

Abstract
The protective/neurotoxic role of fractalkine (CX3CL1) and its receptor CX3C chemokine receptor 1 (CX3CR1) signaling in neurodegenerative disease is an intricate and highly debated research topic and it is becoming even more complicated as new studies reveal discordant results. It appears that the CX3CL1/CX3CR1 axis plays a direct role in neurodegeneration and/or neuroprotection depending on the CNS insult. However, all the above studies focused on the role of CX3CL1/CX3CR1 signaling in pathological conditions, ignoring the relevance of CX3CL1/CX3CR1 signaling under physiological conditions. No approach to date has been taken to decipher the significance of defects in CX3CL1/CX3CR1 signaling in physiological condition. In the present study we used CX3CR1−/−, CX3CR1+/−, and wild-type mice to investigate the physiological role of CX3CR1 receptor in cognition and synaptic plasticity. Our results demonstrate for the first time that mice lacking the CX3CR1 receptor show contextual fear conditioning and Morris water maze deficits. CX3CR1 deficiency also affects motor learning. Importantly, mice lacking the receptor have a significant impairment in long-term potentiation (LTP). Infusion with IL-1β receptor antagonist significantly reversed the deficit in cognitive function and impairment in LTP. Our results reveal that under physiological conditions, disruption in CX3CL1 signaling will lead to impairment in cognitive function and synaptic plasticity via increased action of IL-1β.
Introduction
Microglia are the resident immune cells in the CNS that act as macrophages (Harrison et al., 1998; Aloisi, 2001). Microglia can rapidly respond to the detection of homeostatic disturbances by inducing an immune response, which consists of an upregulation of inflammatory molecules and neurotrophic factors (Miwa et al., 1997; Batchelor et al., 1999; O'Donnell et al., 2002; Nakajima and Kohsaka, 2004). Through this immune response, microglia protect proper brain function and remove cells damaged from an acute injury. When chronic inflammation occurs, prolonged activation of microglia trigger the release of several neurotoxic products and proinflammatory cytokines including IL-1β, IL-6, and tumor necrosis factor α (TNFα) (Colton and Gilbert, 1987).
Microglia are restrained by numerous microenvironmental influences, many of which are produced by neurons (Cardona et al., 2006; Hanisch and Kettenmann, 2007; Lyons et al., 2007). Fractalkine (CX3CL1) is a chemokine that is constitutively expressed by healthy neurons and identified as a novel neuroimmune regulatory protein. CX3CL1 signals to microglia, which inhibits microglial activity under inflammatory conditions (Harrison et al., 1998; Ransohoff et al., 2007). In contrast to other chemokines, CX3CL1 binds to only one receptor, CX3C chemokine receptor 1 (CX3CR1) (Harrison et al., 1998). In the brain, CX3CR1 is exclusively expressed by microglia (Harrison et al., 1998). CX3CL1 can bind to CX3CR1 either as a membrane-bound protein or a soluble ligand following constitutive cleavage by ADAM10 and ADAM17 metalloproteases (Bazan et al., 1997). Interactions between CX3CL1 and CX3CR1 contribute to microglial ability to maintain a resting phenotype. However, when neurons are injured, CX3CL1 levels decrease, which results in microglia recruitment and activation.
Through interactions with microglia, CX3CL1 serves as an endogenous neuronal modulator and controls the overproduction of inducible nitric oxide synthase (iNOS), IL-1β, TNFα, and IL-6. Interestingly, CX3CL1/CX3CR1 signaling has been linked to human neurodegeneration, highlighted by the identification of the V249I and T280M polymorphisms in CX3CR1. These polymorphisms are associated with the neuroinflammatory disorder human age-related macular degeneration (Tuo et al., 2004; Chan et al., 2005). Furthermore, mice that are deficient in CX3CR1 have increased microglial cell expression of IL-1β in response to lipopolysacharide stimulation. This increased IL-1β expression is associated with increased neuronal cell death in the hippocampus (Ransohoff et al., 2007), and CX3CR1-deficent mice have increased susceptibility to neurotoxins such as MPTP (Cardona et al., 2006). However, all of the studies mentioned have focused on the role of CX3CL1/CX3CR1 signaling in pathological conditions, ignoring the relevance of CX3CL1/CX3CR1 signaling under physiological conditions. Our lab has previously established the impact of functional disruption of CX3CR1 in a nonpathological condition. We have demonstrated that CX3CR1−/− mice have decreased hippocampal neurogenesis (Bachstetter et al., 2011). Furthermore, antagonism of CX3CR1 in young rats leads to increased hippocampal protein levels of IL-1β and decreased neurogenesis. We hypothesize that disruption of CX3CL1/CX3CR1 signaling will result in alterations of the physiological activities of the brain. Here, we demonstrate under physiological conditions, disruption of CX3CL1/CX3CR1 signaling leads to impairments in motor learning, cognitive function, and synaptic plasticity through increased inflammation in the CNS.
Materials and Methods
Animals
All experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of South Florida, College of Medicine or the University of Florida, as appropriate. CX3CR1−/− mice were backcrossed to the C57BL/6J background for >10 generations and were obtained from Jackson Laboratories. Colonies of the CX3CR1+/− and CX3CR1−/− mice were maintained at the University of South Florida and genotyping performed as described previously (Bachstetter et al., 2011). Three-month-old male CX3CR1+/− and CX3CR1−/− littermates and C57BL/6J [wild-type (WT)] mice were used in the experiments. Mice were housed in pairs in environmentally controlled conditions (12 h light/dark cycle at 21 ± 1°C) and provided food and water ad libitum. Animals that developed skin lesions were excluded from the Morris water maze test to avoid the development of infection.
Bromodeoxyuridine administration
Three-month-old male (n = 5/genotype) CX3CR1+/− and CX3CR1−/− littermates and C57BL/6 (WT) mice received two intraperitoneal injections within a 12 h interval of bromodeoxyuridine (5-bromo-2-deoxyuridine; BrdU; Sigma) at dose of 50 mg/kg. Animals were killed 24 h after the last injection.
Surgical procedure
Three-month-old male CX3CR1−/− and C57BL/6 (WT) mice were implanted with a brain infusion cannula connected with an osmotic minipump. Before implantation, the pumps were incubated in sterile saline for at least 48 h at 37°C to prime the pumps. For implantation, mice were anesthetized with isofluorane and placed in a stereotaxic frame. A guide cannula was implanted in the left ventricle (anteroposterior, 1.0 mm; mediolateral, −0.5 mm; dorsoventral, −2.5 mm) and connected to the osmotic minipump (Model 1004; Alzet; pumping rate, 0.11 μl/h; total volume, 100 μl), which was inserted subcutaneously. Pumps were weighed before implantation and at the end of the experiment to ensure complete delivery of their content. r-metHu IL-1ra (100 μg/ml/d; kind gift from Amgen) was infused through the cannula, which was connected to the filled minipump. The infusion started on the day of the surgery and continued for 4 weeks. Control animals received the same volume of heat-inactivated IL-1ra (heat inactivated for 45 min in a water bath at 90°C) or artificial CSF (ACSF). Behavioral analysis started 15 d following the beginning of the infusion. Mice that showed signs of distress during the course of behavioral testing or infections during the surgical procedure were not included in the following behavioral studies.
Tissue collection and processing
For immunohistochemistry studies, animals were anesthetized with pentobarbital (50 mg/kg, i.p.). The mice were transcardially perfused with PBS, followed by 4% paraformaldehyde in PBS. The brains were postfixed in 4% paraformaldehyde for 12 h, after which they were transferred into 30% sucrose in PBS for at least 16 h at 4°C. Exhaustive sagittal sections of the left hemisphere were made at 40 μm using a Microm cryostat (Richard-Allan Scientific) and stored in cryoprotectant at 4°C. Animals used for biochemical studies were killed by rapid decapitation. Brain tissues were separately dissected and rapidly frozen in liquid nitrogen before storage at −80°C. Both hippocampi were homogenized using an electric tissue homogenizer in a 1:10 weight-to-volume ratio of ice-cold RIPA buffer (Millipore) containing protease inhibitors and EDTA (Pierce). Following homogenization, sample lysates were centrifuged at 10,000 × g at 4°C for 15 min, and the cleared supernatant was collected.
ELISA
Total protein was measured using BCA assay (Pierce). IL-1β and TNFα concentrations were measured using commercially available ELISA kit (eBioscience) following the manufacturer's protocol.
Western blot analysis
Fifty micrograms of total protein per lane were loaded onto a 10% SDS polyacrylamide gel (Bio-Rad) for electrophoresis. Proteins were transferred onto a nitrocellulose membrane for immunodetection. The membrane was blocked for 1 h in 5% non-fat dry milk (NFDM) in PBS-Tween 20 (PBS-T; 0.1%). Antibodies for rabbit phospho-p38 (Cell Signaling Technology; 1:1000) and mouse β-actin (Sigma; 1:3000) were incubated overnight at 4°C in 1% NFDM in PBS-T. Following washes, anti-rabbit and anti-mouse secondary antibodies (LI-COR Biosciences) were incubated for 1 h at room temperature in 1% NFDM in PBS-T. Membranes were scanned using LI-COR Odyssey Imager, and the raw intensity for each band was measured using LI-COR Odyssey image analysis software.
Immunohistochemistry
Except where specifically indicated, standard staining procedures were conducted on free-floating sections using every sixth section of the entire hippocampus beginning with a random start and including sections before and after the hippocampus to ensure that the entire structure was sampled. Sections were blocked in 10% normal serum from the species that the secondary antibody was raised in, with the addition of 0.1% Triton X-100. Sections were incubated with primary antibody (Abcam anti-Iba1 1:500) diluted in 3% normal serum with 0.1% Triton X-100 overnight at 4°C. For immunohistochemistry, biotinylated secondary antibodies (horse anti-goat; 1:2000) were diluted in 3% normal serum with 0.1% Triton X-100 and incubated for 2 h at room temperature. Enzyme detection was done using avidin-biotin substrate (ABC kit; Vector Laboratories) followed by color development in diaminobenzidine solution (Sigma). For immunodetection of BrdU labeling, protocol was followed as described previously (Gemma et al., 2007; Bachstetter et al., 2011). Briefly, sections were pretreated with 50% formamide/2× SSC (0.3 m NaCl, 0.03 m sodium citrate) at 65°C for 2 h, rinsed in 2 × SSC. BrdU was detected using a mouse anti-BrdU (1:100; Roche; clone B44). Doublecortin (DCX) is a marker of migrating neurons that is expressed for approximately 3 weeks after the cell is born and has been shown to be a reliable indicator of neurogenesis (Rao and Shetty, 2004; Couillard-Despres et al., 2005). For DCX immunodetection, incubation in primary antibody was done for 36 h at 4°C using a polyclonal goat antibody C terminus of human DCX (1:200; Santa Cruz Biotechnology).
Quantification and imaging.
To determine cell numbers, the optical fractionator method of unbiased stereological cell counting techniques (West et al., 1991) was used with a Nikon Eclipse 600 microscope and quantified using Stereo Investigator software (MicroBrightField). The virtual grid (175 × 175) and counting frame (100 × 100) were optimized to count at least 200 cells per animal with error coefficients <0.07. Outlines of the anatomical structures were done using a 10×/0.45 objective, and cell quantification was conducted using a 60×/1.40 objective.
Iba1 quantification.
Slides with Iba1-stained sections were scanned using a Zeiss Mirax slide scanner. Following scanning, digital images were analyzed for Iba1-positive staining using Zeiss Neuroquant IAE analysis software. Briefly, an investigator blind to the study randomly placed a measurement grid within the DG of each hippocampal section. Each measurement grid had the same dimensions and therefore the same measurement area. Iba1-positive cells were defined in the analysis software based on threshold to include both the cell body and processes, while eliminating background values. Data were obtained solely from the randomly placed measurement grid on an average of five sites per animal within the DG. Raw data were normalized to account for variations in the number of sampling sites. Results are given as the average ratio of Iba1-positive area relative to the total measurement area.
Behavioral analysis
Rotarod.
Aged-matched 3-month-old wild-type, CX3CR1−/−, and CX3CR1+/− mice were first tested for overall balance, motor coordination, and motor learning. This test was performed on an accelerating rotarod apparatus (Ugo Basile) with a 3-cm-diameter rod starting at an initial rotation of 4 rpm accelerating to 40 rpm over 5 min. Mice were tested for the time spent on the rod during each of four trials with a 30 min intertrial interval. Each trial was completed when the mouse fell off the rod (distance of 12 cm) onto a spring-cushioned lever.
Open field.
The open field was used as a standard test of general activity. Animals were monitored for 15 min in a 40 cm square open field with video tracking software (ANY-Maze; Stoelting) and under moderate lighting. General activity levels were evaluated by determining the total amount of distance traveled. Anxiety levels were assessed by the pattern of exploration of in the open field (center vs periphery).
Elevated-plus maze.
Anxiety was also assessed through the elevated-plus maze (EPM). The EPM consisted of two well-lit open arms (35 cm) facing each other and two enclosed arms (30.5 cm) also facing each other. Each arm was attached to a common center platform (4.5 cm square) and elevated 40 cm off the floor. The mouse was placed in the center platform and allowed to explore for 5 min. Video tracking software measured movement in each section (ANY-Maze; Stoelting).
Fear conditioning.
Fear conditioning was used to assess memory formation that is especially suited to test for proper hippocampus function. Animals were placed in the fear conditioning apparatus (Panlab) for 2 min. Then, a 30 s acoustic conditioned stimulus (CS; 80 dB tone) was delivered, and a 0.5 mA shock unconditioned stimulus (US) was applied to the floor grid during the last 2 s of the CS. Training consisted of two CS–US pairings, with a 1.5 min interval between each. The mice were placed in the chamber and monitored for freezing to the context 24 h after training (no shocks or conditioned stimulus given). Immediately after the contextual test, mice were placed into a novel context and exposed to the CS for 3 min (cued fear conditioning). Learning was assessed by measuring freezing behavior (i.e., motionless position) for two consecutive seconds. Shock threshold was assessed by placing an animal in the same conditioning chamber as used for associative fear conditioning. Foot shock intensity started at 0.075 mA and was increased by 0.05 mA every 30 s. The shock intensity to induce flinching, jumping, and vocalization were determined, and the shock intensity was recorded. The experiment was terminated at the shock intensity sufficient to induce vocalization.
Morris water maze.
The Morris hidden-platform water maze consisted of a circular pool (1.38 m diameter) filled with nontoxic opaque water at room temperature with an escape platform (10 cm diameter) hidden beneath the water (3 cm). Each mouse was placed in the pool in a pseudorandom order and given 60 s to locate the escape platform. When the mouse found the platform or if the mouse failed to find the platform within 60 s, it was placed on the platform where it remained for 30 s. Each mouse was given four trials per day, with an intertrial interval of 1 h. The time to find the platform (escape latency), the total distance traveled, and the swim speed of the animals were recorded by video tracking software (ANY-Maze; Stoelting). The mice were then towel dried and placed in a cage with a heating pad underneath until dry and returned to their home cage. On Day 7, all mice were subjected to one probe trial in which the platform was removed, and each animal had 60 s to search the pool for the platform. Since wild-type mice did not show improvement in learning ability to find the platform on Day 7, a second probe trial was performed on Day 11, following 3 additional continuous days of training.
Extracellular recordings
Mice were killed by rapid decapitation, and hippocampi were dissected out for electrophysiological experimental paradigms as described previously (Weeber et al., 2002; Beffert et al., 2005; Peters et al., 2006). Field EPSPs (fEPSPs) were obtained from area CA1 stratum radiatum with the use of a glass microelectrode filled with ACSF (2–4 mΩ). fEPSPs were evoked through stimulation of the Schaffer collaterals using a 0.1 ms biphasic pulse delivered every 20 s. After a consistent response to a voltage stimulus was established, threshold voltage for evoking fEPSPs was determined, and the voltage was increased incrementally every 0.5 mV until the maximum amplitude of the fEPSP was reached (input/output curve). All other stimulation paradigms were induced at the same voltage, defined as 50% of the stimulus voltage used to produce the maximum fEPSP amplitude, for each individual slice. Paired-pulse facilitation (PPF) was induced with two paired pulses given with an initial delay of 20 ms, and the time to the second pulse incrementally increased 20 ms until a final delay of 300 ms was reached.
Long-term potentiation recordings.
Slices that received IL-1ra (100 μg/ml; a kind gift from Amgen) were bathed in ACSF containing IL-1ra for at least 15 min before the start of recordings. A fEPSP long-term potentiation (LTP) baseline response was recorded every 20 s for 20 min. The tetanus used to evoke LTP was high-frequency stimulation (HFS), which consisted of two trains of 1 s, 100 Hz pulses, with an intertrain interval of 20 s. Following HFS, fEPSPs were recorded for 60 min. The slopes of fEPSPs were averaged into 1 min bins and graphed. Potentiation was measured as the increase of the mean fEPSP descending slope following TBS normalized to the mean fEPSP descending slope of baseline recordings.
Post-tetanic potentiation recordings.
All hippocampal slices were bathed in ACSF containing 50 μm APV for at least 15 min before recordings. Slices that received IL-1a were bathed in ACSF containing IL-1ra for at least 15 min before the start of recordings. A fEPSP post-tetanic potentiation (PTP) baseline response were recorded every 3 s for 1 min. The tetanus used to induce PTP was HFS, which consisted of a 1 s, 100 Hz pulses. Following HFS, fEPSPs were recorded every 3 s for 5 min. The slopes of fEPSPs were averaged into 12 s bins and graphed. Potentiation was measured as the increase of the mean fEPSP descending slope following PTP normalized to the mean fEPSP descending slope of baseline recordings.
DAPI staining.
Mice were killed, and hippocampi were dissected out for DAPI staining in the exact same manner as for electrophysiological experimental paradigms. Hippocampal slices were placed in a submerged holding chamber containing oxygenated ACSF for no less than 2 h before being transferred to 4% PFA for fixation for ∼16 h. Following fixation, 40 μm sections were made through entire dissected region and mounted on superfrost slides. Visualization of cell nuclei was achieved using DAPI VectaShield hardest mounting media (Vector Laboratories). Fluorescent images were taken using an Olympus FluoView FV1000 confocal microscope. Hippocampal DAPI-positive cells were quantified from 16 bit grayscale images using ImageJ software for each of the three genotypes (n = 5 per group).
Statistical analysis
Statistical analysis comparing the three genotypes was done using a one-way ANOVA or two-way ANOVA with a Tukey–Kramer or Bonferroni post hoc test. Repeated-measures ANOVA was used to analyze rotarod data, followed a Bonferroni post hoc test. Statistical analysis was done in GraphPad Prism software.
Results
Decreased hippocampal neurogenesis in CX3CR1-deficient mice in a gene-dose-dependent manner
Previously, we demonstrated that CX3CR1−/− young mice have a significant decrease in hippocampal neurogenesis compared to heterozygous littermates. To determine whether both alleles are required for normal receptor function, we counted the number of DCX-positive cells in the subgranular zone (SGZ) of the dentate gyrus in CX3CR1−/−, CX3CR1+/−, and wild-type mice. Using the optical fractionator method of design-based stereology, we observed a significant decrease in the number of DCX+ cells in the SGZ of the dentate gyrus in CX3CR1−/− mice compared to CX3CR1+/− and wild-type mice (Fig. 1a; one-way ANOVA, p < 0.0001). Interestingly, loss of one allele is sufficient to impair receptor function (Tukey's post hoc test; WT vs CX3CR1+/−, p < 0.001; WT vs CX3CR1−/−, p < 0.001; CX3CR1+/− vs CX3CR1−/−, p < 0.05). To determine whether this decrease is due to reduced cell proliferation, mice were injected twice (8 h interval) with BrdU (50 mg/kg). Mice were killed the following day. Quantification of the number of BrdU+ cells revealed a significant decrease in the number of BrdU+ cells in the CX3CR1−/− mice compared to the CX3CR1+/− and wild-type mice, indicating reduced cell proliferation (Fig. 1b; Tukey's post hoc test; WT vs CX3CR1+/−, p < 0.05; WT vs CX3CR1−/−, p < 0.001; CX3CR1+/− vs CX3CR1−/−, p < 0.05).
Figure 1.

a, b, CX3CR1−/− mice show decreased hippocampal neurogenesis in a gene-dose-dependent manner. Unbiased stereology revealed a significant decrease in the number of DCX+ cells in the SGZ of adult male CX3CR1−/− and CX3CR1+/− mice compared to wild-type mice (p < 0.0001). a, CX3CR1−/− mice were also significantly different from CX3CR1+/− mice (p < 0.001). Quantification of the number of cells that were proliferating during the preceding 24 h, as determined by the incorporation of BrdU, showed significantly fewer in the CX3CR1−/− and CX3CR1+/− mice compared to wild-types (p < 0.0001). b, CX3CR1−/− mice were also significantly different from CX3CR1+/− mice (p < 0.05). White bars, WT; gray bars, CX3CR1+/− mice; black bars, CX3CR1−/−. All data are presented as mean ± SEM. ***p < 0.0001; **p < 0.001; *p < 0.05.
Motor learning but not motor coordination is impaired in CX3CR1-deficient mice
Coordination and motor skill acquisition were tested using an accelerating rotarod. The amount of time an animal stays on the rotarod is an indicator of its general level of balance and coordination. In general, mice improve their performance over time with training, which is an indicator of motor learning. In the first study, aged-matched littermate wild-type (n = 10), CX3CR1−/− (n = 12), and CX3CR1+/− (n = 12) mice were tested for the time spent on the rod during each of four trials per day for 2 consecutive days. No differences were observed between genotypes during the first day of training (Fig. 2a). On the second day of training (Trials 5–8), wild-type mice remained on the rod for longer periods of time. However, neither the CX3CR1−/− or the CX3CR1+/− mice significantly improvement motor coordination on the second day of training (Fig. 2a; repeated-measures ANOVA, p < 0.0001). Wild-type mice performed significantly better on Trial 8 compared to Trial 1 than did CX3CR1−/− and CX3CR1+/− mice (Fig. 2b), suggesting motor learning deficits in CX3CR1−/− and CX3CR1+/− mice. There were no significant differences on the first trial of testing between all experimental groups, which suggests that there is no difference in baseline motor skills. Together, these data suggest that CX3CR1−/− and CX3CR1+/− mice have deficits in motor learning, indicating that cerebellar or striatal-dependent learning may be compromised in these mice.
Figure 2.

a–c, CX3CR1−/− and CX3CR1+/− mice show motor learning impairment but normal spontaneous activity. a, On the first day of training (Trials 1–4) no difference between genotypes was observed in the learning ability in the rotarod task. On the second day of training (Trials 5–8) as expected, wild-type mice learned the rotarod task as demonstrated by their ability to remain on the rod for longer periods. Neither the CX3CR1−/− nor the CX3CR1+/− mice showed significant improvement in motor coordination with training when compared to wild-type (repeated-measure ANOVA; p < 0.0001). White circles, WT; gray circles, CX3CR1+/−; black circles, CX3CR1−/−. All data are presented as mean ± SEM. *p < 0.01. b, Comparison of the motor performance of each group of mice on Trial 1 versus Trial 8 shows that wild-type performed significantly better than CX3CR1−/− and CX3CR1+/− mice (linear regression; **p < 0.001). c, CX3CR −/− and CX3CR1+/− mice show normal spontaneous locomotor activity. Wild-type, CX3CR1−/− and CX3CR1+/− mice do not show any difference in total distance traveled in the open field. White bar, WT; gray bar, CX3CR1+/−; black bar, CX3CR1−/−. All data are presented as mean ± SEM.
Absence of CX3CR1 receptor does not affect spontaneous locomotor activity or cause excessive anxiety
To examine spontaneous locomotor activity in response to a novel environment, CX3CR1−/−, CX3CR1+/−, and wild-type mice were tested in the open field behavioral task. The open field task monitors activity in a brightly lit, novel environment and is a necessary control for interpretation of associative fear conditioning. Spontaneous locomotor activity was assessed as the total amount of distance traveled in the chamber. Also, anxiety levels can be measured in the open field task through assessment of the distances traveled in the center versus perimeter of the chamber. This task exploits the natural tendency of mice to avoid open areas. There were no significant differences in total distance traveled (Fig. 2c). In addition, no differences were observed between distance traveled in the center zone compared to the perimeter zone and the time spent in each zone (data not shown). Anxiety levels were also measured using the elevated-plus maze task. Mice generally prefer to be in the closed arms of the maze, but their natural curiosity compels them to explore their environment. Their level of anxiety was determined by the amount of time spent exploring the open arms of the maze versus the time spent in the closed arms. CX3CR1−/− and CX3CR1+/− mice spent similar amounts of time in the open arms and in the center zone compared to wild-type mice (data not shown). Together, the absence of the CX3CR1 receptor does not impair spontaneous locomotor activity nor cause excessive anxiety-like behavior.
Impairment of cognitive function in CX3CR1-deficient mice
Hippocampal-dependent associative learning and memory were assessed in CX3CR1−/−, CX3CR1+/−, and wild-type mice using contextual fear conditioning and cued fear conditioning. Animals were trained with a standard two-shock protocol as described previously (Weeber et al., 2002). Freezing to the context (hippocampal dependent) or to the conditioned stimulus in a novel context (hippocampal and amygdala dependent) was used as an index of memory formation. During training, CX3CR1−/− and CX3CR1+/− mice displayed similar freezing behavior compared to wild-type mice (Fig. 3a). When placed in the context 24 h following training, CX3CR1−/− and CX3CR1+/− mice show a decrease in freezing to the context compared to wild-type mice (Fig. 3b; one-way ANOVA followed by Bonferroni post hoc test, p < 0.001). Interestingly, CX3CR1+/− mice were significantly different from the CX3CR1−/− mice, further suggesting that the loss of one allele is sufficient to affect receptor function. When placed in the novel environment and presented with the conditioned stimulus, there were no significant differences between CX3CR1−/−, CX3CR1+/−, and wild-type mice, suggesting that the behavioral deficits may be limited to hippocampal-dependent behaviors (Fig. 3c).
Figure 3.

CX3CR1−/− mice show impairment in contextual fear conditioning and Morris water maze memory function. CX3CR1−/− (black circles) and CX3CR1+/− (gray circles) are compared to littermate wild-type mice (white circles). a, A tone (solid bar) was paired with a foot shock (arrowhead) at 2 and 4 min. Freezing behavior is shown on the day of training for CX3CR1−/−, CX3CR1+/−, and wild-type mice and is comparable in all groups. b, CX3CR1−/− and CX3CR1+/− mice showed significant reduced freezing response compared to wild-type when tested 24 h following training. c, CX3CR1−/− and CX3CR1+/− mice showed normal freezing to cue component compared to controls. **p < 0.01. d, e, The hidden-platform version of the Morris water maze task. d, Mean latency to escape from a pool to hidden platform across training days. e, A first probe test was performed on Day 7, and a second probe test was performed on Day 11 to determine the number of pseudo-platform crossings in the target quadrant (TQ) compared to the opposite quadrant (OP), the left quadrant (LQ), and the right quadrant (RQ). White bar, WT; gray bar, CX3CR1+/−; black bar, CX3CR1−/−. All data are presented as mean ± SEM. p < 0.01. f, Average swim speed. No differences were seen in the overall swim distance between WT, CX3CR1−/− and CX3CR1+/− mice during the probe test.
To further assess hippocampal-dependent learning and memory, the same group of mice was tested in the hidden-platform water maze task. There were no significant differences in escape latencies between experimental groups during training (Fig. 3d). However, both CX3CR1−/− and CX3CR1+/− mice had a significant decrease in the number of target platform crossings during the probe trial compared to wild-type mice (Fig. 3e; one-way ANOVA followed by Bonferroni post hoc, p < 0.05). No differences were observed between experimental groups on the visible platform test (data not shown). These data further support the role of the CX3CR1 receptor to modulate hippocampal-dependent learning and memory.
CX3CR1-deficient mice have impaired long-term potentiation
LTP is the most widely studied cellular model of memory. The observation that CX3CR1−/− and CX3CR1+/− mice have deficiencies in both associative and spatial memory led us to determine whether the hippocampal-dependent memory deficits observed in these mice correlate with a reduction in synaptic plasticity. Both CX3CR1−/− and CX3CR1+/− mice showed significantly reduced hippocampal-dependent LTP compared to wild-type controls (Fig. 4a–c). Interestingly, examination of basal synaptic transmission revealed no differences between experimental groups (data not shown), suggesting that the decrease in LTP may be due to post–high frequency stimulation-dependent cellular mechanisms and may not represent differences in overall synaptic transmission. Additionally, short-term plasticity evaluated by PPF is found to be normal in these mice, further suggesting that presynaptic function is not affected through decreased or absent levels of CX3CR1 (Fig. 4c; one-way ANOVA followed by Tukey's post hoc, p < 0.01).
Figure 4.
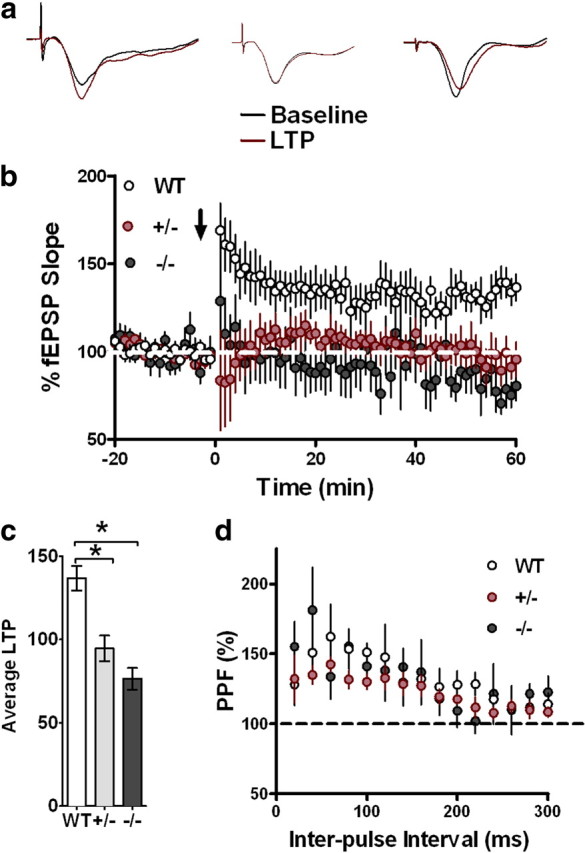
a, b, CX3CR1 deficiency leads to impairments in LTP. LTP was induced with HFS (2 1 s, 100 Hz bursts separated by 20 s; b, arrow) after 20 min of baseline recording, and changes in fEPSP slope are expressed as a percentage of baseline (a). b, Representative fEPSP traces taken from hippocampal slices of (left to right) wild-type, CX3CR1+/− and CX3CR1−/− mice. Both CX3CR1−/− (black) and CX3CR1+/− (red) have deficiencies in LTP of area CA1 compared to wild-type controls (white). c, The last 5 min of fEPSPs slope recordings were averaged for wild-type (n = 12), CX3CR1−/− (n = 10), and CX3CR1+/− (n = 9) mice (**p < 0.05). d, PPF was induced with the use of paired pulses given with an initial delay of 20 ms, and the time to the second pulse was increased 20 ms incrementally until a final delay of 300 ms was reached. There was no significant PPF differences between experimental groups. All data are presented as mean ± SEM. p < 0.01.
It has been shown that hippocampal slice preparations enhance microglial activation (Haynes et al., 2006). Given the well-characterized effects of CX3CR1 signaling in modulating microglial activation, it was important to examine the hippocampal slice preparations for baseline alterations in microglial activation and/or neurotoxicity between WT, CX3CR1+/−, and CX3CR1−/− genotypes. DAPI staining showed no difference between genotype in microglia alteration and or neurotoxicity (data not shown).
CX3CR1-deficient mice have increased microglial activation, hippocampal levels of IL-1β and p38, and cerebellar levels of TNFα
The IL-1 receptor antagonist IL-1ra is the naturally occurring receptor antagonist of IL-1β (Dinarello, 1997; Rothwell et al., 1997). Recently, we demonstrated in young rats that infusion of IL-1ra completely reverses the decrease in hippocampal neurogenesis induced by the coadministration of CX3CR1-blocking antibody (Bachstetter et al., 2011). Next, we wanted to determine whether basal levels of hippocampal IL-1β were altered in CX3CR1−/− mice. We analyzed the hippocampi of these mice for protein expression of IL-1β. Hippocampi from CX3CR1−/− and CX3CR1+/− mice have increased IL-1β expression compared to those from wild-type mice (Fig. 5a; one-way ANOVAs, p < 0.01).
Figure 5.

a, CX3CR1−/− and CX3CR1+/− mice show increased hippocampal protein levels of IL-1β compared to littermate WTs as measured by ELISA. b, Western blot analysis of hippocampi from wild-type, CX3CR1−/−, and CX3CR1+/− mice shows an increase in phospho (p)-p38 protein in CX3CR1−/− and CX3CR1+/− mice compared to that of wild-type (right blot; near-infrared image displayed in gray scale). Relative p-p38 expression normalized to β-actin shows the highest expression in CX3CR1−/− and CX3CR1+/− mice. c, TNFα cerebellar protein levels are significantly increased in CX3CR1−/− and CX3CR1+/− mice compared to wild-type mice as measured by ELISA. d, The area of Iba-1 staining is significant higher in CX3CR1−/− mice compared to wild-type and CX3CR1+/− mice as measured by immunohistochemistry. White bar, WT; gray bar, CX3CR1+/−; black bar, CX3CR1−/−. e, f, Representative photomicrographs of the Iba-1+ cells in the dentate gyrus of wild-type (e) and CX3CR1-deficient mice (f). All data are presented as mean ± SEM. p < 0.01. **p < 0.005; *p < 0.05.
The MAP kinase p38 is a key signal transduction factor involved in the production of IL-1β and TNFα (Guesdon et al., 1993; Rouse et al., 1994; Raingeaud et al., 1995). Due to the importance of p38 in IL-1β signaling, we wanted to determine whether hippocampal levels of phospho-p38 were altered in CX3CR1-deficient mice. Not surprisingly, phospho-p38 protein levels were significantly increased in the hippocampi of CX3CR1−/− and CX3CR1+/− mice compared to wild-type mice (Fig. 5b). CX3CR1−/− mice have impaired motor learning that may represent cerebellar dysfunction. We next determined whether deficiencies in CX3CR1 affected cerebellar TNFα protein levels. TNFα expression was significantly increased in CX3CR1−/− mice compared to wild-types (Fig. 5c). Together, CX3CR1 deficiency leads to increased expression of IL-1β, p38, and TNFα, suggesting increased inflammation of the CNS.
Loss of CX3CR1 leads to overactivation of microglia following an insult. To determine under basal conditions whether lack of CX3CR1 receptor leads to increased activation of microglial cells, we immunostained hippocampal sections from CX3CR1−/−, CX3CR1+/−, and wild-type mice for Iba-1, a marker for both resting and activated microglia. The area of Iba-1 staining is significant higher in CX3CR1−/− compared to wild-type and CX3CR1+/− mice (Fig. 5d; p < 0.05, CX3CR1−/− vs wild-type), reflecting an increased volume of microglial cells reflective of morphology associated with activated microglia.
IL-1ra recovered the LTP deficit of CX3CR1-deficient mice
Increased levels of IL-1β negatively modulate induction and maintenance of LTP (Cunningham et al., 1996; Kelly et al., 2003). Because mice lacking the CX3CR1 receptor have increased levels of IL-1β, we determined whether the IL-1 receptor antagonist, IL-1ra, reverses the impairment in synaptic plasticity observed in the CX3CR1−/− mice. To determine whether IL-1ra can improve synaptic plasticity in CX3CR1-deficient mice, hippocampal slices were perfused with varying concentrations of IL-1ra, and post-tetanic potentiation was induced. In agreement with the LTP results, hippocampal slices from CX3CR1−/− mice had deficits in PTP compared to wild-type mice (Fig. 6a). However, when hippocampal slices from CX3CR1−/− mice were perfused with 100 μg/ml of IL-1ra, the deficit in PTP was rescued (Fig. 6a). Blocking the function of IL-1β completely reversed the impairment in PTP. Our observation that PTP in hippocampal slices from CX3CR1−/− was rescued with perfusion of IL-1ra prompted us to determine whether the long-lasting LTP deficit in CX3CR1−/− mice could be rescued as well. Perfusion of 100 μg/ml of IL-1ra to hippocampal slices from CX3CR1−/− mice completely rescued the deficit in LTP induction and maintenance (Fig. 6b). Together, these results suggest that the deficits seen in CX3CR1−/− are due to increased levels of IL-1β, further implicating a detrimental increase of inflammation in the CNS.
Figure 6.
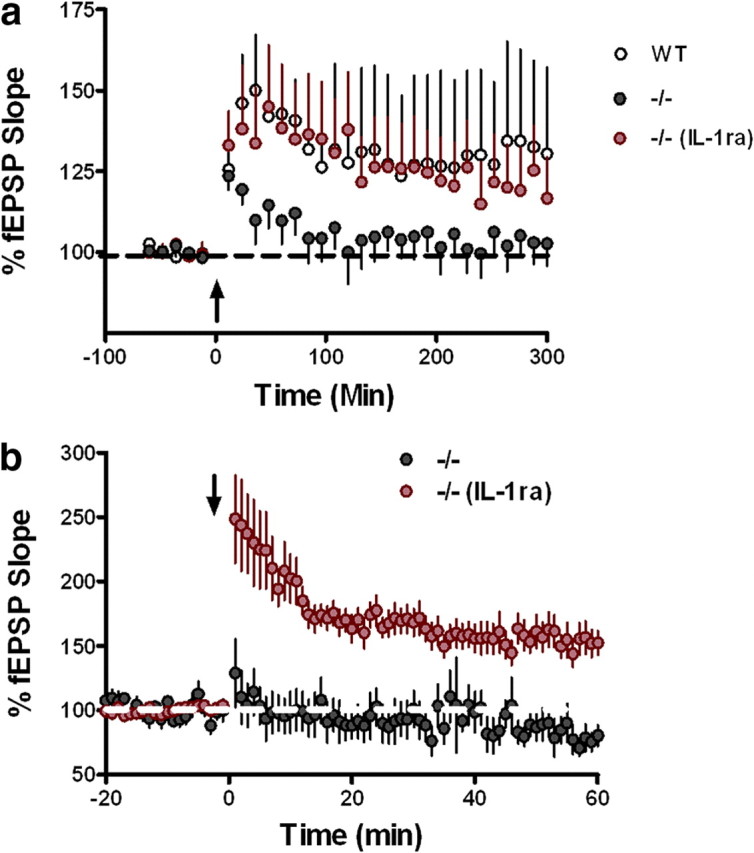
a, b, CX3CR1 deficiency leads to impairments in synaptic plasticity which are rescued through IL-1β antagonism. PTP was induced with HFS (1 s, 100 Hz bursts; arrow) after 1 min of baseline recording. LTP was induced with HFS (2 1 s, 100 Hz bursts separated by 20 s; arrow) after 20 min of baseline recording. Changes in fEPSP slope are expressed as a percentage of baseline. a, The IL-1β antagonist, IL-1ra (100 μg/ml), rescues the PTP deficit (red; n = 7) in hippocampi taken from CX3CR1−/− (black; n = 8) in area CA1 to levels seen in wild-type (white; n = 9). b, The LTP deficit seen in hippocampi taken from CX3CR1−/− (black; n = 10) mice is rescued with the IL-1β antagonist IL-1ra (100 μg/ml) (red; n = 11).
IL-1ra reversed the deficit in hippocampal-dependent cognitive function but not motor learning in CX3CR1-deficient mice
To determine whether the impairment in motor learning and cognitive function was also dependent on IL-1β, we performed a second study in which wild-type and CX3CR1−/− mice were infused with either IL-1ra, heat-inactivated IL-1ra, or ACSF. In agreement with the first study, mice were tested starting from the least invasive task (open field) to the most invasive task (Morris water maze) (McIlwain et al., 2001).
Open field and elevated-plus maze
Although in the first study there were no significant impairments in general locomotor activity or explorative behavior or anxiety levels of the CX3CR1-deficient mice and wild-types, it was crucial to retest the performance of new groups of mice to exclude the possibility that the surgery procedure could impair general motor activity or enhance anxiety levels. Wild-type mice infused with ACSF (n = 11), heat-inactivated IL-1ra (n = 12), IL-1ra (n = 11), or CX3CR1−/− infused with either ACSF (n = 14) or IL-1ra (n = 13) were first tested in the open field behavioral task. Spontaneous locomotor activity was assessed as the total amount of distance traveled in the chamber. As observed in the first study, there were no significant differences between genotypes in total distance traveled. Furthermore, no significant differences were observed between treatments (data not shown). In addition, the distance traveled in the center zone compared to the perimeter zone and the time spent in each zone were similar between groups (data not shown). Anxiety levels were also measured using the elevated-plus maze task. Their levels of anxiety were determined by the amount of time spent exploring the open arms of the maze versus the time spent in the closed arms. As shown previously, all groups spent similar amounts of time in the open arms and in the center zone compared to wild-type mice, and no effect of IL-1ra was observed (data not shown).
Rotarod
Wild-type mice (ACSF, n = 11; heat-inactivated IL-1ra, n = 12; IL-1ra, n = 11) and CX3CR1−/− mice (ACSF, n = 14; IL-1ra, n = 13) were then tested on the rotarod as described above. No differences were observed between genotypes during the first day of training (Fig. 7a). On the second day of training (Trials 5–8), wild-type mice remained on the rod for longer periods of time compared to CX3CR1−/− mice. However, neither CX3CR1−/− treated with IL-1ra or CX3CR1−/− control mice showed significantly improvement in motor coordination on the second day of training (Fig. 7a; two-way ANOVA; trial, p < 0.0001; treatment, p < 0.0001; interaction, p = 0.69). As demonstrated in the first study (Fig. 2a), all wild-type mice performed significantly better on Trial 8 compared to Trial 1 than the CX3CR1−/− control group (Bonferroni post hoc test, WT ACSF, heat-inactivated IL-1ra, and WT IL-1ra vs CX3CR1−/−, p < 0.05). CX3CR1−/− IL-1ra-treated mouse motor performance was not different from the CX3CR1−/− control group (CX3CR1−/− vs CX3CR1−/− IL-1ra, p > 0.05; data not shown), suggesting that IL-1ra does not modulate the motor learning impairment. There were no significant differences on the first trial of testing between all experimental groups, which suggests that there was no difference in baseline motor skills (data not shown). Together, these data suggest that IL-1β does not modulate the deficits in motor learning observed in the CX3CR1−/− mice.
Figure 7.

IL-1ra reversed the deficit in contextual fear conditioning and Morris water maze memory function induced by the loss of CX3CR1 but not the deficit in motor learning. a, CX3CR1−/− ACSF-treated (blue squares) and CX3CR1−/− IL-1ra-treated mice (red squares) are compared to wild-type ACSF-treated mice (open circles), wild-type mice treated with heat-inactivated IL-1ra (black squares), and wild-type mice treated with IL-1ra (green squares). On the first day of training (Trials 1–4), no difference between groups was observed in the learning ability on the rotarod task. On the second day of training (Trials 5–8), all wild-type mouse groups learned the rotarod task as demonstrated by their ability to remain on the rod for longer periods. Neither the CX3CR1−/− ACSF-treated nor CX3CR1−/− IL-1ra-treated mice showed significant improvement in motor coordination with training when compared to wild-type. b, A tone (solid bar) was paired with a foot shock (arrowhead) at 2 and 4 min. Freezing behavior is shown on the day of training for CX3CR1−/− ACSF-treated mice (blue squares), CX3CR1−/− IL-1ra-treated mice (red squares), wild-type ACSF-treated mice (white circles), wild-type treated with heat-inactivated IL-1ra (black squares), and wild-type IL-1ra-treated mice (green squares) and is comparable in all groups. c, CX3CR1−/− control mice showed significantly reduced freezing compared to all wild-type groups when tested 24 h after training. d, CX3CR1−/− treated with IL-1ra (red squares) showed a freezing response similar to that of wild-type groups. d–f, The hidden-platform version of Morris water maze task. Mean latency to escape from a pool to hidden platform across training days (d). A probe test was performed on Day 10 to determine the number of pseudo-platform crossings in the target quadrant (TQ) compared to the opposite quadrant (OP) (e). White bar, WT ACSF-treated mice; light gray bar, wild-type treated with heat-inactivated IL-1ra; dark gray bar, wild-type treated with IL-1ra; black bar, CX3CR1−/− ACSF-treated mice; black/white bar, CX3CR1−/− IL-1ra-treated mice. f shows the time spent in the target platform zone. CX3CR1−/− ACSF-treated mice (black bar) spent significantly less time in the target zone compared to wild-type controls [white bar, wild-type ACSF-treated mice; light gray bar, wild-type heat-inactivated IL-1ra-treated mice; dark gray bar, wild-type IL-1ra-treated mice]. CX3CR1−/− mice treated with IL-1ra spent the same amount of time in the target quadrant compared to wild-type controls. All data are presented as mean ± SEM. p < 0.01. *p < 0.05 (CX3CR1−/− control vs wild-type controls); ##p < 0.001 (CX3CR1−/− IL-1ra vs CX3CR1−/−).
Contextual fear conditioning
To address the role of IL-1β in the cognitive impairment observed in CX3CR1-deficient mice, we trained wild-type mice (three subgroups, ACSF, heat-inactivated IL-1ra, and IL-1ra) and CX3CR1−/− mice (two subgroups, ACSF and IL-1ra) on contextual fear conditioning and auditory cue fear conditioning as described above. During training, all groups showed similar levels of freezing in response to the first and second pairings of tone and mild foot shock (Fig. 7b). This indicates that all groups have the capability of freezing and that the acquisition of fear memory is indistinguishable between the two groups. When placed in the training context 24 h following training, CX3CR1−/− mice showed a significant reduction in the amount of freezing compared to wild-type mice (Fig. 7c; p < 0.0005; Bonferroni post hoc test, WT ACSF vs CX3CR1−/−, p < 0.05; wild-type heat-inactivated IL-1ra vs CX3CR1−/−, p < 0.001). Interestingly, IL-1ra completely reversed the deficit in contextual memory in CX3CR1−/− mice (Bonferroni post hoc test, p < 0.001), suggesting that IL-1β modulates the deficit in contextual memory due to the loss the loss of CX3CR1. Wild-type mice treated with IL-1ra were not significantly different from CX3CR1−/− controls or wild-type controls. In contrast, all groups exhibited identical responses to the presentation of the auditory CS when presented in a different context (data not shown). These data suggest that IL-1β action is specific for hippocampal-dependent cognitive function. To control for possible differences in response to the foot shock, a shock threshold test was performed. No differences were observed in the perception of the foot shock delivery as assessed by determining the stimulus amplitude necessary to evoke flinching, jumping, or a vocalization (data not shown).
Morris water maze
Following the observation that IL-1 directly modulated the impairment in contextual memory in CX3CR1-deficient mice, we sought to determine whether IL-1β also modulated the deficit observed previously in spatial learning (Fig. 3). To answer this question, 8 d following fear conditioning, the same animals were trained to locate a submerged platform in a circular pool filled with opaque water as described in the first study, a probe trial was performed after 9 d of training matching the data shown from study 1. No difference were observed between groups in the latencies to find the platform during training, indicating that all animals have normal acquisition of spatial information (Fig. 7d). We then analyzed memory retention with a probe trial where the platform was removed and mice were allowed to free-swim. CX3CR1−/− control mice had a significant decrease in the number of target platform crossings during the probe trial compared to wild-type control mice (Fig. 7e; two-way ANOVA, p = < 0.0001; followed by Bonferroni post hoc, WT ACSF and WT heat-inactivated IL-1ra vs CX3CR1−/− control, p < 0.05). Importantly IL-1ra was able to completely reverse the deficit in spatial memory observed in CX3CR1-deficient mice (CX3CR1−/− control vs CX3CR1−/− IL-1ra, p < 0.05). No differences were observed between experimental groups on the visible platform test (data not shown). In addition, we analyzed the time spent in the target platform zone. CX3CR1−/− mice spent significantly less time in the target zone compared to wild-type controls (Fig. 7f, two-way ANOVA, p < 0.0001; followed by Bonferroni post hoc test, wild-type ACSF or wild-type heat-inactivated IL-1ra vs CX3CR1−/− control IL-1ra, p < 0.05). CX3CR1−/− mice treated with IL-1ra spent the same amount of time in the target zone compared to control (CX3CR1−/− control vs CX3CR1−/− IL-1ra, p < 0.001). No difference was observed in the number of crossing or the time spent in the target quadrant between CX3CR1−/− control mice and wild-type mice treated with IL-1ra. These data further support the role of the CX3CR1 receptor in modulating hippocampal-dependent learning and memory and that IL-1β modulates the impairment in hippocampus-dependent cognitive function.
Discussion
For the first time, the present study tests the hypothesis that CX3CL1 signaling through CX3CR1 may be necessary for normal synaptic function. We demonstrated a physiological role of the chemokine receptor CX3CR1 in regulating cognitive function and synaptic plasticity. Furthermore, we showed that genetic disruption of CX3CR1 impairs hippocampal neurogenesis, motor learning, associative memory, spatial memory, and the induction of LTP. Importantly, these deficits appear to be gene-dose dependent and related to increased levels of IL-1β.
These novel discoveries provide important insight to understanding the involvement of the CX3CR1 receptor under physiological conditions. Previously, we showed that CX3CR1−/− mice have decrease hippocampal neurogenesis compared to heterozygote littermates (Bachstetter et al., 2011). We now demonstrate a gene-dose-dependent effect of CX3CR1 deficiency as demonstrated by heterozygous CX3CR1 mice that exhibit an intermediate phenotype. This effect was observed in associative and spatial memory formation, which indicates that the loss of a single allele is sufficient to impair the receptor function.
Recently, a gene-dose effect of CX3CR1 receptor was reported that related to fibrillar Aβ deposition in APP-PS1 mice crossed with CX3CR1−/−, CX3CR1+/−, or CX3CR1+/+ mice (Lee et al., 2010). Reduction in fibrillar Aβ deposition in the APP-PS1 mouse model of AD was gene-dose dependent on CX3CR1 deficiency, with CX3CR1+/− mice displaying an intermediate phenotype. The present study is in agreement with those previous results and underscores the profound implication of this discovery toward interpretation of all previous studies using only CX3CR1−/− and CX3CR1+/− mice.
We report a novel physiological role for the CX3CR1 receptor in the regulation of hippocampal-dependent memory formation. The impairment of LTP and neurogenesis likely represents the mechanism responsible for the defect observed in hippocampal-dependent associative and spatial memory formation. However, multiple mechanisms can account for the impairment in cognitive function and synaptic plasticity observed in the CX3CR1-defient mice.
CX3CL1 has been implicated as a endogenous neuronal modulator, which controls the overproduction of iNOS, IL-1β, TNFα, and IL-6 (Biber et al., 2007). CX3CL1 levels are downregulated with neuronal injury, which results in microglial recruitment, activation, and increased production of TNFα, IL-1β, and p38 MAP kinase. IL-1β is a proinflammatory cytokine, which is constitutively expressed in the CNS, particularly in the hippocampus, and is synthesized by neurons and glial cells following neuronal injury (Rothwell et al., 1997; Dinarello, 1998). Binding studies demonstrate that the hippocampus contains the highest density of IL-1β binding sites (Farrar et al., 1987; Takao et al., 1990). These findings suggest that the effects of IL-1β might be specific to hippocampus, which may affect hippocampal-dependent learning and memory processes. Furthermore, there may be a causal relationship between the decrease of LTP and increase of IL-1β expression.
In the present study, we demonstrate that hippocampal protein levels of IL-1β and phospho-p38 are significantly increased in mice lacking CX3CR1 receptor. This increase was accompanied by a change in morphology, consistent with the activation of hippocampal microglia cells, observed as an increased area of Iba-1 staining. Furthermore, the increase of these protein levels was accompanied by a decrease in LTP in CX3CR1-deficient mice. The deficit in LTP was gene-dose dependent, with CX3CR1+/− mice demonstrating an intermediate phenotype. Importantly, both the deficits in LTP and cognitive function were rescued with administration of IL-1ra, which blocked the IL-1 receptor I. Under physiologic circumstances, IL-1β seems to be required for normal learning and memory processes. This assertion is derived from the observation that mice with targeted deletion of IL-1RI exhibit impairments in memory and synaptic plasticity (Hirsch et al., 1996). Similarly, the administration of IL-1ra impairs fear conditioning (Pugh et al., 1999; Rachal et al., 2001) and performance in the Morris water maze in young rats (Yirmiya et al., 2002). In agreement with the literature, our results show that wild-type mice treated with IL-1ra are not different from CX3CR1−/−-deficient mice in the response to contextual freezing and Morris water maze task. Thus, physiologic levels of IL-1β might be critical for normal memory formation in the hippocampus and the increased IL-1β levels observed with age might impair hippocampal-dependent learning and memory.
In addition to the impairment of memory function, CX3CR1-deficient mice have impairments in motor learning. Interesting, cerebellar levels of TNFα were significantly increased in mice lacking CX3CR1 receptor. This result is in agreement with previous reports that show that TNFα modulates age-dependent deficits in motor learning (Cartford et al., 2002). Also, we demonstrate an important, physiological role of the CX3CR1 receptor in normal memory formation and synaptic plasticity. Furthermore, our results demonstrate that disruption of CX3CR1 signaling triggers a downstream increase in IL-1β, which appear to be the key modulator responsible for the detrimental effect of loss of CX3CL1/CX3CR1 signaling on cognitive function.
We clearly demonstrate that under nonpathological conditions, disruption of CX3CL1/CX3CR1 signaling results in increased expression of IL-1β and p38, which is accompanied by the loss of synaptic plasticity and memory formation. Furthermore, these effects appear to be gene-dose dependent. This observation needs to be considered when interpreting data associated with disease conditions or under conditions where heterozygous mice were used as controls for CX3CR1−/− mice. However, this discovery does not negate all of the important findings to date with regard to the role of CX3CR1/CX3CL1 signaling in many disease conditions.
There is substantial literature in the area of CX3CR1/CX3CL1 signaling, yet no consensus as to the role of this pathway as either neuroprotective or neurodegenerative has been agreed upon. In AD mouse models, CX3CR1−/− microglia have increased phagocytosis, which helps to clear Aβ (Lee et al., 2010). Deletion of the CX3CR1 gene in a triple transgenic (3×Tg) mouse model of AD is protective against neuronal loss (Fuhrmann et al., 2010). However, CX3CR1-deficient 3×Tg mice were examined at an age before the development of either extracellular Aβ deposition or intracellular MAPT (microtubule-associated protein tau) aggregation. Furthermore, CX3CR1−/− mice crossed with the AD mouse model CRND8 had lower brain levels of Aβ40 and Aβ42 and reduced amyloid deposits, which are related to increased microglia proliferation and phagocytosis (Liu et al., 2010). Finally, CX3CR1−/− mice crossed with mice overexpressing hTau have increased hyperphosphorylated tau and increased toxicity, likely related to the increase p38 and IL-1β (Bhaskar et al., 2010) expression. However, the mechanisms underlying CX3CR1 signaling that lead to neurotoxicity remain unclear. In conclusion, this study demonstrates that CX3CR1 receptors play a physiological role in normal hippocampal-dependent cognitive function and synaptic plasticity. In addition, the present study provides mechanistic links between CX3CR1 and the release of IL-1β and phospho-p38, ultimately implicating an increase in inflammation with the deficits observed in synaptic plasticity and cognition.
Footnotes
This work was supported by United States Public Health Service Grant AG-04418 and the VA Medical Research Service (P.C.B.). Amgen (Thousand Oaks, CA) provided the IL-1Ra as a kind gift.
The authors declare no competing financial interests.
References
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, Brewster KW, Hudson CE, Cole MJ, Harrison JK, Bickford PC, Gemma C. Fractalkine and CX(3)CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging. 2011;32:2030–2044. doi: 10.1016/j.neurobiolaging.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal “on” and “off” signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Cartford MC, Gemma C, Bickford PC. Eighteen-month-old Fischer 344 rats fed a spinach-enriched diet show improved delay classical eyeblink conditioning and reduced expression of tumor necrosis factor alpha (TNFalpha) and TNFbeta in the cerebellum. J Neurosci. 2002;22:5813–5816. doi: 10.1523/JNEUROSCI.22-14-05813.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O'Neill LA, Lynch MA, O'Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–265. doi: 10.1016/s1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- Farrar WL, Kilian PL, Ruff MR, Hill JM, Pert CB. Visualization and characterization of interleukin 1 receptors in brain. J Immunol. 1987;139:459–463. [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Bachstetter AD, Cole MJ, Fister M, Hudson C, Bickford PC. Blockade of caspase-1 increases neurogenesis in the aged hippocampus. Eur J Neurosci. 2007;26:2795–2803. doi: 10.1111/j.1460-9568.2007.05875.x. [DOI] [PubMed] [Google Scholar]
- Guesdon F, Freshney N, Waller RJ, Rawlinson L, Saklatvala J. Interleukin 1 and tumor necrosis factor stimulate two novel protein kinases that phosphorylate the heat shock protein hsp27 and beta-casein. J Biol Chem. 1993;268:4236–4243. [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Irikura VM, Paul SM, Hirsh D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc Natl Acad Sci U S A. 1996;93:11008–11013. doi: 10.1073/pnas.93.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J Biol Chem. 2003;278:19453–19462. doi: 10.1074/jbc.M301938200. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R. The use of behavioral test batteries: effects of training history. Physiol Behav. 2001;73:705–717. doi: 10.1016/s0031-9384(01)00528-5. [DOI] [PubMed] [Google Scholar]
- Miwa T, Furukawa S, Nakajima K, Furukawa Y, Kohsaka S. Lipopolysaccharide enhances synthesis of brain-derived neurotrophic factor in cultured rat microglia. J Neurosci Res. 1997;50:1023–1029. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1023::AID-JNR13>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- O'Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia. 2002;39:85–97. doi: 10.1002/glia.10081. [DOI] [PubMed] [Google Scholar]
- Peters MM, Hill KE, Burk RF, Weeber EJ. Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol Neurodegener. 2006;1:12. doi: 10.1186/1750-1326-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CR, Nguyen KT, Gonyea JL, Fleshner M, Wakins LR, Maier SF, Rudy JW. Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res. 1999;106:109–118. doi: 10.1016/s0166-4328(99)00098-4. [DOI] [PubMed] [Google Scholar]
- Rachal PC, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25:29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Rothwell N, Allan S, Toulmond S. The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100:2648–2652. doi: 10.1172/JCI119808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- Takao T, Tracey DE, Mitchell WM, De Souza EB. Interleukin-1 receptors in mouse brain: characterization and neuronal localization. Endocrinology. 1990;127:3070–3078. doi: 10.1210/endo-127-6-3070. [DOI] [PubMed] [Google Scholar]
- Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG, Chew EY, Chan CC. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optional fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Winocur G, Goshen I. Brain interleukin-1 is involved in spatial memory and passive avoidance conditioning. Neurobiol Learn Mem. 2002;78:379–389. doi: 10.1006/nlme.2002.4072. [DOI] [PubMed] [Google Scholar]