

Abstract
Mutations in the L1 gene cause severe brain malformations and mental retardation. We investigated the potential roles of L1 in the regulation of choline acetyltransferase (ChAT) and in the development of septal cholinergic neurons, which are known to project to the hippocampus and play key roles in cognitive functions. Using stereological approaches, we detected significantly fewer ChAT-positive cholinergic neurons in the medial septum and vertical limb of the diagonal band of Broca (MS/VDB) of 2-week-old L1-deficient mice compared to wild-type littermates (1644 ± 137 vs. 2051 ± 165, P = 0.038). ChAT protein levels in the septum were 53% lower in 2-week-old L1-deficient mice compared to wild-type littermates. ChAT activity in the septum was significantly reduced in L1-deficient mice compared to wild-type littermates at 1 (34%) and 2 (40%) weeks of age. In vitro, increasing doses of L1-Fc induced ChAT activity in septal neurons with a significant linear trend (*P = 0.0065). At 4 weeks of age in the septum and at all time points investigated in the caudate-putamen (CPu), the number of ChAT-positive neurons and the levels of ChAT activity were not statistically different between L1-deficient mice and wild-type littermates. The total number of cells positive for the neuronal nuclear antigen (NeuN) in the MS/VDB and CPu was not statistically different in L1-deficient mice compared to wild-type littermates, and comparable expression of the cell cycle marker Ki67 was observed. Our results indicate that L1 is required for the timely maturation of septal cholinergic neurons and that L1 promotes the expression and activity of ChAT in septal neurons.
Keywords: Caudate-putamen, cell adhesion molecule L1, choline acetyltransferase, cholinergic neurons, L1-deficient mice, medial septum, stereology, striatum, vertical limb of diagonal band of Broca
Introduction
The cell adhesion molecule L1 is a member of the immunoglobulin superfamily, and it is predominantly expressed by postmitotic neurons of the central nervous system (Rathjen and Schachner 1984; Maness and Schachner 2007). By activating diverse mechanisms such as homophilic and heterophilic adhesion, as well as signal transduction, L1 plays key roles in cell adhesion, neuronal migration and survival, neurite outgrowth, axonal fasciculation, synaptic plasticity, and cognitive function (Sandi 2004; Maness and Schachner 2007).
In humans, mutations in the L1 gene cause obvious malformations to the brain, including ventricular dilatation and the abnormal formation of major axonal tracts (e.g., corticospinal tract, corpus callosum) (Brümmendorf et al. 1998; Kamiguchi et al. 1998). In vitro, L1 mutations influence neurite outgrowth (Moulding et al. 2000; Cheng and Lemmon 2004) and L1-deficient mice have brain anomalies similar to those described in humans with L1 mutations. L1-null mice also have impaired distribution of tyrosine hydroxylase positive neurons in areas of the mesencephalon and diencephalon, mapping of retinal ganglion cell axons to the superior colliculus, and fasciculation of thalamocortical and corticothalamic fibers (Cohen et al. 1997; Dahmé et al. 1997; Fransen et al. 1998; Demyanenko et al. 1999, 2001; Rolf et al. 2001; Demyanenko and Maness 2003; Ohyama et al. 2004).
L1 mutations are associated with some types of mental retardation in humans and it is has been demonstrated that L1 influences hippocampal function and behavior in rodents (Wong et al. 1995; Arami et al. 1996; Law et al. 2003; Sandi 2004; Maness and Schachner 2007). A clear relationship between L1 expression and the alteration of neurotransmitter systems critical to cognitive functions has yet to be established. Gamma-aminobutyric acid (GABA)-ergic neurotransmission is reduced in the hippocampus of L1-deficient mice and this may have functional consequences in vivo (Saghatelyan et al. 2004). Septal cholinergic innervation to the hippocampus is known to be essential to cognitive functions (Sarter and Parikh 2005). Here, we investigated whether the acetylcholine-synthesizing enzyme choline acetyltransferase (ChAT) and the development of septal cholinergic neurons are regulated by L1.
We focus on septal cholinergic neurons for several reasons, including the report on septal malformations in L1-deficient mice (Demyanenko et al. 1999) and the delayed growth of the medial septum in acallosal mice (Wahlsten and Bulman–Fleming 1994). Furthermore, we previously found that L1 is strongly expressed by developing septal neurons in vitro (Frappé et al. 2004) and by regenerating cholinergic septohippocampal axons in vivo (Aubert et al. 1998). L1-deficient mice have a reduced number of pyramidal and granular cells in the hippocampus (Demyanenko et al. 1999), which are normally innervated by septal cholinergic axons (Frotscher and Leranth 1985). Taken together, these studies suggest that L1 could be involved in the proper development of septal cholinergic neurons and that an abnormal maturation of these neurons may contribute to the known defective development of hippocampal neurons in L1-deficient mice.
We report that L1 is critical for the timely maturation of septal cholinergic neurons and ChAT expression and activity in vivo. We also provide direct evidence that L1 stimulates ChAT activity in vitro. The absence of L1 in transgenic mice did not influence the number or size of total neurons in the septum and CPu, or the cholinergic development of striatal neurons. The role of L1 in the regulation of ChAT may be of significance in cognitive impairments observed in L1-deficient cases and in the design of strategies aiming to treat mental retardation and disorders with cholinergic deficits, such as Alzheimer's disease.
Materials and Methods
Animals and tissue preparation
All experimental procedures were approved by the Animal Care Committee of Sunnybrook Research Institute and conformed to the guidelines set by the Canadian Council on Animal Care and the Animals for Research Act of Ontario.
L1-deficient mice
L1 expression in mutant mice was abolished by the insertion of a tetracycline-controlled transactivator in the second exon of the L1 gene (L1/tTA knock-in) (Rolf et al. 2001; Dihné et al. 2003; Ohyama et al. 2004; Saghatelyan et al. 2004; Bernreuther et al. 2006). L1-deficient males (L1−/y), heterozygous females (L1+/−), and wild-type littermates were generated by crossing heterozygous female mice (L1+/−) on a C57BL/6J/129SvJ genetic background with wild-type 129X1/SvJ male mice (JAX mice, ME).
Genotyping was performed by polymerase chain reaction (PCR). Briefly, genomic tail DNA was isolated and the L1 mutant allele was detected by a 454 base pair DNA fragment generated by PCR using a 5′ primer that anneals to the tTA sequence (5′-TAC ATG CCA ATA CAA TGT AGG CTG C) and a 3′ primer in the L1 sequence (5′-GGA ATT TGG AGT TCC AAA CAA GGT GAT C). The wild-type L1 allele was detected by a 351 base pair PCR product generated using the primers 5′ (5′-AGA GGC CAC ACG TAC CGC AGC ATC) and 3′ (5′-GGA ATT TGG AGT TCC AAA CAA GGT GAT C) in the L1 sequence. PCR results were confirmed by immunoblot and immunocytochemistry analyses in brain tissue, assuring that L1 is abolished in L1-deficient mice.
L1-deficient mice and their wild-type littermates were used at the following postnatal ages: 1 day and 1, 2, 4, and 8 weeks. As reported by other groups, L1-deficient mice were significantly smaller than their wild-type littermates, and had significantly lower body weight at 1 day and at 1, 2, and 4 weeks (but not at 8 weeks) postnatally (Fig. 1).
Figure 1.
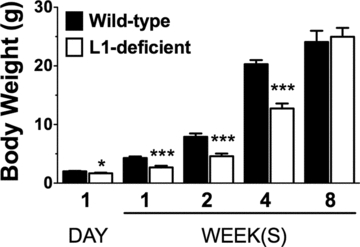
Body weight of L1-deficient and wild-type mice during postnatal development. L1-deficient mice have significantly lower body weight compared to wild-type mice at 1 day (*P = 0.021, n = 5), 1 week (***P < 0.001, n = 5), 2 weeks (***P < 0.001, n = 8), and 4 weeks (***P < 0.001, n = 11) of age. No statistical difference was observed at 8 weeks of age.
At 1 day and at 1, 2, 4, and 8 weeks postnatally, mice were deeply anesthetized with a mixture of ketamine (150 mg/kg) and xylazine (10 mg/kg) and perfused intracardially with saline for 1 min. For ChAT activity (all time points) and immunoblot analyses (2- and 4-week-old mice), the brain was quickly removed from the skull and the septum and caudate-putamen (CPu) were dissected on ice. The isolated tissue was frozen in liquid nitrogen and stored at −70°C. Tissues were homogenized in 1:10 wet w/v ice-cold lysis buffer (20 mM Tris-Cl, 0.25 M sucrose, 1 mM EDTA, 1 mM EGTA, pH 7.4) containing a protease inhibitor cocktail (Calbiochem, La Jolla, CA). The homogenates were centrifuged at 12,000 g for 15 min at 4°C, and the supernatant was used for ChAT activity assays and western blot analyses. The protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad, Heracles, CA). For immunohistochemistry and stereological analyses, deeply anesthetized mice at 2 and 4 weeks were perfused intracardially with saline, followed by a fixative composed of 12.5% picric acid and 2% paraformaldehyde in 0.1 M phosphate buffer. Brains were removed, postfixed overnight, and cryoprotected in a 30% sucrose solution.
Culture of primary septal neurons
To demonstrate that L1 can increase ChAT activity in a dose-dependent manner, we used well-characterized rat primary septal neurons (Burgess and Aubert 2006; Burgess et al. 2009). Timed-pregnant Sprague Dawley rats were obtained from Charles River Laboratories (St. Constant, Quebec, Canada). They were housed individually and received food and water ad libitum for 2 days prior to embryo retrieval.
Embryos at gestation day 17 were retrieved from Sprague Dawley rats and cells from the septal area of the basal forebrain were prepared as detailed previously (Burgess and Aubert 2006; Burgess et al. 2009), with slight modifications from Hefti et al. (1989) and Pongrac and Rylett (1998). Briefly, septal cells were plated in 10% serum. After 1 h, the medium containing unattached cells was removed and replaced by serum-free medium supplemented with N-2 and containing L1-Fc [0, 5, 25, 50 μM] (Loers et al. 2005). Cells were maintained in culture for 4 days, yielding ∼98% pure neuronal population.
Western blot analysis
Standards and samples (10 μg protein/sample) were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in 5% skim milk for 1 h and then incubated for 2 h with a rabbit anti-L1 antibody [1:2000] (a generous gift from Dr. Stallcup et al. 1985) in Tris-buffered saline and tween (TBST). For the detection of ChAT, the membrane was blocked with 0.1% BSA for 1 h and then incubated overnight with the goat anti-ChAT antibody [1:2000] (AP144P, Chemicon, Temecula, CA) in TBST. The membranes were rinsed and incubated for 2 h at room temperature with the appropriate horseradish peroxidase conjugated antibodies (Jackson ImmunoResearch, West Grove, PA) directed against rabbit [1:100,000] in 5% skim milk, or goat [1:20,000] in 0.1% BSA. Immunoreactive signals were detected using the enhanced chemiluminescence system (Millipore, Bedford, MA).
To quantify the relative amount of ChAT protein, the blots were stripped and reprobed with an antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [1:2000] (Biodesign, Saco, ME) for 1 h, followed by a horseradish peroxidase conjugated antibody [1:2000]. The ChAT and GAPDH bands were quantified using the Genetools Analysis Software (Syngene, Cambridge, UK). ChAT immunoreactivity was normalized to GAPDH and the relative amount of ChAT protein in L1-deficient mice was expressed as a percent of ChAT present in wild-type littermates.
ChAT activity
ChAT activity was measured as previously described (Burgess and Aubert 2006; Burgess et al. 2009), using the method of Fonnum (1969), modified by Tucek (1978). Briefly, each sample dissected from septal and striatal regions was homogenized, diluted, and incubated with [14C] acetyl CoA at 37°C for 30 min. Homogenates prepared from septal cells in vitro were incubated for 50 min. The reaction was then stopped, and the newly formed [14C]acetylcholine was extracted, counted, and expressed as nanomoles of acetylcholine produced per milligram of protein per hour (nmol ACh/mg prot/h). The final value for each sample represents an average of duplicates.
Immunostaining
Coronal brain sections were cut at 50 μm on a freezing microtome, collected serially in 96-well plates filled with cryoprotectant, and stored at −20°C. Sections from L1-deficient mice and their wild-type littermates were processed simultaneously using standard immunostaining procedures for fluorescence microscopy (Aubert et al. 1998) and stereology (Ypsilanti et al. 2008).
For immunofluorescence staining, sections were rinsed in 0.1 M Tris-buffered saline (TBS, pH 7.4) and incubated in TBS with 5% normal donkey serum and 0.25% Triton X-100 for 1 h at room temperature. For the combined detection of ChAT and L1, the goat anti-ChAT antibody [1:100] (AB144P, Chemicon) and the rabbit anti-L1 antibody [1:1000] (a generous gift from Dr. Stallcup et al. 1985) were used overnight at 4°C. Sections were rinsed and incubated with donkey anti-goat and donkey anti-rabbit secondary antibodies [1:200] (Jackson ImmunoResearch) coupled to biotin and indocarbocyanine (Cy3), respectively, for 2 h at room temperature in the dark. Sections were rinsed and incubated for 2 h in the dark with streptavidin-Alexa 488 and the nucleic acid staining cyanine dye monomer TO-PRO-3 iodide (2 μM). Sections were rinsed and mounted on presubbed slides, allowed to dry briefly, and coverslipped with a 10% solution of polyvinyl alcohol containing 2.5% 1,4-diazabicyclo-2,2,2-octane (PVA/DABCO, both from Sigma, St. Louis, MO).
To evaluate possible postnatal cell proliferation and the level of neuronal maturity in the MS/VDB and CPu of L1-null and wild-type mice at 2 and 4 weeks, brain sections were processed for immunostaining with Ki67 and the neuronal nuclear antigen (NeuN). Sections were incubated overnight at 4°C with the following primary antibodies: rabbit anti-Ki67 [1:1000] (Novocrasta, Newcastle, UK), mouse anti-NeuN [1:100] (Chemicon), and goat anti-ChAT antibody [1:100] (AB144P, Chemicon). Sections were rinsed and incubated for 2 h at room temperature in the dark with the appropriate secondary antibodies [1:200] from Jackson ImmunoResearch: donkey anti-rabbit-Cy3, donkey anti-mouse-indodicarbocyanine (Cy5), and donkey anti-goat-biotin followed by streptavidin-Alexa 488 [1:200] for 2 h at room temperature in the dark. Sections were rinsed and mounted as described above.
For brightfield and stereological analyses, sections were incubated in 0.6% hydrogen peroxide for 20 min and blocked for 1 h. For ChAT staining, the blocking buffer and solution to dilute the primary antibody contained 5% donkey serum and 0.25% TritonX-100. For NeuN staining, these solutions contained 5% donkey serum, 1% BSA, and 0.1% TritonX-100. Following an overnight incubation at 4°C with the goat anti-ChAT antibody AB144P [1:200] (Chemicon) and mouse anti-NeuN antibody [1:400] (Chemicon), sections were treated with a biotinylated secondary antibody [1:250] (Jackson ImmunoResearch Laboratory) for 2 h followed by the avidin-biotin complex (ABC Elite kit, Vector Laboratories, Burlingame, CA). Sections were then treated with a solution containing 3,3′-diaminobenzidine (DAB; Sigma and Vector Laboratories), 0.01% nickel ammonium sulfate, and 0.005% hydrogen peroxide until a brown reaction developed. The reaction was stopped and sections were mounted on gelatin-coated slides, dehydrated, and coverslipped with Pro-texx (Lerner Laboraories, Pittsburgh, PA).
Confocal microscopy, image analysis, and presentation of the results
Fluorescent labeling was detected with a confocal microscope equipped with argon and helium/neon lasers with excitation wavelength of 488, 543, and 633 nm (Zeiss Axiovert 100M, LSM510; Carl Zeiss, Don Mills, Canada).
Brightfield labeling was captured with a Zeiss Axioplan 2 microscope coupled to a DEI-750 CE video camera (Optronics, Goleta, CA), a software-driven Ludl X-Y-Z motorized stage (Ludl Electronic products, Hawthorne, NY), and a stereology system using the software Stereo Investigator 5.05.4 (optical fractionator and vertical nucleator probes) and the Virtual Slice module (MBF Bioscience, Williston, VT).
Montages of the figures were made in Adobe Photoshop CS5 (Adobe Systems Inc., San Jose, CA). GraphPad Prism 5 (GraphPad Software, San Diego, CA) was used for the presentation of scatter plots and bar graphs.
Stereological analyses in the septum (MS/VDB) and CPu
The rostro-caudal span of sections included in the stereological analyses of ChAT-positive and NeuN-positive neurons started at the appearance of the lateral ventricle and ended at the midline crossing of the anterior commissure. These boundaries covered the entire MS/VDB and the corresponding portion of the CPu analyzed.
Cholinergic cell number and size (ChAT-positive neurons)
A systematic series of one in three sections was randomly selected, totaling on average of eight sections per animal. Histological slides were coded and the sterological analysis was done blindly with regard to the identity of the animals. The number of cholinergic (ChAT-positive) neurons (N) was estimated with the optical fractionator probe (Stereo Investigator, MBF Bioscience) (West 1993) and based on the number of cell bodies (cell tops) counted using a 100× objective, according to the equation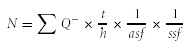 where ∑Q− is the number of particles counted, t is the section thickness calculated by the software at each sampling site, h is the counting frame height (h = 17 μm), asf is the area sampling fraction (asf = area of counting frame/area of sampling grid = 50 μm × 50 μm/80 μm × 80 μm), and ssf is the section sampling fraction (ssf = 1/3). On average, 158 septal and 171 striatal ChAT-positive neurons were counted per animal. The cells marked for counting had a stochastic pattern within the disector height (z-axis), as visualized with the software. The coefficient of error (CE Gundersen) for the estimations of cholinergic cell numbers was similar in L1-deficient mice and their wild-type littermates, averaging 0.077 in the MS/VDB and 0.078 in the CPu.
where ∑Q− is the number of particles counted, t is the section thickness calculated by the software at each sampling site, h is the counting frame height (h = 17 μm), asf is the area sampling fraction (asf = area of counting frame/area of sampling grid = 50 μm × 50 μm/80 μm × 80 μm), and ssf is the section sampling fraction (ssf = 1/3). On average, 158 septal and 171 striatal ChAT-positive neurons were counted per animal. The cells marked for counting had a stochastic pattern within the disector height (z-axis), as visualized with the software. The coefficient of error (CE Gundersen) for the estimations of cholinergic cell numbers was similar in L1-deficient mice and their wild-type littermates, averaging 0.077 in the MS/VDB and 0.078 in the CPu.
The vertical nucleator probe (Stereo Investigator, MBF Bioscience) was used to estimate the largest cross-sectional profile area of each ChAT-positive neuron whose cell top fulfilled the three-dimensional counting rules of the optical fractionator. Briefly, at the largest cross-sectional profile of the cell, a set of four rays is extended from a point within the cell and radiate with a random orientation in four opposite directions toward the edge of the profile. The four intersections with the cell boundary are marked. The area of the profiles (A) was estimated according to the equation 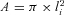 , where li is the average of the lengths of the intercepts.
, where li is the average of the lengths of the intercepts.
Total cell number (NeuN-positive neurons)
The number of total number of NeuN-positive neurons (N) in the septum and CPu was assessed as described above for ChAT-positive neurons but with the following parameters: number of particles (NeuN-positive nuclei) counted (∑Q−), the section thickness (t), the counting frame height (h = 17 μm), the area sampling fraction (asf = area of counting frame/area of sampling grid = 50 μm × 50 μm/250 μm × 250 μm for MS/VDB, 40 μm × 40 μm /350 μm × 350 μm for CPu), and the section sampling fraction (ssf = 1/6). On average, 330 septal and 634 striatal neurons were counted per animal. The coefficient of error (CE Gundersen) for the estimations of NeuN-positive cell numbers was similar in L1-deficient mice and their wild-type littermates, averaging 0.0675 in the MS/VDB and 0.0517 in the CPu.
Statistical Analysis
Statistical analyses were done with GraphPad Prism 5 (GraphPad Software). Data in bar graphs are given as the mean ± SEM. Comparisons within one age group were made with paired t-tests, matching L1-null mice with respective wild-type littermates. Significance was noted at P < 0.05. One-way ANOVA was used to compare the mean values of ChAT activity in response to increasing doses of L1-Fc followed by a Tukey's multiple comparison test and a linear trend posttest.
Results
Evaluation of L1's expression in the brain of wild-type and L1-deficient mice
Protein extracts from the brain of wild-type mice revealed the typical L1 bands at 140 and 200–220 kDa, which were absent in L1-deficient mice (Fig. 2A). Cholinergic neurons, immunoreactive for ChAT (red), were found in the MS/VDB of L1-expressing (green) 2-week-old wild-type mice (Fig. 2B and C). L1 immunostaining was not detected in L1-deficient mice (Fig. 2D).
Figure 2.
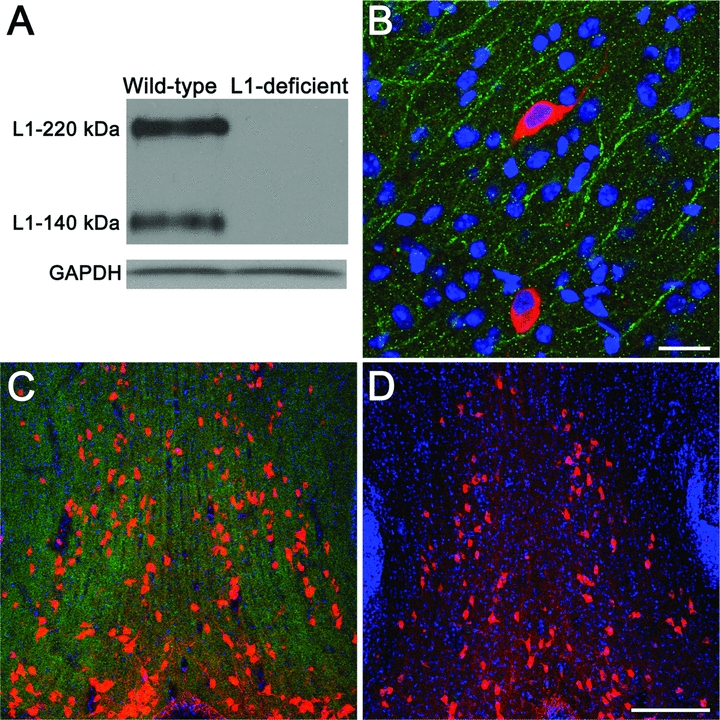
Evaluation of L1's expression in the brain of wild-type and L1-deficient mice. (A) Western blot analysis of whole-brain extracts from 2-week-old wild-type littermates and L1-deficient mice, confirming the lack of the typical 140, 200–220 kDa bands in L1-deficient mice. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) immunoreactivity controlled for the amount of total protein loaded in wild-type and L1-deficient mice. (B, C) Choline acetyltransferase (ChAT)-positive neurons (red), located in the L1-rich (green) medial septal nucleus and the vertical limb of diagonal band of Broca (MS/VDB) of 2-week-old wild-type mice. (D) ChAT-positive neurons (red) in the MS/VDB of 2-week-old L1-deficient mice, which are null for L1 immunoreactivity (absence of green). The nuclear dye TO-PRO-3 iodide (blue) was used as a counterstain (B–D). Scale bars, B = 20 μm, C and D = 200 μm.
ChAT-positive neurons in the MS/VDB and CPu of L1-deficient mice
ChAT-positive neurons of the MS/VDB and the CPu were easily detectable and of similar appearance in L1-deficient compared to wild-type mice at 2 (Figs. 3A–D) and 4 (not shown) weeks of age. Most L1-deficient mice had enlarged lateral ventricles (Fig. 3C) compared to wild-type littermates (Fig. 3A) but the appearance of the MS/VDB and CPu was not strikingly different between L1-deficient and wild-type mice.
Figure 3.
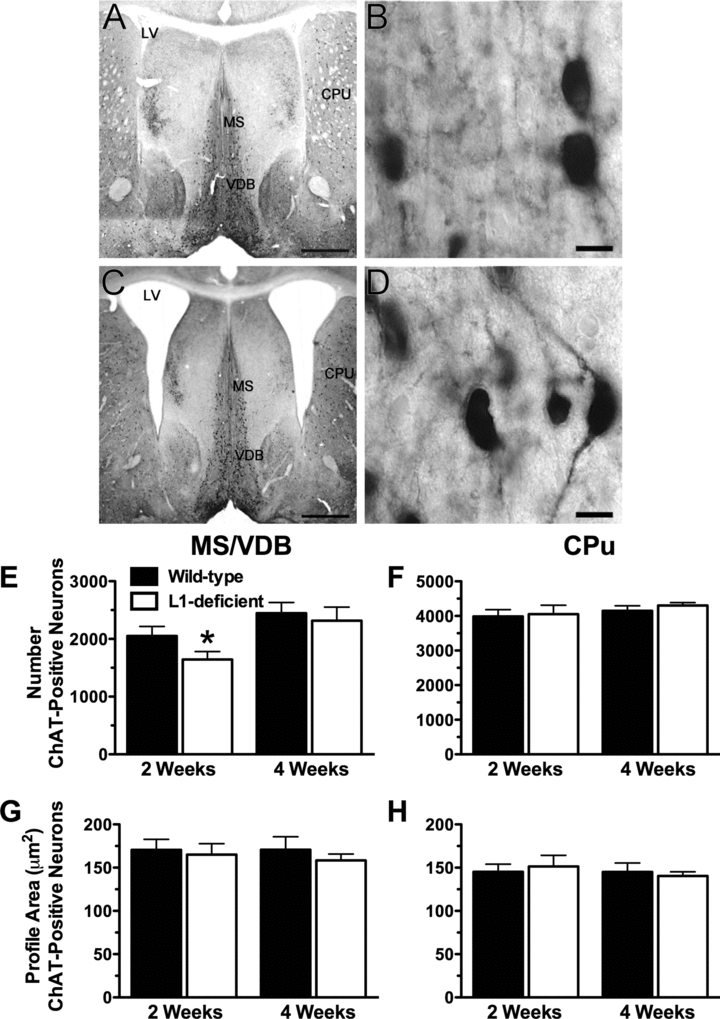
ChAT-positive neurons in L1-deficient mice. (A–D) ChAT-positive cells are observed in the medial septal nuclei and the vertical limb of the diagonal band (MS/VDB) and in the caudate-putamen (CPu) of wild-type (A, B) and L1-deficient (C, D) mice at 2 weeks of age. Most L1-deficient mice (e.g., C) had enlarged lateral ventricles (LV). Scale bars: A and C = 1 mm; B and D = 10 μm. (E–H) Stereological estimations of ChAT-positive cell number and size in the MS/VDB and CPu in L1-deficient mice and wild-type littermates at 2 and 4 weeks of age. (E) A significantly lower number of ChAT-positive neurons was found in the MS/VDB of 2-week-old L1-deficient mice compared to their littermate controls (*P = 0.038). (F) The number of ChAT-positive neurons in the CPu was not statistically different in L1-deficient mice compared to their littermate controls. (G, H) The mean profile area of cholinergic neurons in the MS/VDB (G) and CPu (H) was not statistically different in L1-deficient mice compared to their littermate controls.
Estimated by the optical fractionator probe, the total number of ChAT-positive neurons in the MS/VDB of 2-week-old L1-deficient mice was 20% lower than in wild-type littermates (Fig. 3E, *P = 0.038, n = 4). In contrast, the number of ChAT-positive neurons in the CPu of 2-week-old L1-deficient mice and wild-type littermates was not statistically different (P = 0.590, n = 3) (Fig. 3F). At 4 weeks, the number of ChAT-positive neurons was not statistically different in L1-deficient mice compared to wild-type littermates in the MS/VDB (P = 0.604, n = 5, Fig. 3E) and in the CPu (P = 0.440, n = 4 Fig. 3F).
Using the nucleator probe on the same ChAT-positive neurons that were counted with the optical fractionator, the maximal cross-sectional area of ChAT-positive neurons was not statistically different in L1-deficient mice compared to wild-type littermates in the septum at 2 (P = 0.737) and 4 weeks (P = 0.424) (Fig. 3E) and in the CPu at 2 (P = 0.589) and 4 weeks (P = 0.432) (Fig. 3F).
Regulation of ChAT by L1
To further investigate the role of L1 in the regulation of a cholinergic phenotype, we measured the levels of ChAT activity and ChAT protein in L1-deficient mice at postnatal day 1 and at 1, 2, 4, and 8 weeks of age compared to wild-type littermates (Figs. 4A–C). We also tested whether ChAT activity can be induced by increasing doses of L1-Fc in primary septal neurons (Fig. 4D).
Figure 4.
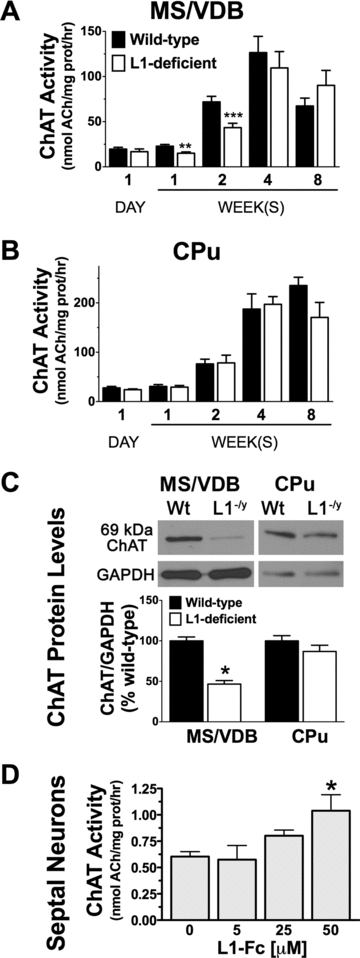
Regulation of ChAT by L1. (A) ChAT activity in MS/VDB was significantly lower in L1-deficient mice compared to wild-type littermates at 1 day (**P = 0.004) and 2 weeks (***P = 0.0003) of age. (B) In the CPu, ChAT activity was not statistically different in L1-deficient mice compared to wild-type littermates. (C) Western blot analyses revealed a 53% decrease in ChAT in L1-deficient (L1−/y) mice compared to wild-type (Wt) littermates in the MS/VDB (*P = 0.028, n = 3) but not in the CPu. (D) In primary septal neurons, L1-Fc (50 μM) significantly increased ChAT activity (*P = 0.039). ChAT activity was measured in duplicates from three (5 and 15 μM L1-Fc) to six (0 and 50 μM L1-Fc) independent experiments.
ChAT activity was reduced by 34% (**P = 0.004, n = 5) and 40% (***P = 0.0003, n = 9) in the septum of 1- and 2-week-old L1-deficient mice compared to wild-type littermates (Fig. 4A). ChAT activity in the septum of L1-deficient mice recovered over time and it was not significantly different compared to wild-type littermates at 4 weeks (P = 0.066, n = 6) and 8 weeks of age (P = 0.240, n = 4). In the CPu, ChAT activity was not statistically different in L1-deficient mice compared to wild-type mice at 1 day (P = 0.334, n = 5), 1 week (P = 0.789, n = 5), 2 weeks (P = 0.941, n = 5), 4 weeks (P = 0.854, n = 3), and 8 weeks (P = 0.127, n = 4) of age (Fig. 4B).
Western blot analyses revealed that the levels of ChAT protein in the septum of 2-week-old L1-deficient mice are 53% lower than in wild-type littermates (Fig. 4C, *P = 0.028, n = 3). In the CPu, the amount of ChAT protein was not statistically different in L1-deficient mice compared to wild-type littermates at 2 weeks of age (Fig. 4C, P = 0.381, n = 3).
To test whether the presence of L1 can activate ChAT, a range of L1-Fc concentrations [5, 25, and 50 μM] was applied to primary septal neurons in culture (Fig. 5D). Our analysis indicates a significant linear trend between ChAT activity and increasing doses of L1-Fc (P = 0.0065). A significant increase in ChAT activity was found at 50 μM compared to 0 μM L1-Fc (Fig. 4D, *P = 0.039, n = 6).
Figure 5.
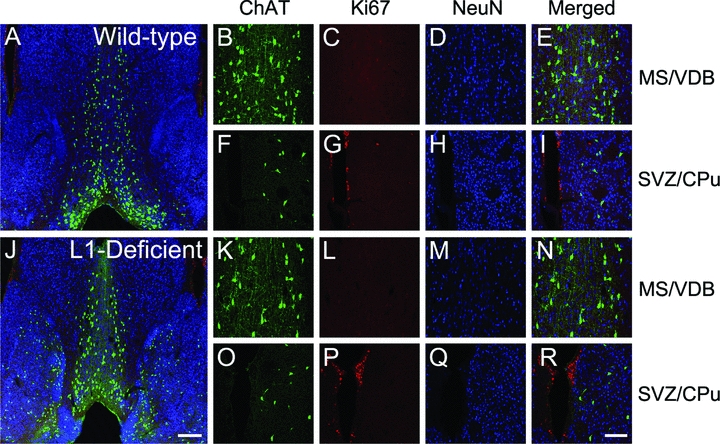
Markers of cell proliferation and maturation in 2-week-old mice. Immunostaining for ChAT (green), cell proliferation marker Ki67 (red), and mature neuronal nuclei antigen (NeuN, blue) in wild-type (A–I) and L1-deficient (J–R) mice at the level of the MS/VDB (enlarged in panels B–E and K–L), and the suventricular zone (SVZ) adjacent to CPu (enlarged in panels F–I and O–R), with the CPu being to the right of the SVZ. Ki67 staining indicates the typical cell proliferation in the SVZ of both wild-type and L1-deficient mice. Ki67-positive cells were not found in areas enriched in ChAT-positive neurons such as the MS/VDB and CPu in wild-type and L1-deficient mice and where NeuN-positive cells were present and quantified in Figure 6. Scale bars: A and J = 200 μm, B–I and K–R = 100 μm.
Cell proliferation and numbers of mature neurons in the MS/VDB and CPu of L1-null mice
Immunostaining for the cell cycle marker Ki67 revealed the typical labeling of proliferating cells in subventricular zone of wild-type and L1-deficient mice at 2 (Fig. 5) and 4 (not shown) weeks of age. Cells in the MS/VDB and CPu were negative for Ki67 (Fig. 5C and L), indicating that cell division no longer occurs in these regions by 2 weeks of age in wild-type and L1-deficient mice.
NeuN-positive cells in the MS/VDB and CPu (Figs. 5D, H, M, Q, and 6A–D) were quantified in L1-deficient mice and wild-type littermates at 2 and 4 weeks of age (Fig. 6E and F). The number of NeuN-positive neurons in the MS/VDB and CPu was not statistically different in L1-deficient mice compared to wild-type littermates (Fig. 6E and F).
Figure 6.
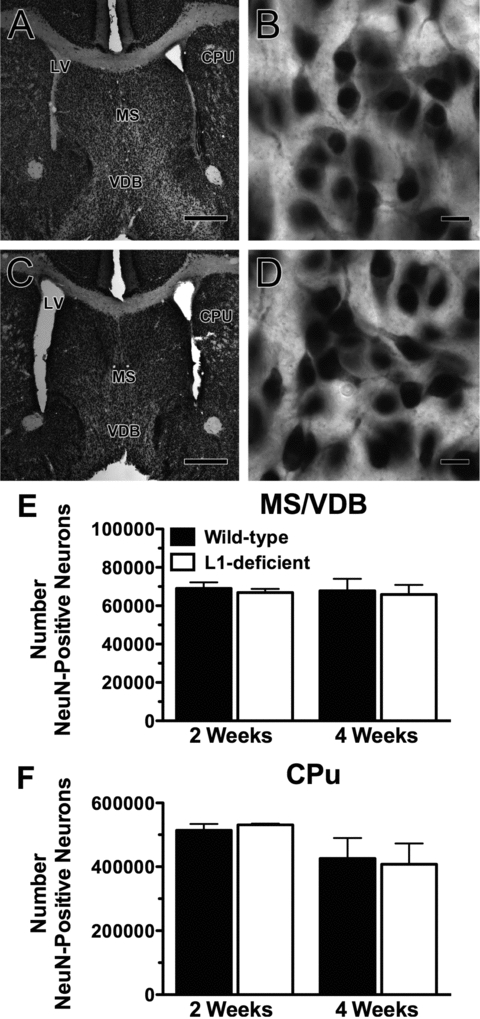
Quantification of the number of mature neurons in the MS/VDB and CPu. (A–D) Neurons stained for the mature NeuN in wild-type (A, B) and L1-deficient mice (C, D) at 2 weeks of age at the level of the MS/VDB, CPu, and lateral ventricles (LV). Scale bars: A and C = 500 μm; B and D = 10 μm. Stereological estimations of the total number of NeuN-positive cells in the MS/VDB (E) and CPu (F), with no statistical difference found between wild-type and L1-deficient mice at 2 and 4 weeks of age.
Discussion
The present study reveals a novel role for L1 in the temporal maturation of septal cholinergic neurons and in the regulation of ChAT. Specifically, L1-deficient mice had significantly less (20%) ChAT-positive neurons in the MS/VDB compared to their littermate controls at 2 weeks of age. Significant reductions in the levels of ChAT protein (53%) and ChAT activity (40%) in the MS/VDB of L1-deficient mice compared to wild-type littermates at 2 weeks of age were also found. Using stereological analyses, all ChAT-positive cells are counted regardless of the levels of ChAT protein or enzyme activity, which likely explains the smaller difference found in the number of ChAT-positive neurons (20%) compared to the 53% reduction in total ChAT protein and the 40% reduction of active ChAT in 2-week-old L1-deficient mice compared to wild-type littermates.
By 4 weeks of age, the number of ChAT-positive neurons and levels of ChAT activity in the MS/VDB were no longer statistically different in L1-deficient compared to control mice, suggesting that L1 is involved in the maturation of a cholinergic phenotype and not in the survival of cholinergic neurons, a role which is attributed to nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) (Chen et al. 1997; Ward and Hagg 2000). Much remains to be investigated to elucidate the full impact of L1 on the development, maturation, and function of the cholinergic septohippocampal system. For example, given the fact that less septal cholinergic neurons and lower levels of ChAT protein and activity are detected in 2-week-old L1-deficient mice, along with the well-characterized role of L1 in axonal growth, guidance, and synaptic plasticity (Maness and Schachner 2007), there is a strong possibility that septohippocampal axonal projections will not develop and mature normally in absence of L1. This could ultimately result in deficits in cholinergic neurotransmission in the hippocampus, explaining some learning and memory impairments detected in adult L1-deficient mice Maness and Schachner 2007.
The absence of the cell cycle marker in the septum at 2 and 4 weeks of age ruled out the possibility of abnormal cell division in the MS/VDB in L1-deficient mice. It remains to be established whether the lower number of ChAT-positive cells estimated in the MS/VDB of 2-week-old L1-deficient mice compared to wild-type littermates reflects a lower detection of the ChAT protein rather than less cholinergic neurons per se. This could be solved by repeating the stereological analysis in the MS/VDB with other cholinergic markers, for example, vesicular acetylcholine transporter, choline uptake transporter, tropomyosin tyrosine kinase A (TrkA), and the p75 neurotrophin receptor (p75NTR)—provided that the expression level of these proteins is not influenced by L1, which remains to be established.
The cholinergic development of striatal neurons was not affected in L1-deficient mice. Septal and striatal cholinergic neurons are generated at similar embryonic stages, but temporal differences in their phenotypic maturation exist during the postnatal period (Semba and Fibiger 1988; Phelps et al. 1989; Gould et al. 1991) and may contribute to their different response to L1 during development. A detailed evaluation of the spatiotemporal pattern of L1's expression in relation to the phenotypic cholinergic maturation of septal and striatal neurons may explain why septal and not striatal neurons have cholinergic deficiencies in 2-week-old L1-deficient mice. Furthermore, in vitro studies demonstrated that L1 transiently regulates the differentiation of neural precursor cells derived from the lateral and medial ganglionic eminences, which give rise to striatal neurons (Dihné et al. 2003). Therefore, it remains possible that the analyses of L1-deficient and wild-type mice at other time points during development would reveal differences in the status of striatal cholinergic neurons.
The number of NeuN-positive cells observed in the septum and CPu was not statistically different in L1-deficient compared to wild-type mice at 2 and 4 weeks postnatally. Therefore, the delay in neuronal maturation in the septum was observed for ChAT-positive neurons and not for the large population NeuN-positive neurons.
The comparable mean maximal crossed-sectional area of cholinergic neurons in the MS/VDB and CPu between L1-deficient and wild-type mice suggests that L1 is not required for the maintenance of the size of cholinergic neurons detected at 2 and 4 weeks postnatally. Molecules classically considered to be essential in the development of cholinergic neurons belong to the neurotrophin family, for example, NGF and BDNF (Chen et al. 1997; Ward and Hagg 2000). NGF and BDNF have well-established actions on cholinergic function, for example, by increasing ChAT activity, and acetylcholine synthesis and release (Alderson et al. 1990; Nonner et al. 1996; Oosawa et al. 1999; Auld et al. 2001). Previous studies in NGF- and BDNF-deficient mice reported that, at given rostro-caudal levels of the brain, the surface area or the diameter of cholinergic neurons is decreased compared to wild-type mice (Chen et al. 1997; Ward and Hagg 2000). Very little is known about how cell adhesion molecules regulate ChAT and the development of cholinergic neurons. The neural cell adhesion molecule (NCAM) was recently implicated in the development of septohippocampal cholinergic neurons (Tereshchenko et al. 2011) and previous work from Acheson and Rutishauser and from our group provides evidence that NCAM can stimulate ChAT activity through cell adhesion (Acheson and Rutishauser 1988), signaling of the fibroblast growth factor receptor (FGFR) (Burgess et al. 2009), and potentiation of BDNF binding and signaling upon removal of polysialic acid (Burgess and Aubert 2006).
L1 was first described as the NGF-inducible large external glycoprotein (NILE) (Bock et al. 1985; Prince et al. 1991). L1's expression is clearly induced by NGF (Salton et al. 1983) but the mechanisms linking L1, NGF, and ChAT expression remain to be established. Blocking TrkA or p75NTR is known to abolish NGF-induced ChAT (Nonner et al. 2000). In contrast, NGF-induced L1 expression can occur in absence of p75NTR (Walsh et al. 1998) or independently of the high-affinity NGF receptor (Itoh et al. 1995). Our in vitro data clearly show that L1-Fc induces ChAT activity and future studies will investigate potential mechanisms. It is possible that L1's activation of ChAT is carried out in part through FGFR (Maness and Schachner 2007), which is known to be a strong ChAT activator (Grothe et al. 1989), which is similar to what we have found with C3d, an NCAM mimetic peptide (Burgess et al. 2009).
In conclusion, L1 regulates the expression of ChAT, it influences levels of ChAT activity, and it is required for the proper development of septal cholinergic neurons in the first 2 postnatal weeks. It remains to be established whether improving cholinergic neurotransmission can rescue cognitive deficits in mice lacking L1. The promoting effects of L1 on ChAT activity and on the development of cholinergic neurons are of significance in the design of therapeutic strategies aiming to alleviate mental retardation and disorders of cholinergic deficits, as in Alzheimer's disease.
Acknowledgments
This work was funded by Natural Science and Engineering Research Council of Canada (NSERCC), Canadian Institutes of Health Research (CIHR Funding Reference Number 93603), Canadian Neurotrauma Research Program (CNRP), Canada Foundation for Innovation (CFI), Ontario Innovation Trust (OIT) (IA), Ontario Neurotrauma Foundation (ONF) Fellowship (IF), Ontario Mental Health Foundation (OMHF) Studentship (AB), and Fundação para a Ciência e a Tecnologia post-doctoral fellowship SFRH/BPD/14581/2003 (MTGdaC). M. Schachner is a New Jersey Professor of Spinal Cord Research. The authors would also like to thank A. Tandon for his critical reading of the article; S. Bell for editing and proof-reading the article; MBF Bioscience and Geoff Greene for stereology support; A. Ypsilanti and S. Rideout for assistance in experiments; and G. Loers, I. Jakovcevski, and P. Putthoff for a generous supply of L1-Fc, reagents, and mice. We are grateful to W. B. Stallcup for the L1 antibody. We appreciated the expert assistance of G. Knowles at the Centre for Cytometry and Scanning Microscopy, and E. Yang at the Proteomics Core Facility of the Toronto Angiogenesis Research Centre, both located at the Sunnybrook Research Institute and supported by a CIHR Multi-User Equipment & Maintenance Grant.
References
- Acheson A, Rutishauser U. Neural cell adhesion molecule regulates cell contact-mediated changes in choline acetyltransferase activity of embryonic chick sympathetic neurons. J. Cell Biol. 1988;106:479–486. doi: 10.1083/jcb.106.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderson RF, Alterman AL, Barde YA, Lindsay RM. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. 1990;5:297–306. doi: 10.1016/0896-6273(90)90166-d. [DOI] [PubMed] [Google Scholar]
- Arami S, Jucker M, Schachner M, Welzl H. The effect of continuous intraventricular infusion of L1 and NCAM antibodies on spatial learning in rats. Behav. Brain Res. 1996;81:81–87. doi: 10.1016/s0166-4328(96)00046-0. [DOI] [PubMed] [Google Scholar]
- Aubert I, Ridet JL, Schachner M, Rougon G, Gage FH. Expression of L1 and PSA during sprouting and regeneration in the adult hippocampal formation. J. Comp. Neurol. 1998;399:1–19. [PubMed] [Google Scholar]
- Auld DS, Mennicken F, Quirion R. Nerve growth factor rapidly induces prolonged acetylcholine release from cultured basal forebrain neurons: differentiation between neuromodulatory and neurotrophic influences. J. Neurosci. 2001;21:3375–3382. doi: 10.1523/JNEUROSCI.21-10-03375.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernreuther C, Dihné M, Johann V, Schiefer J, Cui Y, Hargus G, Schmid JS, Xu J, Kosinski CM, Schachner M. Neural cell adhesion molecule L1-transfected embryonic stem cells promote functional recovery after excitotoxic lesion of the mouse striatum. J. Neurosci. 2006;26:11532–11539. doi: 10.1523/JNEUROSCI.2688-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock E, Richter-Landsberg C, Faissner A, Schachner M. Demonstration of immunochemical identity between the nerve growth factor-inducible large external (NILE) glycoprotein and the cell adhesion molecule L1. EMBO J. 1985;4:2765–2768. doi: 10.1002/j.1460-2075.1985.tb04001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brümmendorf T, Kenwrick S, Rathjen FG. Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr. Opin. Neurobiol. 1998;8:87–97. doi: 10.1016/s0959-4388(98)80012-3. [DOI] [PubMed] [Google Scholar]
- Burgess A, Aubert I. Polysialic acid limits choline acetyltransferase activity induced by brain-derived neurotrophic factor. J. Neurochem. 2006;99:797–806. doi: 10.1111/j.1471-4159.2006.04110.x. [DOI] [PubMed] [Google Scholar]
- Burgess A, Saini S, Weng YQ, Aubert I. Stimulation of choline acetyltransferase by C3d, a neural cell adhesion molecule ligand. J. Neurosci. Res. 2009;87:609–616. doi: 10.1002/jnr.21888. [DOI] [PubMed] [Google Scholar]
- Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J. Neurosci. 1997;17:7288–7296. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Lemmon V. Pathological missense mutations of neural cell adhesion molecule L1 affect neurite outgrowth and branching on an L1 substrate. Mol. Cell Neurosci. 2004;27:522–530. doi: 10.1016/j.mcn.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Cohen NR, Taylor JSH, Scott LB, Guillery RW, Soriano P, Furley AJW. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr. Biol. 1997;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- Dahmé M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat. Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Maness PF. The L1 cell adhesion molecule is essential for topographic mapping of retinal axons. J. Neurosci. 2003;23:530–538. doi: 10.1523/JNEUROSCI.23-02-00530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J. Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Shibata Y, Maness PF. Altered distribution of dopaminergic neurons in the brain of L1 null mice. Dev. Brain Res. 2001;126:21–30. doi: 10.1016/s0165-3806(00)00129-2. [DOI] [PubMed] [Google Scholar]
- Dihné M, Bernreuther C, Sibbe M, Paulus W, Schachner M. A new role for the cell adhesion molecule L1 in neural precursor cell proliferation, differentiation, and transmitter-specific subtype generation. J. Neurosci. 2003;23:6638–6650. doi: 10.1523/JNEUROSCI.23-16-06638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F. Radiochemical micro assays for the determination of choline acetyltransferase and acetylcholinesterase activities. Biochem J. 1969;115:465–472. doi: 10.1042/bj1150465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen E, D'Hooge R, van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SK. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum. Mol. Genet. 1998;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- Frappé I, Wang C, Caines G, Rideout-Gros S, Aubert I. Cell adhesion molecule L1 promotes neurite outgrowth of septal neurons. J. Neurosci. Res. 2004;75:667–677. doi: 10.1002/jnr.20026. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Leranth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J. Comp. Neurol. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolf NJ, Butcher LL. Postnatal development of cholinergic neurons in the rat: I. forebrain. Brain Res. Bull. 1991;27:767–789. doi: 10.1016/0361-9230(91)90209-3. [DOI] [PubMed] [Google Scholar]
- Grothe C, Otto D, Unsicker K. Basic fibroblast growth factor promotes in vitro survival and cholinergic development of rat septal neurons: comparison with the effects of nerve growth factor. Neuroscience. 1989;31:649–661. doi: 10.1016/0306-4522(89)90430-2. [DOI] [PubMed] [Google Scholar]
- Hefti F, Hartikka J, Sanchez-Ramos J. Dissociated cholinergic neurons of the basal forebrain in culture. In: Shahar A, de Vellis J, Vernadakis A, Haber B, editors. A dissection and tissue culture manual of the nervous system. New York: Allan R. Liss Inc; 1989. pp. 172–182. [Google Scholar]
- Itoh K, Brackenbury R, Akeson RA. Induction of L1 mRNA in PC12 cells by NGF is modulated by cell-cell contact and does not require the high-affinity NGF receptor. J. Neurosci. 1995;15:2504–2512. doi: 10.1523/JNEUROSCI.15-03-02504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Hlavin ML, Yamasaki M, Lemmon V. Adhesion molecules and inherited diseases of the human nervous system. Annu. Rev. Neurosci. 1998;21:97–125. doi: 10.1146/annurev.neuro.21.1.97. [DOI] [PubMed] [Google Scholar]
- Law JW, Lee AY, Sun M, Nikonenko AG, Chung SK, Dityatev A, Schachner M, Morellini F. Decreased anxiety, altered place learning, and increased CA1 basal excitatory synaptic transmission in mice with conditional ablation of the neural cell adhesion molecule L1. J. Neurosci. 2003;23:10419–10432. doi: 10.1523/JNEUROSCI.23-32-10419.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loers G, Chen S, Grumet M, Schachner M. Signal transduction pathways implicated in neural recognition molecule L1 triggered neuroprotection and neuritogenesis. J. Neurochem. 2005;92:1463–1476. doi: 10.1111/j.1471-4159.2004.02983.x. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Moulding HD, Martuza RL, Rabkin SD. Clinical mutations in the L1 neural cell adhesion molecule affect cell-surface expression. J. Neurosci. 2000;20:5696–5702. doi: 10.1523/JNEUROSCI.20-15-05696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Barrett JN. Neurotrophin effects on survival and expression of cholinergic properties in cultured rat septal neurons under normal and stress conditions. J. Neurosci. 1996;16:6665–6675. doi: 10.1523/JNEUROSCI.16-21-06665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Barrett JN. Brief exposure to neurotrophins produces a calcium-dependent increase in choline acetyltransferase activity in cultured rat septal neurons. J. Neurochem. 2000;74:988–999. doi: 10.1046/j.1471-4159.2000.0740988.x. [DOI] [PubMed] [Google Scholar]
- Ohyama K, Tan-Takeuchi K, Kutsche M, Schachner M, Uyemura K, Kawamura K. Neural cell adhesion molecule L1 is required for fasciculation and routing of thalamocortical fibres and corticothalamic fibres. Neurosci. Res. 2004;48:471–475. doi: 10.1016/j.neures.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Oosawa H, Fujii T, Kawashima K. Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J. Neurosci. Res. 1999;57:381–387. [PubMed] [Google Scholar]
- Phelps PE, Brady DR, Vaughn JE. The generation and differentiation of cholinergic neurons in rat caudate-putamen. Dev. Brain Res. 1989;46:47–60. doi: 10.1016/0165-3806(89)90142-9. [DOI] [PubMed] [Google Scholar]
- Pongrac JL, Rylett RJ. Optimization of serum-free culture conditions for growth of embryonic rat cholinergic basal forebrain neurons. J. Neurosci. Methods. 1998;84:69–76. doi: 10.1016/s0165-0270(98)00099-5. [DOI] [PubMed] [Google Scholar]
- Prince JT, Alberti L, Healy PA, Nauman SJ, Stallcup WB. Molecular cloning of NILE glycoprotein and evidence for its continued expression in mature rat CNS. J. Neurosci. Res. 1991;30:567–581. doi: 10.1002/jnr.490300315. [DOI] [PubMed] [Google Scholar]
- Rathjen FG, Schachner M. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 1984;3:1–10. doi: 10.1002/j.1460-2075.1984.tb01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolf B, Kutsche M, Bartsch U. Severe hydrocephalus in L1-deficient mice. Brain Res. 2001;891:247–252. doi: 10.1016/s0006-8993(00)03219-4. [DOI] [PubMed] [Google Scholar]
- Saghatelyan AK, Nikonenko AG, Sun M, Rolf B, Putthoff P, Kutsche M, Bartsch U, Dityatev A, Schachner M. Reduced GABAergic transmission and number of hippocampal perisomatic inhibitory synapses in juvenile mice deficient in the neural cell adhesion molecule L1. Mol. Cell Neurosci. 2004;26:191–203. doi: 10.1016/j.mcn.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Salton SR, Shelanski ML, Greene LA. Biochemical properties of the nerve growth factor-inducible large external (NILE) glycoprotein. J. Neurosci. 1983;3:2420–2430. doi: 10.1523/JNEUROSCI.03-12-02420.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi CS. Cognitive impairment and cell adhesion molecules. Nat. Rev. Neurosci. 2004;5:917–930. doi: 10.1038/nrn1555. [DOI] [PubMed] [Google Scholar]
- Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat. Rev. Neurosci. 2005;6:48–56. doi: 10.1038/nrn1588. [DOI] [PubMed] [Google Scholar]
- Semba K, Fibiger HC. Time and origin of cholinergic neurons in the rat basal forebrain. J. Comp. Neurol. 1988;269:87–95. doi: 10.1002/cne.902690107. [DOI] [PubMed] [Google Scholar]
- Stallcup WB, Beasley LL, Levine JM. Antibody against nerve growth factor-inducible large external (NILE) glycoprotein labels nerve fiber tracts in the developing rat nervous system. J. Neurosci. 1985;5:1090–1101. doi: 10.1523/JNEUROSCI.05-04-01090.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshchenko Y, Morellini F, Dityatev A, Schachner M, Irintchev A. NCAM ablation in mice causes hippocampal dysplasia and loss of septal cholinergic neurons. J. Comp. Neurol. 2011;519:2475–2492. doi: 10.1002/cne.22636. [DOI] [PubMed] [Google Scholar]
- Tucek S. Choline acetyltransferase. Acetylcholine synthesis in neurons. London: Chapman and Hall; 1978. pp. 1–258. [Google Scholar]
- Wahlsten D, Bulman-Fleming B. Retarded growth of the medial septum: a major gene effect in acallosal mice. Dev. Brain Res. 1994;77:203–214. doi: 10.1016/0165-3806(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Walsh GS, Petruccelli K, Kawaja MD. p75-deficient sensory axons are immunoreactive for the glycoprotein L1 in mice overexpressing nerve growth factor. Brain Res. 1998;798:184–194. doi: 10.1016/s0006-8993(98)00409-0. [DOI] [PubMed] [Google Scholar]
- Ward NL, Hagg T. BDNF is needed for postnatal maturation of basal forebrain and neostriatum cholinergic neurons in vivo. Exp. Neurol. 2000;162:297–310. doi: 10.1006/exnr.1999.7346. [DOI] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol. Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- Wong EV, Kenwrick S, Willems P, Lemmon V. Mutations in the cell adhesion molecule L1 cause mental retardation. Trends Neurosci. 1995;18:168–172. doi: 10.1016/0166-2236(95)93896-6. [DOI] [PubMed] [Google Scholar]
- Ypsilanti AR, Girão da Cruz MT, Burgess A, Aubert I. The length of hippocampal cholinergic fibers is reduced in the aging brain. Neurobiol. Aging. 2008;29:1666–1679. doi: 10.1016/j.neurobiolaging.2007.04.001. [DOI] [PubMed] [Google Scholar]