

Abstract
The detrimental effects of hypoxic damage to central nervous system lead to energy depletion, free radical formation, lipid peroxidation (LPO), and increased calcium. We hypothesized that in vitro tacrolimus (FK-506) and cyclosporine A (CsA) could be protective against hypoxic damage in spinal cord. Dorsal columns were isolated from the spinal cord of adult rats and injured by exposure to hypoxic condition for 1 h, and treated with FK-506 (0.1 μM) and CsA (0.1 μM). After injury, reperfusion was carried out for 2 h. Tissues were collected, processed for biochemical assays, and 2,3,5-triphenyltetrazolium chloride (TTC) staining. Spinal cord hypoxia caused a significant decrease (P < 0.001) in mitochondrial ATP (30.64%) and tissue reduced glutathione (GSH) (60.14%) content. Conversely, a significant increase (P < 0.001) in tissue LPO level (57.77%) and myeloperoxidase (MPO) activity (461.24%) was observed in hypoxic group. Mitochondrial swelling was also significantly increased in hypoxic group (90.0%). Treatment with either FK-506 or CsA showed that significant neuroprotective effects (P < 0.05–0.01) were measured in various parameters in hypoxic groups. FK-506 and CsA treatment showed increase in ATP by 11.19% and 16.14% while GSH content increased by 66.46% and 77.32%, respectively. Conversely, LPO content decreased by 18.97% and 24.06% and MPO level by 42.86% and 18.66% after FK-506 and CsA treatment. Calcium uptake was also decreased in mitochondria as exhibited by the increase in absorbance by 11.19% after FK-506 treatment. TTC staining also showed increased viability after FK-506 and CsA treatment. In conclusion, present study demonstrates the neuroprotective effect of FK-506 and CsA treatment against spinal cord hypoxia induced damage is mediated via their antioxidant actions.
Keywords: ATP, hypoxia, mitochondrial swelling, neuroprotection, spinal cord
Introduction
Spinal hypoxia leads to severe neurological damage and dysfunction. There is no current effective therapeutic treatment, due to the limited capacity for axonal regeneration and myriad inhibitory cues. Immediately after the primary damage, other events cause secondary damages including energy depletion, excitotoxicity, increased calcium influx, free radical formation, and lipid peroxidation (LPO; Mobley and Agrawal 2003). Basic strategy of treatment after spinal hypoxia is the protection of the remaining healthy spinal neurons from secondary damage that triggers multifarious degenerative processes. Systemic and cellular level responses, which regulate many physiological and pathological processes, are disrupted due to poor oxygen supply (Bunn and Poyton 1996). Mitochondrial dysfunction as a result of poor oxygen supply plays an important role in the pathophysiology of hypoxic cell death (Kroemer et al. 1998). Mitochondrial dysfunction leads to cellular oxidative stress and cell death. Preserving the mitochondrial integrity may be considered one of the basic prophylactic cruxes for reducing the spinal cord injury.
Cyclosporin A (CsA, immunophillin) and FK-506 (Tacrolimus) are the most commonly used immunomodulatory agents. These immunosuppressants inhibit a calcium-dependent phosphoserine–phosphothreonine protein phosphatase, calcineurin, and thereby inhibit calcineurin-mediated secondary neuronal damage (Sullivan et al. 2000; Diaz–Ruiz et al. 2005). FK-506 is reported to exhibit both neuroprotective and neuroregenerative properties through various mechanisms. Some mechanisms include the ability to cross the blood brain barrier (BBB; Gold et al. 1997; Steiner et al. 1997), inhibitory action against cytochrome c release, antiinflammatory action (Furuichi et al. 2004), and suppressing the death protein BAD (Wang et al. 1999). Sheehan et al. (2006) reported the potentiating effect of CsA and FK506 on neurite outgrowth as well as the protective effect against apoptotic cell death in a human postmitotic neuronal cell line. Recently, it has been reported that pretreatment with cyclosporin derivative NIM811 improves the function of synaptic mitochondria by improving mitochondrial respiratory control ratios in spinal cord contusion in rats and by scavenging free radical generation in mitochondria (McEwen et al. 2007).
In the present study, we have investigated the neuroprotective effects of CsA and FK-506 in oxidative injury of spinal cord white matter.
Material and Methods
Drugs and chemicals
FK-506 and CsA were purchased from Calbiochem. Calcium chloride, ATP Kit, N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid, sucrose, EDTA, Tris-HCl, ficoll, percholl, HEPES buffer, rotenone, antimycin, sulphosalicylic acid, DTNB, butylated hydroxyl toluene (BHT), ortho-phosphoric acid (OPA), thiobarbituric acid (TBA), and ortho-dianisidine were procured from Sigma Aldrich Saint Louis, MO, USA.
Animals
Male Wistar rats (250–300 gm) were used. All animal procedures were approved by the Institutional Animal Care and Use committee (IACUC) and University of Nebraska Medical Center, NE, USA. Rats were housed at the animal care facility at the animal house of University of Nebraska Medical Center, NE, USA.
Isolation of spinal cord
Rats were anesthetized with pentobarbital (40 mg/kg, i.p.). When no flexor withdrawal was seen in response to noxious foot pinch, anesthesia was considered complete. Laminectomy was performed from T4–T8 region, and then the spinal cord was exposed and dissected out. Immediately, it was placed in cold (4–7°C) Ringer's solution (124 mM NaCl, 5 mM KCl, 1.2 mM K2HPO4, 1.3 mM MgSO4, 2 mM CaCl2, 20 mM glucose, and 26 mM NaHCO3, pH 7.4), dura matter and the dorsal roots were excised in cold Ringer's solution bubbled continuously with 95% O2–5% CO2.
Experimental protocol
Animals were divided into four groups. Group I was sham, in which the harvested spinal cord was incubated in oxygenated (95%O2–5%CO2) Ringer's solution at 37 ± 0.5°C for 1 h. Group II was hypoxic group, in which the cord was incubated in hypoxic (95%N2–5%CO2) Ringer's solution at 37 ± 0.5°C for 1 h. Group III was FK-506-treated group (FK-506 + Hypoxia), in which the spinal cord was incubated in hypoxic Ringer's solution in the presence of FK-506 (0.1 μM) at 37 ± 0.5°C for 1 h. Group IV was treated with CsA (CsA + Hypoxia), in which the spinal cord was incubated in hypoxic Ringer's solution with CsA (0.1 μM) at 37 ± 0.5°C for 1 h. After hypoxia, reperfusion was carried out for 2 h with oxygenated (95%O2–5%CO2) Ringer's solution. After the completion of all treatments, tissues from various groups were processed for various biochemical estimations. Tissues were homogenized (10%) in ice-cold homogenization medium (5 mM HEPES with 0.32 M sucrose, 1 mM MgCl2, 2 mM EGTA, and 0.1 mM PMSF). The homogenates were centrifuged at 10,000 ×g for 10 min at 4°C and supernatant was collected for LPO, reduced glutathione (GSH), and myeloperoxidase (MPO) activity.
Isolation and purification of mitochondria
Mitochondria were isolated and purified according to the method of Rendon and Masmoudi (1985). Spinal cords were finely minced briefly and a 20% homogenate (w/v) was made in isolation buffer (0.32 M sucrose, 1 mM EDTA K+, 10 mM Tris-HCl, pH 7.4). The homogenate was centrifuged at 1100 ×g for 5 min. The resulting supernatant was again centrifuged at 17,000 ×g for 10 min to yield the crude mitochondrial pellet containing synaptosomes (P2 fraction). P2 fraction was washed by resuspending in isolation buffer and centrifuged at 17,000 ×g for 20 min. The pellet was resuspended in isolation buffer and manually homogenized. The suspension was layered onto 7.5% ficoll medium on 13% of ficoll medium and centrifuged at 100,000 ×g for 30 min. The 7.5% and 13% ficoll media contained w/v ficoll in isolation buffer. Mitochondria were collected from the bottom of the tube and washed again in isolation buffer and were stored in –70°C for further biochemical analysis.
ATP quantitation
The isolated mitochondria (1 mg/mL of protein) were resuspended in a reaction mixture containing 0.25 M sucrose, 1 mM MgCl2, 10 mM HEPES, and 1 mM EDTA. The suspension was centrifuged at 5000 ×g for 10 min. The supernatant was incubated with luciferin-luciferase (5 mg/mL) and the bioluminescence was measured by Fluostar Optima microplate reader (BMG Labtech). The results were expressed as pmol of ATP per mg protein.
Measurement of mitochondrial swelling
Ca2+-induced mitochondrial swelling of the deenergized mitochondria was done by the method of Halestrap and Davidson (1990). Mitochondria (25 μg of protein) were added 1.1 mL of isotonic buffer containing 150 mM KSCN, 5 mM Tris, 0.5 μl rotenone/mL, and 0.5 μg antimycin/mL (pH 7.2) at 30°C. Swelling was initiated by addition of Ca2+ (100 μM) to the cuvette and the absorbance was monitored for 5 min at 540 nm. Change in absorbance was monitored as percent change compared with the control values.
Lipid peroxidation (LPO)
LPO was determined by the procedure of Uchiyama and Mihara (1978). Briefly, 0.25 mL of tissue homogenate was mixed with 25 μl of 10 mM BHT. OPA (3 mL of 1% solution) and TBA (1 mL of 0.67% solution) were added and the mixture was incubated at 90°C for 45 min. The absorbance was measured at 535 nm. The rate of LPO was expressed as nanomoles of TBA reactive substance (TBARS) formed per hour per gram of tissue using a molar extinction coefficient of 1.56 × 105 M−1 cm−1.
Reduced glutathione (GSH)
GSH content was determined by the method of Jollow et al. (1974). Ten percent tissue homogenate was mixed with 4.0% sulphosalicylic acid (w/v) in a 1:1 ratio (v/v). The samples were incubated at 4°C for 1 h. The assay mixture contained 0.1 mL of supernatant, 1.0 mM DTNB, and 0.1 M PB (pH 7.4). The yellow color developed was read immediately at 412 nm in a spectrophotometer (BioRad). The GSH content was calculated as nmol GSH mg−1 protein, using molar extinction coefficient of 1.36 × 103 M−1 cm−1.
Myeloperoxidase (MPO)
MPO was evaluated by the method of Bradley et al. (1982). A total of 0.1 mL of tissue homogenate was added to 1.45 mL of o-dianisidine in 1 mL methyl alcohol, 98 mL 50 mM phosphate buffer (pH 6.0), and 1 mL of 0.05% H2O2 solution as a substrate for MPO enzyme. The change in absorbance was measured at 460 nm using BioRad spectrophotometer. One unit of MPO activity was defined as the quantity able to convert 1 μmol H2O2 per min at 25°C and was expressed as unit per gram tissue.
TTC staining
A 1.5-mm-thick cross-section of spinal cord tissue was rapidly cut after completion of hypoxia or hypoxia + treatment in various groups. The sections were stained by incubating them in a solution of 2% of 2,3,5-triphenyltetrazolium chloride (TTC) at 37 ± 0.5°C for 15 min. For imaging, the sections were scanned by a high-resolution scanner (CanoScanLiDE25). The total mean infract area of each section was measured. Infarct size of the different groups was normalized to the sham and expressed as a percentage and an average value from the four slices was presented.
Statistical analysis of data
The statistical analysis of data was done using analysis of variance (ANOVA) with post hoc analysis. The Tukey–Kramer post hoc test was applied to serve as significant among groups. The significance of results was ascertained at P < 0.05. All the data are presented as mean ± SE (n = 6) of the means.
Results
ATP quantitation
Mitochondrial ATP content decreased significantly (P < 0.001) in hypoxic group by 30.64% when compared with sham group values. However, as a result of drugs treatment, a significant (P < 0.05–0.01) restoration was seen in ATP level in the hypoxic groups treated with FK-506 (FK-506 + Hypoxic) and CsA (CsA + Hypoxic) by 11.19% and 16.14%, respectively, when compared to hypoxic group (Fig. 1).
Figure 1.
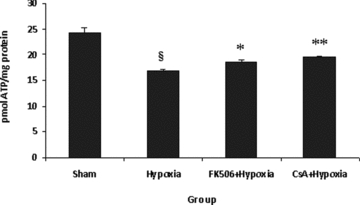
ATP content in various groups. Values are expressed as pmol ATP per mg protein (mean ± SE, n = 6). Significant difference §P < 0.001 when compared with sham; *P < 0.05, **P < 0.01 when compared with hypoxic group. Hypoxia resulted in significant decrease (P < 0.001) in ATP content, which was significantly increased (P < 0.05–0.01) by the treatment of FK-506 and cyclosporine A (CsA) in hypoxic group. Both the drugs showed same ATP-restoring effect. There was no difference between both the groups.
Calcium uptake in mitochondria
Hypoxia/reperfusion resulted in significant mitochondrial swelling (P < 0.001) evident by a decrease in absorbance (90.9%) in the hypoxic group (Fig. 2). It was observed that FK-506 treatment at a concentration 0.1 μM caused a significant decrease (P < 0.05) in mitochondrial swelling in the FK-506 + Hypoxia group. There was also a significant decrease (P < 0.05) in mitochondrial swelling with the CsA-treated hypoxic group, as shown by 14.83% decrease in absorbance. When a comparison was made, FK-506 decreased mitochondrial swelling more efficiently than CsA (Fig. 2).
Figure 2.
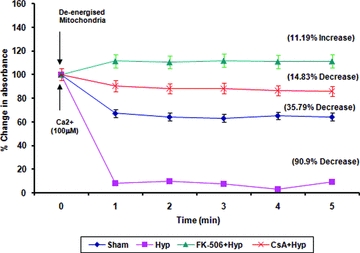
Calcium uptake in spinal cord mitochondria. Mitochondria (25 mg/reaction) were deenergized via incubation in isotonic buffer at room temperature for 3 min. After incubation, calcium (100 μM) was added to the reaction mixture and absorbance was measured at 540 nm for 5 min at every 1-min time interval. Results are shown as percent change in absorbance corresponding to the absorbance of deenergized mitochondria.
Tissue LPO level
As a result of spinal hypoxia/reperfusion, a significant increase (P < 0.001) in LPO level was observed in the hypoxic group as compared with sham values. Drug treatment with either FK-506 or CsA resulted in a significant (P < 0.01–0.001) decrease in the level of LPO in the FK-506 + Hypoxia and CsA + Hypoxia groups when compared to hypoxic group (Fig. 3). No significant difference was observed between both the drug-treated groups.
Figure 3.
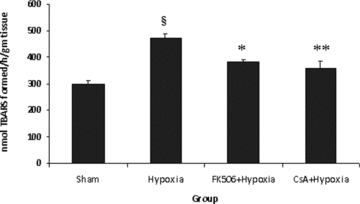
Rate of lipid peroxidation (LPO) in different groups. Values are expressed as mean ± SE (n = 6). Significant difference §P < 0.001 when compared with sham; *P < 0.01, **P < 0.001 when compared with hypoxic group. Hypoxia resulted in significant increase (P < 0.001) in LPO, which was significantly decreased by the treatment of FK-506 and CsA in hypoxic group. Both the drugs showed the same LPO-lowering effect.
Tissue GSH content
Spinal hypoxic damage significantly (P < 0.001) depleted the GSH content in mitochondria in the hypoxic group over sham values (Fig. 4). As a result of FK-506 and CsA treatment, a significant (P < 0.01) restoration in mitochondrial GSH content was seen in the FK-506 + Hypoxia and CsA + Hypoxia groups when compared with the hypoxic group. No significant difference was observed between both the drug-treated groups.
Figure 4.
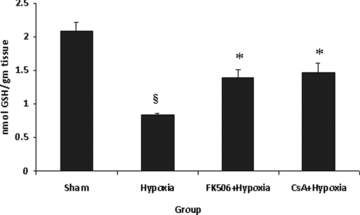
Reduced glutathione (GSH) content in various groups. Values are expressed as mean ± SE (n = 6). Significant difference §P < 0.001 when compared with sham; *P < 0.01 when compared with hypoxic group. Hypoxia resulted in significant decrease (P < 0.001) in GSH content, which was significantly increased (P < 0.01) by the treatment of FK-506 and CsA in hypoxic group. There is no difference between drugs group.
Tissue MPO activity
A significant (P < 0.001) increase in MPO activity was seen in the hypoxic group compared to sham values. It was observed that treatment with either FK-506 (P < 0.001) or CsA (P < 0.01) caused a significant decrease in MPO activity in the FK-506 + Hypoxia and CsA + Hypoxia groups as compared to the hypoxic group (Fig. 5). It was observed that FK-506 significantly (P < 0.01) decreased MPO activity (42.86%) than CsA (18.66%).
Figure 5.
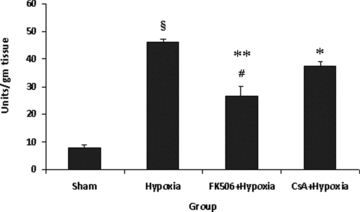
Myeloperoxidase (MPO) activity in various groups. Values are expressed as mean ± SE (n = 6). Significant difference §P < 0.001 when compared with sham; *P < 0.01, **P < 0.001 when compared with hypoxic group and #P < 0.01 when a comparison was made between both the drugs. Hypoxia resulted in significant increase (P < 0.001) in MPO activity, which was significantly decreased by the treatment of FK-506 and CsA in hypoxic group. FK-506 showed significantly (P < 0.01) more MPO lowering effect than CsA.
Tissue viability
TTC staining in sham did not show any detectable lesion (Fig. 6). On the contrary, sections obtained from hypoxic group showed detectable lesions as white patches. The lesions were present in both white and gray matter. It was observed that FK-506 and CsA treatment significantly decreased the lesions (P < 0.05) as compared to hypoxic group.
Figure 6.
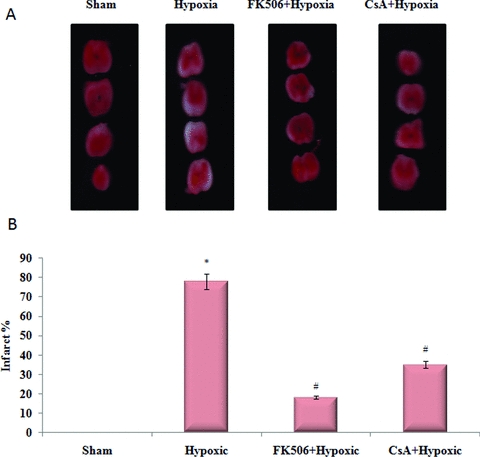
Neuroprotective effect of FK-506 and CsA in spinal cord hypoxia. (A) Representative photographs of spinal cord sections stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC). White patches show the infraction. (B) Infract (%) measurement in different groups. There is significant increase in infract size in hypoxic group as compared to sham (P < 0.001). Treatment of FK-506 and CsA significantly reduces the infract size (P < 0.05) as compared to hypoxic group.
Discussion
The present study demonstrated that in vitro hypoxic/reperfusion spinal injury exacerbates myriad cascade of events involving energy depletion, mitochondrial swelling, increased LPO with decrease in GSH levels, and increased MPO level. Intervention with FK-506 and CsA restored the depleted ATP stores, inhibition of mitochondrial swelling, and decrease in oxidant indices with substantial restoration of endogenous GSH. These results confirm that the protective actions of FK-506 and CsA are mediated via their antioxidant actions.
Reactive oxygen species (ROS) generation is the key component of the secondary neuronal damage in the spinal cord and brain injury (Hall 1989; Aksenova et al. 2002). The disastrously increased ROS formed during spinal hypoxia attach to membrane polyunsaturated fatty acids, thereby inflicting LPO and also increasing membrane permeability because the tissue is very sensitive and vulnerable (Lewen et al. 2000; Yousuf et al. 2005, Atif et al. 2009). We observed a marked increase in LPO level in hypoxic spinal cord that could be attributed to the ROS action. Under normal conditions, ROS are generated in the mitochondria, which are rapidly scavenged by the various enzymatic and nonenzymatic antioxidants. Reduced GSH is the primary line of defense against ROS. GSH is consumed by glutathione peroxidase enzyme during H2O2 elimination and therefore in an environment where there is oxidative stress, intracellular GSH content is depleted. GSH and other thiol-containing proteins are important in cellular defense against hypoxic damage. In the present study, GSH content was found to be depleted in the hypoxic group.
ROS-mediated oxidative damage participates in the exacerbation of intracellular calcium levels leading to mitochondrial swelling, which plays a critical role in several forms of neuronal death (Coyle and Puttfarcken 1993; Choi 1995; Leist and Nicotera 1998; Okabe et al. 2000; Xiong et al. 2007). Mitochondrial swelling is an indicator of the opening of the mitochondrial permeability transition pores (MPTP), which results in depolarization of mitochondrial membrane potential. Our findings too demonstrated a significant increase in Ca2+-induced swelling in hypoxic mitochondria. Mitochondrial swelling subsequently results in altered oxidative phosphorylation, eventually affecting ATP production (Halestrap 2005). CNS has limited intrinsic energy reserves and requires a constant supply of oxygen and nutrients. Energy metabolism change or ATP depletion leads to depolarization and failure of energy conduction. A significant decrease in ATP level in the hypoxic spinal cord as a result of 1-h spinal hypoxia was observed in this study. There are various reports that indicate that intracellular ATP production is the prime target of hypoxia/ischemia-induced free radicals (Halestrap 2005).
In the present study, MPO activity was assessed for the index of tissue oxidative load, which is considered as one of the hallmark indicator of necrotic cell death (Erman et al. 2005). We observed that hypoxic spinal cord showed increase in MPO activity. Neutrophil and microglia activation have been shown responsible for increased MPO activity (Taoka and Okajima 1998; Erman et al. 2005; Fleming et al. 2006) during CNS injuries. In this in vitro model, since there is no blood infusion, so the probable source of MPO is microglia alone.
We used two neuroimmunophilins (FK-506 and CsA) to understand their effects on spinal cord hypoxic injury induced secondary neuronal damage in spinal cord. It was observed that both FK-506 and CsA significantly reduced the level of LPO and MPO activity in the hypoxic group. This could be due to the ability of FK-506 and CsA to inhibit microglia activation by inhibiting calcineurin, which activates transcription factor NF-AT and thereby eventually decreasing MPO and LPO level in the hypoxic spinal cord (Taoka and Okajima 1998; Erman et al. 2005). However, study by Mun and Ha (2010) has shown that CsA treatment of glioma leads to increase ROS production and neurological side effects. FK-506 has been reported to protect the spinal cord by targeting microglia cells (Guzmán–Lenis et al. 2008) after excitotoxicity. CsA and FK-506 have been used as an immunosuppressant in traumatic or ischemic CNS damage and it was shown that these neuroprotectants inhibit microglia cells activation (Hailer 2008). FK-506 is also reported to block NF-κB, turning off the gene of ICAM-I, thereby limiting the inflammatory damage and infarct size during ischemia/reperfusion (Squadrito et al. 2000). Nishinaka et al. (1993) reported that FK-506 exerted a protection on ischemia/reperfusion-induced damage in canine heart, which was suggested due to the ability of FK-506 to reduce superoxide radical formation.
It was observed that FK-506 and CsA treatment significantly restored GSH content in the hypoxic groups. There is a correlation between the level of LPO and GSH content; both are inversely proportional to each other. The inversely proportional LPO and GSH content could help explain the mechanism by which FK-506 and CsA reduced peroxidative membrane damage by inhibiting microglia activation and thereby maintaining GSH content.
FK-506 and CsA treatment markedly decreased mitochondrial swelling in hypoxic mitochondria. ATP content was also found to be increased with FK-506 and CsA treatment. It has been reported that ROS generation plays a central role in altering mitochondrial membrane integrity, which leads to opening of MPTP and increased ion influx, that is, calcium (Peng and Jou 2004). MPTP opening results in uncoupling respiration from ATP synthesis, organelle swelling, disruption of the outer membrane, and release of different apoptogenic factors into the cytosol (Green and Reed 1998; Kroemer et al. 1998). It has been reported that CsA binds to cyclophilin D and prevents opening of mitochondrial transition pore (Mbye et al. 2008; Panickar et al. 2009). However, the exact mechanism by which FK-506 and CsA exert their ROS-scavenging potential is not well understood yet.
Hypoxia results in tissue damage as observed by white patches in gray and white matter. FK-506 and CsA treatment showed significant reduction of white patches, which confers less tissue damage and increase viability. FK506 was, however, more effective over CsA in reducing white patches.
On the basis of the findings above, it could be speculated that FK-506 and CsA inhibited ROS-mediated mitochondrial swelling and restored ATP synthesis, as evidenced by the decrease in mitochondrial swelling and restoration of ATP content and reduction in the tissue damage as observed by TTC staining.
The present results suggested that ROS culminate in irreversible secondary damage in the spinal cord. Independent treatment with FK-506 and CsA showed neuroprotective effect against ROS-mediated mitochondrial dysfunctions. Further studies on combinatorial effect of CsA and FK-506 may provide a more effective strategy in the management of ROS-mediated secondary neuronal damage in spinal cord.
References
- Aksenova M, Butterfield DA, Zhang SX, Underwood M, Geddes JW. Increased protein oxidation and decreased creatine kinase BB expression and activity after spinal cord contusion injury. J. Neurotrauma. 2002;19:491–502. doi: 10.1089/08977150252932433. [DOI] [PubMed] [Google Scholar]
- Atif F, Yousuf S, Agrawal SK. S-allyl L-cysteine diminishes cerebral ischemia-induced mitochondrial dysfunctions in hippocampus. Brain Res. 2009;1265:128–137. doi: 10.1016/j.brainres.2008.12.077. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Christensen RD, Rothstein G. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood. 1982;60:618–622. [PubMed] [Google Scholar]
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol. Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium: still center-stage in hypoxic–ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Diaz-Ruiz A, Vergara P, Perez-Severiano F, Segovia J, Guizar-Sahagun G, Ibarra A, Rios C. Cyclosporin-A inhibits constitutive nitric oxide synthase activity and neuronal and endothelial nitric oxide synthase expressions after spinal cord injury in rats. Neurochem. Res. 2005;30:245–251. doi: 10.1007/s11064-005-2447-0. [DOI] [PubMed] [Google Scholar]
- Erman T, Sule YM, Iskender GA, Suzan Z, Hakan D, Metin T. Effects of antithrombin iii on myeloperoxidase activity, superoxide dismutase activity, and malondialdehyde levels and histopathological findings after spinal cord injury in the rat. Neurosurgery. 2005;56:828–835. doi: 10.1227/01.neu.0000157004.19427.35. [DOI] [PubMed] [Google Scholar]
- Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Furuichi Y, Noto T, Li JY, Oku T, Ishiye M, Moriguchi A, Aramori I, Matsuoka N, Mutoh S, Yanagihara T. Multiple modes of action of tacrolimus (FK506) for neuroprotective action on ischemic damage after transient focal cerebral ischemia in rats. Brain Res. 2004;1014:120–130. doi: 10.1016/j.brainres.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Gold BG. FK506 and the role of immunophilins in nerve regeneration. Mol. Neurobiol. 1997;15:285–306. doi: 10.1007/BF02740664. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Guzmán-Lenis MS, Vallejo C, Navarro X, Casas C. Analysis of FK506-mediated protection in an organotypic model of spinal cord damage: heat shock protein 70 levels are modulated in microglial cells. Neuroscience. 2008;155:104–113. doi: 10.1016/j.neuroscience.2008.04.078. [DOI] [PubMed] [Google Scholar]
- Hailer NP. Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 2008;84:211–233. doi: 10.1016/j.pneurobio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Halestrap A. Biochemistry: a pore way to die. Nature. 2005;434:578–579. doi: 10.1038/434578a. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporine is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Central nervous system trauma and stroke, physiological and pharmacological evidence for the involvement of oxygen radicals and lipid peroxidation. Free. Radic. Biol. Med. 1989;6:303–313. doi: 10.1016/0891-5849(89)90057-9. [DOI] [PubMed] [Google Scholar]
- Jollow DJ, Mitchell JR, Zampaglione N, Gillete JR. Bromobenzene induced liver necrosis: protective role of glutathione and evidence for 3, 4-bromobenzene oxide as the hepatotoxic intermediate. Pharmacology. 1974;11:161–169. doi: 10.1159/000136485. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Leist M, Nicotera P. Apoptosis, excitotoxicity, and neuropathology. Exp. Cell Res. 1998;239:183–201. doi: 10.1006/excr.1997.4026. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J. Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp. Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Mun KH, Ha EY. Effects of cyclosporine on the antioxidant status and oxidative stress in the glioma cells. Transplant. Proc. 2010;42:983–984. doi: 10.1016/j.transproceed.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Mcewen ML, Sullivan PG, Springer JE. Pretreatment with the cyclosporin derivative, NIM811 improves the function of synaptic mitochondria following spinal cord contusion in rats. J. Neurotrauma. 2007;24:613–624. doi: 10.1089/neu.2006.9969. [DOI] [PubMed] [Google Scholar]
- Mobley LW3rd, Agrawal SK. Role of calcineurin in calcium-mediated hypoxic injury to white matter. Spine J. 2003;3:11–18. doi: 10.1016/s1529-9430(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Nishinaka Y, Sugiyama S, Yokota M, Saito H, Ozawa T. Protective effect of FK506 on ischemia/reperfusion induced myocardial damage in canine heart. J. Cardiovasc. Pharmacol. 1993;21:448–454. doi: 10.1097/00005344-199303000-00015. [DOI] [PubMed] [Google Scholar]
- Okabe E, Tsujimoto Y, Kobayashi Y. Calmodulin and cyclic ADP-ribose interaction in Ca2+ signaling related to cardiac sarcoplasmic reticulum: superoxide anion radical-triggered Ca2+ release. Antioxid. Redox Signal. 2000;2:47–54. doi: 10.1089/ars.2000.2.1-47. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Polansky MM, Anderson RA. Cinnamon polyphenols attenuate cell swelling and mitochondrial dysfunction following oxygen-glucose deprivation in glial cells. Exp. Neurol. 2009;216:420–427. doi: 10.1016/j.expneurol.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Peng TI, Jou M. Mitochondrial swelling and generation of reactive oxygen species induced by photoirradiation are heterogeneously distributed. Ann. N. Y. Acad. Sci. 2004;1011:112–122. doi: 10.1007/978-3-662-41088-2_12. [DOI] [PubMed] [Google Scholar]
- Rendon A, Masmoudi A. Purification of non-synaptic and synaptic mitochondria and plasma membranes from rat brain by a rapid Percoll gradient procedure. J. Neurosci. Methods. 1985;14:41–51. doi: 10.1016/0165-0270(85)90113-x. [DOI] [PubMed] [Google Scholar]
- Sheehan J, Eischeid A, Saunders R, Pouratian N. Potentiation of neurite outgrowth and reduction of apoptosis by immunosuppressive agents: implications for neuronal injury and transplantation. Neurosurg. Focus. 2006;20:E9. doi: 10.3171/foc.2006.20.5.10. [DOI] [PubMed] [Google Scholar]
- Squadrito F, Altavilla D, Squadrito G, Saitta A, Deodato B, Arlotta M, Minutoli L, Quartarone C, Ferlito M, Caputi AP. Tacrolimus limits polymorphonuclear leucocyte accumulation and protects against myocardial ischaemia—reperfusion injury. J. Mol. Cell. Cardiol. 2000;32:429–440. doi: 10.1006/jmcc.1999.1089. [DOI] [PubMed] [Google Scholar]
- Steiner JP, Connolly MA, Valentine HL, Hamilton GS, Dawson TM, Hester L, Snyder SH. Neurotrophic actions of non-immunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporine A. Nat. Med. 1997;3:421–428. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp. Neurol. 2000;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K. Spinal cord injury in the rat. Prog. Neurobiol. 1998;56:341–358. doi: 10.1016/s0301-0082(98)00049-5. [DOI] [PubMed] [Google Scholar]
- Uchiyama M, Mihara M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Chu XP, Simon RP. Acid sensing ion channels–novel therapeutic targets for ischemic brain injury. Front. Biosci. 2007;12:1376–1386. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- Yousuf S, Salim S, Ahmad M, Ahmed AS, Ansari MA, Islam F. Protective effect of Khamira Abresham Uood Mastagiwala against free radical induced damage in focal cerebral ischemia. J. Ethnopharmacol. 2005;99:179–184. doi: 10.1016/j.jep.2004.12.035. [DOI] [PubMed] [Google Scholar]