

Abstract
Non-alcoholic fatty liver disease (NAFLD) is currently evolving as the most common liver disease worldwide. It may progress to liver cirrhosis and liver cancer and is poised to represent the most common indication for liver transplantation in the near future. The pathogenesis of NAFLD is multifactorial and not fully understood, but it represents an insulin resistance state characterized by a cluster of cardiovascular risk factors including obesity, dyslipidemia, hyperglycemia, and hypertension. Importantly, NAFLD also has evolved as independent risk factor for cardiovascular disease. Unfortunately thus far no established treatment does exist for NAFLD. The bile acid-activated nuclear farnesoid X receptor (FXR) has been shown to play a role not only in bile acid but also in lipid and glucose homeostasis. Specific targeting of FXR may be an elegant and very effective way to readjust dysregulated nuclear receptor-mediated metabolic pathways. This review discusses the body's complex response to the activation of FXR with its beneficial actions but also potential undesirable side effects.
1. Introduction
One characteristic of our modern civilization is the easy and unlimited access to unhealthy and caloric dense food. A typical American diet furnishes the liver with ~20 g of fat each day, equivalent to one-half of the total triglyceride content of the liver. In combination with little need for physical activity due to technological advances, one consequence of our sedentary and excessive lifestyle is non-alcoholic fatty liver disease (NAFLD).
NAFLD is a major health problem affecting up to 60 million Americans and evolving as the most common liver disease worldwide [1, 2]. This is several-fold higher than other common chronic liver diseases such as hepatitis C and alcohol-related liver disease. While the majority of subjects with NAFLD are obese, the condition can occur in the absence of obesity or other features of the metabolic syndrome. In patients with diabetes and morbid obesity the prevalence of NAFLD has been shown to be as high as 62% and 96%, respectively [3, 4].
The earliest stage of NAFLD is fatty liver that is defined as the presence of cytoplasmic triglyceride droplets in more than 5% of hepatocytes [5]. Although often self-limited, in 12–40% it can progress to non-alcoholic steatohepatitis (NASH) [6]. NASH is distinguished from simple fatty liver by the presence of hepatocyte injury such as hepatocyte ballooning and apoptosis, an inflammatory infiltrate, and/or collagen deposition. Over a time period of 10–15 years, 15% of patients with NASH will progress to liver cirrhosis [7]. Once cirrhosis has developed in the absence of viral hepatitis, hepatic decompensation occurs at a rate of 4% annually while the ten-year risk of developing liver cancer is 10% [7, 8]. Although liver cancer secondary to NASH typically develops in the setting of cirrhosis, carcinogenesis can occur in the absence of advanced liver disease. It is thus not surprising that NAFLD is poised to become the primary indication for liver transplantations. Like other causes of chronic liver disease, NASH recurs following liver transplantation almost universally [9].
2. Basic Pathophysiological Concepts and Treatment of NAFLD
The pathogenesis of NAFLD is multifactorial and only partially understood. Fatty liver arises in the setting of an imbalance between triglyceride formation/acquisition and removal (Figure 1). Assembly of triglycerides and lipid droplet formation requires fatty acids that can derive from diet, de novo synthesis, or adipose tissue. Dietary fat packed in chylomicrons is hydrolyzed releasing free fatty acids of which approximately 20% are delivered to the liver [8]. Carbohydrate-enriched diets promote de novo synthesis of free fatty acids via insulin-stimulated activation of sterol regulatory element-binding protein-1c [10, 11]. In addition, glucose facilitates lipogenesis via activation of carbohydrate responsive element-binding protein [12]. In the fasting state, a decline of insulin levels stimulates adipocyte triglyceride hydrolase thereby releasing free fatty acids that are transported to the liver [13]. In the liver, free fatty acids can be (i) used for energy and ketone body production via mitochondrial β-oxidation, (ii) esterified and stored as triglycerides in lipid droplets, or (iii) packaged with apolipoprotein B into very low-density lipoproteins that are secreted into the circulation. As the liver extracts approximately 20% of free fatty acids from the circulation, the daily input of triglycerides from diet and fatty acids from adipocyte tissue is equivalent to the entire triglyceride content of the liver [14]. Once the capacity of the liver to store fatty acids in form of triglycerides is overwhelmed, NASH, differentiated from a fatty liver by the presence of increased cell injury, apoptosis, inflammation, and fibrosis, starts to develop. A detailed review of the steps involved in the progression of NAFLD to NASH and cirrhosis has been recently published [15].
Figure 1.
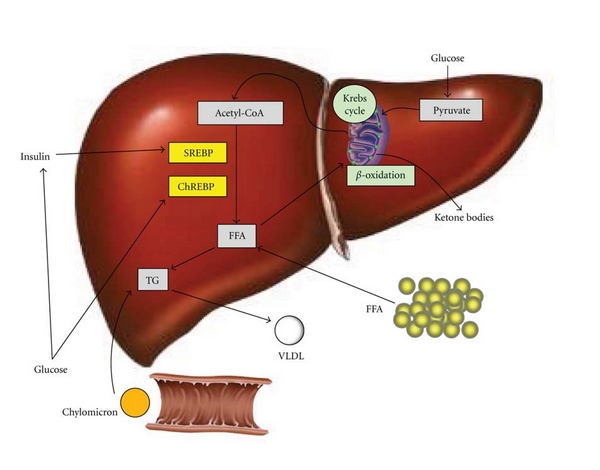
Hepatic triglyceride (TG) formation, acquisition, and removal. Fatty liver is a result of an imbalance between free fatty acids (FFAs), and TG input and FFA and TG output. FFA derives from peripheral tissue, endogenous synthesis or diet in form of chylomicrons. Carbohydrate intake increases glucose and insulin levels thereby promoting lipogenesis through the activation of transcription factors sterol regulatory element-binding protein 1c (SREBP) and carbohydrate responsive element-binding protein (ChREBP). Reducing the FFA burden include β-oxidation in mitochondria, storage as TG, or export as very low-density lipoprotein (VLDL).
Treatment of NAFLD should either prevent disease progression to liver cirrhosis or reverse already established NASH, respectively. Despite many advances in our understanding of the pathogenesis of NAFLD, there is currently no established treatment available. Life-style changes and exercise to reduce body weight and treatment of concomitant diabetes and dyslipidemia are accepted first-line therapy but have not been shown to convincingly reduce the risk of disease progression [16]. Therefore exploring new avenues for treatment of this common disease is crucial.
3. The Bile Acid-Activated Nuclear Farnesoid X Receptor (FXR)
Nuclear receptors are a group of transcription factors that consist of 48 members in humans. They have a common structure consisting of a ligand-independent activation domain for interaction with cofactors, a central DNA binding domain, and a unique ligand binding domain allowing receptor dimerization and coregulator interactions. Most nuclear receptors function either as homodimers or as heterodimers with the retinoid X receptor. Binding of the ligand promotes conformational changes facilitating the release of corepressors and resulting in conformational changes of chromatin enabling access of the transcriptional machinery to the respective promoters. Upon ligand activation, the corepressor complex dissociates and the coactivator complex is recruited allowing start of transcription. Control of nuclear transcriptional activity is also thought to occur by posttranslational modifications [17–19].
In 1995 a protein was discovered that was interacting with the human retinoic X receptor and named retinoic X receptor-interacting protein 14 [20]. Because it was activated by an intermediate of the mevalonate pathway, farnesol, it was renamed to farnesoid X receptor [21]. Another four years later, three independent groups [22–24] discovered bile acids as endogenous ligands for FXR. From an evolutionary point of view the FXR gene is highly conserved suggesting that it plays an important role in many species. At the tissue level, FXR is expressed predominantly in the liver, intestine, kidney, and adrenal gland. Expression in heart and adipose tissue is low [25]. The generation of mice with Fxr gene ablation identified FXR as a master regulator in bile acid homeostasis [26]. Subsequently novel functions of FXR have been identified including protecting the intestinal barrier and modulating the innate immunity [27, 28] and tumorigenesis [29, 30]. The most important roles of FXR are likely in regulating metabolic processes.
4. FXR as Key Player in Multiple Metabolic Processes
For a long time, physiological effects of bile acids have mainly been attributed to their physicochemical properties [31]. In the last couple of years it has been evident that bile acids act like signaling molecules [32] regulating not only their own homeostasis during the enterohepatic circulation but also triglyceride, cholesterol, and glucose metabolism.
4.1. Bile Acid Metabolism
A major physiological role of FXR in bile acid metabolism is to protect hepatocytes from the deleterious effects of increased bile acid levels by inhibiting endogenous bile acid synthesis and accelerating bile acid biotransformation and excretion. In this regard, FXR-mediated effects occur in a tightly coordinated fashion at the level of the hepatocyte and enterocyte and have been reviewed in detail elsewhere [33].
4.2. Triglyceride and Cholesterol Metabolism
It has been known for years that bile acids can modulate lipid metabolism in humans. Reducing the transhepatic flux of bile acids decreases low-density lipoprotein cholesterol and increases high-density lipoprotein cholesterol and very low-density lipoprotein triglyceride levels. Opposite effects are observed when the bile acid pool is expanded [34–36]. Studies in mice with Fxr gene ablation or administering FXR agonists provided key information demonstrating a central role of FXR in lipid homeostasis.
As illustrated in Figure 2, FXR activation of short heterodimer partner is required to suppress sterol regulatory element-binding protein 1c expression [37]. As sterol regulatory element-binding protein 1c is known to regulate several genes involved in fatty acid and triglyceride formation [11], FXR-mediated repression of sterol regulatory element-binding protein 1c inhibits triglyceride and fatty acid synthesis and secretion. Interestingly, recent studies support the concept that FXR-independent mechanisms may also contribute [38]. In addition to decreasing lipogenesis, activation of FXR facilitates the clearance of very low-density lipoproteins and chylomicrons. This is achieved by increasing the expression of the very low-density lipoprotein receptor [39], a protein that enhances lipoprotein lipase-mediated triacylglycerol hydrolysis. Very low-density lipoprotein assembly is controlled by FXR via repressing the expression of microsomal triglyceride transfer protein and apolipoprotein B [38]. FXR also activates syndecan-1, a transmembrane protein that binds remnant particles before their transfer to receptors [40]. Activation of lipoprotein lipase, a key enzyme involved in the lipolysis of triglyceride rich lipoproteins, is also FXR-dependent. This involves activation of apolipoproteins C-II and AIV [41–43] and inhibiting the expression of apolipoprotein C-III [44] and angiopoetin-like 3 [37], respectively. Another effect of FXR activation is the induction of peroxisome proliferator-activated receptor α that promotes fatty acid β-oxidation [45]. Collectively these findings support the concept that FXR activation decreases plasma triglyceride levels by suppressing hepatic lipogenesis and triglyceride secretion and increasing the clearance of triglyceride-rich lipoproteins from blood. These observations therefore support the concept that FXR activation may have a beneficial effect in patients with NAFLD by decreasing hepatic lipogenesis.
Figure 2.
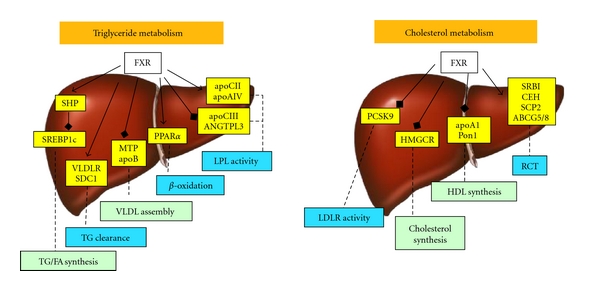
Effect of FXR activation on triglyceride and cholesterol metabolism in the liver. FXR agonists result in a variety of responses modulating triglyceride (TG) and cholesterol metabolism. Activation of FXR inhibits triglyceride (TG)/fatty acid (FA) synthesis facilitated by suppressing sterol regulatory element-binding protein 1c (SREBP1c) via activation of short heterodimer partner (SHP). FXR controls assembly of very low-density lipoprotein (VLDL). FXR may increase the clearance of TG by stimulating lipoprotein lipase (LPL) activity as well as the hepatic uptake of remnants and low-density lipoprotein by inducing syndecan 1 (SDC1) and the VLDL receptor (VLDLR). FXR agonists may modulate LDL receptor activity via inhibition of proprotein convertase subtilisin/kexin 9 (PCSK9) and activate the reverse cholesterol transport pathway (RTC). FXR activation also impairs high-density lipoprotein (HDL) formation and suppresses cholesterol synthesis. apoA1, apoB, apoCII, apoCIII, apoAIV: apolipoprotein A1, B, CII, CIII, AIV; ANGTPL3: angiopoetin like 3; ABCG5/8: adenosine triphosphate binding cassette subfamily G member 5/8; CEH: cholesterylester hydrolase; HMGCR: 3-hydroxy-3-methylglutaryl coenzyme A reductase; MTP: microsomal triglyceride transfer protein; PON1: paraoxonase 1; SRBI: scavenger receptor B1; SCP2: sterol carrier protein 2.
Activation of FXR also modulates the reverse cholesterol transport, a pathway that promotes cholesterol delivery from the periphery to the liver for biliary disposal and fecal elimination. In this scenario, the selective uptake of high-density lipoprotein cholesteryl ester via scavenger receptor BI [46], intracellular cholesteryl ester hydrolysis facilitated by neutral cholesteryl ester hydrolase [47], as well as the canalicular routing of cholesterol by sterol carrier protein 2 [48] for biliary excretion via adenosine triphosphate binding cassette subfamily G member 5/8 [49] are positively regulated by FXR [50]. In addition but controversial, FXR appears to suppress apolipoprotein A-I expression [46, 50, 51], the primary protein constituent of high-density lipoprotein defining its size and shape. This may be of particular importance as it could influence the capability of high-density lipoprotein to remove cholesterol from peripheral cells, activating the lecithin-cholesterol acyl transferase enzyme and delivering the resulting cholesteryl ester to the liver. Another target of FXR is paraoxonase 1, a protein produced in the liver with phospholipase A2 activity that may be important for inactivation of proatherogenic lipids produced by oxidative modification of low-density lipoprotein. FXR-mediated repression of paraoxonase 1 involves the induction of fibroblast growth factor 19, its subsequent binding to the fibroblast growth factor receptor 4, and activation of the c-Jun N-terminal kinase pathway [52, 53]. FXR also regulates the expression of phospholipid transfer protein [54] that is responsible for the transfer of phospholipids and cholesterol from low to high-density lipoprotein and suppresses 3-hydroxy-3-methyl-glutaryl-CoA reductase likely involving sterol regulatory element-binding protein 2 [55]. Finally, FXR represses proprotein convertase subtilisin/kexin 9 [56], a protein that promotes the intracellular degradation of the low-density lipoprotein receptor by interfering with its recycling to the plasma membrane. In summary, these findings raise concern that activation of FXR may alter the cholesterol metabolism in a way that increases the susceptibility to atherosclerosis and thus limit its application in patients with NAFLD.
4.3. Glucose Homeostasis
In addition to their pleiotropic effects on lipid metabolism, bile acids also affect glucose homeostasis. This is supported by an improved glycemic control in patients with diabetes mellitus response to cholestyramine [57]. Several studies addressed the role of bile acids and FXR activation in glucose metabolism, but the underlying mechanisms are far from being understood. It appears clear that FXR exerts a role in glucose homeostasis [58]. In the state of Fxr gene ablation, the failure to suppress gluconeogenesis and a reduced peripheral glucose disposal led to glucose intolerance [59–61]. A potential molecular basis for these observations is the suppression of hepatic phosphenoyl-pyruvate carboxykinase and glucose 6-phosphatase [60, 62]. Reduced plasma levels of free fatty acids in response to FXR activation (see above) may explain the increased insulin sensitivity in the liver. Of note, FXR activation was shown to enhance insulin-stimulated glucose uptake as well as insulin signaling in adipocytes [61]. It should be noted that bile acids also modulate glucose homeostasis in an FXR-independent fashion through cell signaling pathways [63]. Collectively these findings suggest that FXR activation might prove useful in the treatment of hyperglycemia and hyperlipidemia that are present in patients with NAFLD.
4.4. Hepatic Inflammation and Fibrogenesis
Inflammation and collagen deposition in the liver are key histopathological features of NASH. FXR appears to antagonize hepatic inflammatory processes by antagonizing the nuclear factor kappa B pathway [64]. Another protective FXR mechanism involves induction of antimicrobial factors in the intestine [65]. As FXR is expressed in rodent hepatic stellate cells that play a critical role in hepatic fibrosis, it is not surprising that FXR agonists protect against liver fibrosis [66]. This appears to be mediated by a decreased hepatic expression of various profibrotic growth factors including transforming growth factor β1, tissue inhibitor of metalloproteinase 1, α1(I) collagen, α smooth muscle actin, matrix metalloproteinase 2 and α2(I) collagen, and microRNA-29a [67–69]. However, if this mechanism is also operational in humans with a lower expression level of FXR remains to be determined [70]. These data suggest that targeting FXR may impact progression from NAFLD to NASH.
5. FXR and Atherosclerosis
As demonstrated earlier in this article and illustrated in Figure 3, activation of FXR seems to be associated with both anti- and proatherogenic properties. In addition to its impact on dyslipidemia and hyperglycemia, FXR may also directly act at the levels of the arterial wall. Potential beneficial effects of FXR activation against atherosclerosis include suppressing the vasoconstrictive peptide endothelin-1 [71]. Induced expression of intracellular adhesion molecule 1 and vascular cell adhesion molecule 1, however, promotes atherosclerosis by recruiting macrophages to the endothelium [72]. The role of FXR in the initiation and progression of atherosclerosis has been studied in mice with Fxr gene ablation that were backcrossed into atherosclerosis-susceptible strains with either deletion of the low-density lipoprotein receptor or apolipoprotein E, respectively [73, 74]. These studies produced discrepant results whereas more recent experimentations employing an FXR agonist uniformly demonstrated protection against diet-induced aortic plaque formation [75, 76]. Translating these findings to humans is not straightforward as humans carry most cholesterol in LDL compared to the mouse that lacks cholesteryl ester transfer protein activity and thus transports most cholesterol in high-density lipoprotein [77]. In knowledge of these limitations, it would be most logical to carry out future studies in low-density lipoprotein receptor deficient mice that overexpress human cholesteryl ester transfer protein [78].
Figure 3.
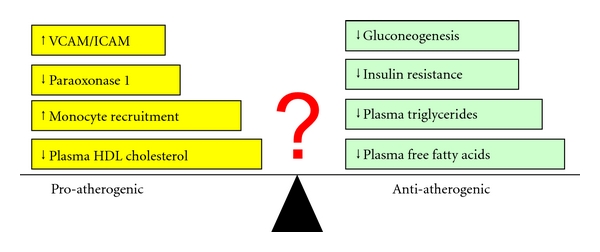
Pro- and antiatherogenic effects of FXR activation. With regard to atherosclerosis, activation of FXR may be associated with beneficial and potential negative effects. Unless tested in humans, one cannot predict with certainty whether pro- or antiatherogenic effects are dominant and development of specific FXR modulators may help to avoid some or most of the negative effects. VCAM: vascular cell adhesion protein. ICAM: intracellular adhesion molecule.
6. Summary and Perspective
FXR plays a key role in the transcriptional control of a myriad of target genes that control metabolic pathways relevant to NAFLD. By virtue of that role FXR is critically involved in the development and progression of NAFLD. Targeting FXR therefore offers exciting new perspectives for the treatment of NAFLD. However, when interpreting data obtained in cell culture and rodent models of human disease, attention needs to be paid to differences between these models and humans. One particular challenge in designing FXR agonists is separating the desired therapeutic effects from the undesirable side effects. The design of organ- or gene-specific FXR ligands may enhance the specificity and reduce side effects of this therapeutic approach. An increased understanding of the effect of cellular signaling of FXR and its coregulator proteins has the potential to aid in discovering novel selective therapeutic modulators and the development of new and more effective therapeutics. Finally one also needs to consider that the response to modulation of the FXR receptor may differ among patient with NAFLD and NASH.
Despite all the concerns raised, it is anticipated that targeting FXR will result in a more specific and individually tailored therapy that could revolutionize the management of NAFLD. Support comes from studies in rats with diabetes mellitus and fatty liver disease that received the FXR agonist INT-747 for two months [79]. This intervention decreased glucose levels and dyslipidemia, protected against body weight gain, and improved insulin resistance. It is thus very encouraging that INT-747 also has shown to improve insulin resistance in patients with diabetes mellitus and NAFLD [80]. Based on this study with a limited number of patients, an ongoing large multicenter trial enrolling 280 patients at eight U.S. centers comprising the NIDDK-sponsored NASH Clinical Research Network is under way, the results of which are eagerly awaited.
References
- 1.Adams LA, Angulo P. Recent concepts in non-alcoholic fatty liver disease. Diabetic Medicine. 2005;22(9):1129–1133. doi: 10.1111/j.1464-5491.2005.01748.x. [DOI] [PubMed] [Google Scholar]
- 2.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. New England Journal of Medicine. 2010;363(14):1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 3.Giday SA, Ashiny Z, Naab T, Smoot D, Banks A. Frequency of nonalcoholic fatty liver disease and degree of hepatic steatosis in African-American patients. Journal of the National Medical Association. 2006;98(10):1613–1615. [PMC free article] [PubMed] [Google Scholar]
- 4.Jimba S, Nakagami T, Takahashi M, et al. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabetic Medicine. 2005;22(9):1141–1145. doi: 10.1111/j.1464-5491.2005.01582.x. [DOI] [PubMed] [Google Scholar]
- 5.Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. American Journal of Physiology, Endocrinology and Metabolism. 2005;288:E462–E468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 6.de Alwis NMW, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. Journal of Hepatology. 2008;48(1):S104–S112. doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 7.Targher G, Arcaro G. Non-alcoholic fatty liver disease and increased risk of cardiovascular disease. Atherosclerosis. 2007;191(2):235–240. doi: 10.1016/j.atherosclerosis.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Ekstedt M, Franzén LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 9.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. Journal of Clinical Investigation. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metabolism. 2006;4(2):107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 14.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuchs M, Sanyal AJ. Non-alcoholic fatty liver disease: a pathophysiological perspective. In: Arias IM, Alter HJ, Boyer JL, et al., editors. The liver. Biology and Pathobiology. Pa, USA: Wiley-Blackwell; 2009. pp. 719–741. [Google Scholar]
- 16.Satapathy SK, Sanyal AJ. Novel treatment modalities for nonalcoholic steatohepatitis. Trends in Endocrinology and Metabolism. 2010;21(11):668–675. doi: 10.1016/j.tem.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Arrese M, Karpen SJ. Nuclear receptors, inflammation, and liver disease: insights for cholestatic and fatty liver diseases. Clinical Pharmacology and Therapeutics. 2010;87(4):473–478. doi: 10.1038/clpt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsia EY, Goodson ML, Zou JX, Privalsky ML, Chen H-W. Nuclear receptor coregulators as a new paradigm for therapeutic targeting. Advanced Drug Delivery Reviews. 2010;62(13):1227–1237. doi: 10.1016/j.addr.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berrabah W, Aumercier P, Lefebvre P, Staels B. Control of nuclear receptor activities in metabolism by post-translational modifications. FEBS Letters. 2011;585(11):1640–1650. doi: 10.1016/j.febslet.2011.03.066. [DOI] [PubMed] [Google Scholar]
- 20.Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Molecular Endocrinology. 1995;9(1):72–85. doi: 10.1210/mend.9.1.7760852. [DOI] [PubMed] [Google Scholar]
- 21.Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81(5):687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 22.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bite acids. Science. 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 23.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284(5418):1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Molecular Cell. 1999;3(5):543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. Journal of Biological Chemistry. 2003;278(1):104–110. doi: 10.1074/jbc.M209505200. [DOI] [PubMed] [Google Scholar]
- 26.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 27.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60(4):463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 28.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. Journal of Immunology. 2009;183(10):6251–6261. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 29.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Research. 2007;67(3):863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 30.Modica S, Murzilli S, Salvatore L, Schmidt DR, Moschetta A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Research. 2008;68(23):9589–9594. doi: 10.1158/0008-5472.CAN-08-1791. [DOI] [PubMed] [Google Scholar]
- 31.Hofmann AF, Small DM. Detergent properties of bile salts: correlation with physiological function. Annual Review of Medicine. 1967;18:333–376. doi: 10.1146/annurev.me.18.020167.002001. [DOI] [PubMed] [Google Scholar]
- 32.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. Journal of Lipid Research. 2009;50(8):1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trauner M, Halilbasic E. Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology. 2011;140(4):1120–1125. doi: 10.1053/j.gastro.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 34.Grundy SM, Ahrens EH, Salen G. Interruption of the enterohepatic circulation of bile acids in man: comparative effects of cholestyramine and ileal exclusion on cholesterol metabolism. The Journal of Laboratory and Clinical Medicine. 1971;78(1):94–121. [PubMed] [Google Scholar]
- 35.Nestel PJ, Grundy SM. Changes in plasma triglyceride metabolism during withdrawal of bile. Metabolism: Clinical and Experimental. 1976;25(11):1259–1268. doi: 10.1016/s0026-0495(76)80009-1. [DOI] [PubMed] [Google Scholar]
- 36.Angelin B, Einarsson K, Hellstrom K, Leijd B. Effects of cholestyramine and chenodeoxycholic acid on the metabolism of endogenous triglyceride in hyperlipoproteinemia. Journal of Lipid Research. 1978;19(8):1017–1024. [PubMed] [Google Scholar]
- 37.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes and Development. 2004;18(2):157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sirvent A, Claudel T, Martin G, et al. The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Letters. 2004;566(1–3):173–177. doi: 10.1016/j.febslet.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 40.Anisfeld AM, Kast-Woelbern HR, Meyer ME, et al. Syndecan-1 expression is regulated in an isoform-specific manner by the farnesoid-X receptor. Journal of Biological Chemistry. 2003;278(22):20420–20428. doi: 10.1074/jbc.M302505200. [DOI] [PubMed] [Google Scholar]
- 41.Fruchart-Najib J, Baugé E, Niculescu L-S, et al. Mechanism of triglyceride lowering in mice expressing human apolipoprotein A5. Biochemical and Biophysical Research Communications. 2004;319(2):397–404. doi: 10.1016/j.bbrc.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Kast HR, Nguyen CM, Sinal CJ, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Molecular Endocrinology. 2001;15(10):1720–1728. doi: 10.1210/mend.15.10.0712. [DOI] [PubMed] [Google Scholar]
- 43.Kardassis D, Roussou A, Papakosta P, Boulias K, Talianidis I, Zannis VI. Synergism between nuclear receptors bound to specific hormone response elements of the hepatic control region-1 and the proximal apolipoprotein C-II promoter mediate apolipoprotein C-II gene regulation by bile acids and retinoids. Biochemical Journal. 2003;372(2):291–304. doi: 10.1042/BJ20021532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125(2):544–555. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 45.Torra IP, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor α gene via activation of the farnesoid X receptor. Molecular Endocrinology. 2003;17(2):259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs M, Ivandic B, Müller O, et al. Biliary cholesterol hypersecretion in gallstone-susceptible mice is associated with hepatic up-regulation of the high-density lipoprotein receptor SRBI. Hepatology. 2001;33(6):1451–1459. doi: 10.1053/jhep.2001.24373. [DOI] [PubMed] [Google Scholar]
- 47.Zhao B, Song J, Ghosh S. Hepatic overexpression of cholesteryl ester hydrolase enhances cholesterol elimination and in vivo reverse cholesterol transport. Journal of Lipid Research. 2008;49(10):2212–2217. doi: 10.1194/jlr.M800277-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Fuchs M, Lammert F, Wang DQH, Paigen B, Carey MC, Cohen DE. Sterol carrier protein 2 participates in hypersecretion of biliary cholesterol during gallstone formation in genetically gallstone-susceptible mice. Biochemical Journal. 1998;336(1):33–37. doi: 10.1042/bj3360033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu K, Lee MH, Hazard S, et al. Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. American Journal of Human Genetics. 2001;69(2):278–290. doi: 10.1086/321294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert G, Amar MJA, Guo G, Brewer HB, Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. Journal of Biological Chemistry. 2003;278(4):2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 51.Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. Journal of Clinical Investigation. 2002;109(7):961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gutierrez A, Ratliff EP, Andres AM, Huang X, McKeehan WL, Davis RA. Bile acids decrease hepatic paraoxonase 1 expression and plasma high-density lipoprotein levels via FXR-mediated signaling of FGFR4. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(2):301–306. doi: 10.1161/01.ATV.0000195793.73118.b4. [DOI] [PubMed] [Google Scholar]
- 53.Shih DM, Kast-Woelbern HR, Wong J, Xia YR, Edwards PA, Lusis AJ. A role for FXR and human FGF-19 in the repression of paraoxonase-1 gene expression by bile acids. Journal of Lipid Research. 2006;47(2):384–392. doi: 10.1194/jlr.M500378-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Urizar NL, Dowhan DH, Moore DD. The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression. Journal of Biological Chemistry. 2000;275(50):39313–39317. doi: 10.1074/jbc.M007998200. [DOI] [PubMed] [Google Scholar]
- 55.Hubbert ML, Zhang Y, Lee FY, Edwards PA. Regulation of hepatic insig-2 by the farnesoid X receptor. Molecular Endocrinology. 2007;21(6):1359–1369. doi: 10.1210/me.2007-0089. [DOI] [PubMed] [Google Scholar]
- 56.Langhi C, Le May C, Kourimate S, et al. Activation of the farnesoid X receptor represses PCSK9 expression in human hepatocytes. FEBS Letters. 2008;582(6):949–955. doi: 10.1016/j.febslet.2008.02.038. [DOI] [PubMed] [Google Scholar]
- 57.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Annals of Internal Medicine. 1994;121(6):416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 58.Duran-Sandoval D, Mautino G, Martin G, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53(4):890–898. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 59.Cariou B. The farnesoid X receptor (FXR) as a new target in non-alcoholic steatohepatitis. Diabetes & Metabolism. 2008;34(6):685–691. doi: 10.1016/S1262-3636(08)74605-6. [DOI] [PubMed] [Google Scholar]
- 60.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. Journal of Clinical Investigation. 2006;116(4):1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cariou B, van Harmelen K, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. Journal of Biological Chemistry. 2006;281(16):11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improve hyperglycemia and hyperlipidemia in a diabetic mouse. Proceedings of the National Academy of Sciences. 2006;103:1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cellular Signalling. 2008;20(12):2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 64.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology. 2008;48(5):1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(10):3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fiorucci S, Antonelli E, Rizzo G, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127(5):1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 67.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. Journal of Hepatology. 2009;51(2):380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 68.Kong B, Luyendyk JP, Tawfik O, Guo GL. Faresoid X receptor deficiency induced nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. Journal of Pharmacology and Experimental Therapeutics. 2009;328:116–122. doi: 10.1124/jpet.108.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Zhang Y, Kuruba R, et al. Roles of microRNA-29a in the antifibrotic effect of farnesoid X receptor in hepatic stellate cells. Molecular Pharmacology. 2011;80(1):191–200. doi: 10.1124/mol.110.068247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fickert P, Fuchsbichler A, Moustafa T, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. American Journal of Pathology. 2009;175(6):2392–2405. doi: 10.2353/ajpath.2009.090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He F, Li J, Mu Y, et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circulation Research. 2006;98(2):192–199. doi: 10.1161/01.RES.0000200400.55539.85. [DOI] [PubMed] [Google Scholar]
- 72.Qin P, Tang X, Elloso MM, Harnish DC. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-κB, and p38. American Journal of Physiology, Heart and Circulatory Physiology. 2006;291(2):H741–H747. doi: 10.1152/ajpheart.01182.2005. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Wang X, Vales C, et al. FXR deficiency reduces atherosclerosis in Ldlr -/- mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:2316–2321. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
- 74.Hanniman EA, Lambert G, McCarthy TC, Sinal CJ. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. Journal of Lipid Research. 2005;46(12):2595–2604. doi: 10.1194/jlr.M500390-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Hartman HB, Gardell SJ, Petucci CJ, Wang S, Krueger JA, Evans MJ. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE -/- mice. Journal of Lipid Research. 2009;50(6):1090–1100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mencarelli A, Renga B, Distrutti E, Fiorucci S. Antiatherosclerotic effect of farnesoid X receptor. American Journal of Physiology, Heart and Circulatory Physiology. 2009;296(2):H272–H281. doi: 10.1152/ajpheart.01075.2008. [DOI] [PubMed] [Google Scholar]
- 77.Hogarth CA, Roy A, Ebert DL. Genomic evidence for the absence of a functional cholesteryl ester transfer protein gene in mice and rats. Comparative Biochemistry and Physiology. B. 2003;135(2):219–229. doi: 10.1016/s1096-4959(03)00046-0. [DOI] [PubMed] [Google Scholar]
- 78.Harada LM, Carrilho AJF, Oliveira HCF, Nakandakare ER, Quintão ECR. Regulation of hepatic cholesterol metabolism in CETP+/-/ LDLr+/- mice by cholesterol feeding and by drugs (cholestyramine and lovastatin) that lower plasma cholesterol. Clinical and Experimental Pharmacology and Physiology. 2006;33(12):1209–1215. doi: 10.1111/j.1440-1681.2006.04512.x. [DOI] [PubMed] [Google Scholar]
- 79.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. Journal of Lipid Research. 2010;51(4):771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanyal AJ, Mudaliar S, Henry RR, et al. A new therapy for nonalcoholic fatty liver disease and diabetes? INT-747–the first FXR hepatic therapeutic study. Hepatology. 2009;50:p. 389A. [Google Scholar]