 21st Century: A Tool for Decisionmaking. Washington, DC, USA: National Academy Press; 2000. [PubMed] [Google Scholar]
- 5.Plotkin SA, Furukawa T, Zygraich N, Huygelen C. Candidate cytomegalovirus strain for human vaccination. Infection and Immunity. 1975;12(3):521–527. doi: 10.1128/iai.12.3.521-527.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clinical Infectious Diseases. 2004;39(2):233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 7.Plotkin SA, Smiley ML, Friedman HM, et al. Towne-vaccine-induced prevention of cytomegalovirus disease after renal transplants. The Lancet. 1984;1(8376):528–530. doi: 10.1016/s0140-6736(84)90930-9. [DOI] [PubMed] [Google Scholar]
- 8.Adler SP, Starr SE, Plotkin SA, et al. Immunity induced by primary human cytomegalovirus infection protects against secondary infection among women of childbearing age. Journal of Infectious Diseases. 1995;171(1):26–32. doi: 10.1093/infdis/171.1.26. Erratum In Journal of Infectious Diseases, vol. 171, no. 4, p.1080, 1995. [DOI] [PubMed] [Google Scholar]
- 9.Hahn G, Rose D, Wagner M, Rhiel S, McVoy MA. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Townelong as BACs and site-directed mutagenesis using a PCR-based technique. Virology. 2003;307(1):164–177. doi: 10.1016/s0042-6822(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 10.Tamashiro JC, Spector DH. Terminal structure and heterogeneity in human cytomegalovirus strain AD169. Journal of Virology. 1986;59(3):591–604. doi: 10.1128/jvi.59.3.591-604.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha TAN, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. Journal of Virology. 1996;70(1):78–83. doi: 10.1128/jvi.70.1.78-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prichard MN, Penfold MET, Duke GM, Spaete RR, Kemble GW. A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Reviews in Medical Virology. 2001;11(3):191–200. doi: 10.1002/rmv.315. [DOI] [PubMed] [Google Scholar]
- 13.Bradley AJ, Lurain NS, Ghazal P, et al. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. Journal of General Virology. 2009;90, part 10:2375–2380. doi: 10.1099/vir.0.013250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Shenk T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. Journal of Virology. 2005;79(16):10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui X, Meza BP, Adler SP, McVoy MA. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine. 2008;26(45):5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler SP, Hempfling SH, Starr SE, Plotkin SA, Riddell S. Safety and immunogenicity of the Towne strain cytomegalovirus vaccine. Pediatric Infectious Disease Journal. 1998;17(3):200–206. doi: 10.1097/00006454-199803000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Stinski MF. Cytomegalovirus and its replication. In: Fields BN, Knipe DM, editors. Fundamental Virology. 2nd edition. New York, NY, USA: Ravem Press; 1991. pp. 929–950. [Google Scholar]
- 18.Yu D, Smith GA, Enquist LW, Shenk T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. Journal of Virology. 2002;76(5):2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McVoy MA, Nixon DE, Adler SP. Circularization and cleavage of guinea pig cytomegalovirus genomes. Journal of Virology. 1997;71(6):4209–4217. doi: 10.1128/jvi.71.6.4209-4217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui X, McGregor A, Schleiss MR, McVoy MA. Cloning the complete guinea pig cytomegalovirus genome as an infectious bacterial artificial chromosome with excisable origin of replication. Journal of Virological Methods. 2008;149(2):231–239. doi: 10.1016/j.jviromet.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamashiro JC, Filpula D, Friedmann T, Spector DH. Structure of the heterogeneous L-S junction region of human cytomegalovirus strain AD169 DNA. Journal of Virology. 1984;52(2):541–548. doi: 10.1128/jvi.52.2.541-548.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham C, Gatherer D, Hilfrich B, et al. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. Journal of General Virology. 2010;91, part 3:605–615. doi: 10.1099/vir.0.015891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunn W, Chou C, Li H, et al. Functional profiling of a human cytomegalovirus genome. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy E, Yu D, Grimwood J, et al. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini A, Liu H, Zhu H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. Journal of Virology. 2001;75(4):1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolan A, Cunningham C, Hector RD, et al. Genetic content of wild-type human cytomegalovirus. Journal of General Virology. 2004;85, part 5:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 27.Blankenship CA, Shenk T. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. Journal of Virology. 2002;76(23):12290–12299. doi: 10.1128/JVI.76.23.12290-12299.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davison AJ, Dolan A, Akter P, et al. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. Journal of General Virology. 2003;84, part 1:17–28. doi: 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 29.Akter P, Cunningham C, McSharry BP, et al. Two novel spliced genes in human cytomegalovirus. Journal of General Virology. 2003;84(5):1117–1122. doi: 10.1099/vir.0.18952-0. [DOI] [PubMed] [Google Scholar]
- 30.Hahn G, Revello MG, Patrone M, et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. Journal of Virology. 2004;78(18):10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patrone M, Secchi M, Fiorina L, Ierardi M, Milanesi G, Gallina A. Human cytomegalovirus UL130 protein promotes endothelial cell infection through a producer cell modification of the virion. Journal of Virology. 2005;79(13):8361–8373. doi: 10.1128/JVI.79.13.8361-8373.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui X, McGregor A, Schleiss MR, McVoy MA. The impact of genome length on replication and genome stability of the herpesvirus guinea pig cytomegalovirus. Virology. 2009;386(1):132–138. doi: 10.1016/j.virol.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou F, Li Q, Gao SJ. A sequence-independent in vitro transposon-based strategy for efficient cloning of genomes of large DNA viruses as bacterial artificial chromosomes. Nucleic Acids Research. 2009;37(1, article e2) doi: 10.1093/nar/gkn890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macagno A, Bernasconi NL, Vanzetta F, et al. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. Journal of Virology. 2010;84(2):1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saccoccio FM, Sauer AL, Cui X, et al. Peptides from cytomegalovirus UL130 and UL131 proteins induce high titer antibodies that block viral entry into mucosal epithelial cells. Vaccine. 2011;29(15):2705–2711. doi: 10.1016/j.vaccine.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]