

Abstract
Background
Esophageal adenocarcinoma is often considered to arise from a clonal stem like population of cells, potentially responsible for its poor prognosis. TGF-β and Notch signaling pathways play important roles in regulating self-renewal of stem cells, cell-fate determination. Both pathways are frequently implicated in gastrointestinal carcinogenesis. However, their contributions to esophageal adenocarcinoma remain unclear.
Methods
We evaluated TGF-β and Notch signaling components in normal esophagus, Barrett's esophagus and adenocarcinoma tissues and cell lines by IHC and immunoblotting; Hes-1 transcription was assayed using a Hes-1 luciferase reporter.
Results
We demonstrate loss of Smad4 (p<0.05), and β2SP (p<0.01) in 5/10 Barrett's and 17/22 adenocarcinoma tissue sections. Concomitantly, dramatically raised levels of Notch signaling components Hes1 and Jagged1 occur in adenocarcinomas tissues and cell lines, compared to normal tissues. In normal esophagus, Oct3/4 positive cells are located in the basal layer (2-3 per cluster), representing a pool of progenitor cells. We observed an expansion of this pool of Oct3/4 positive cells in esophageal adenocarcinoma (15 per cluster). Furthermore, a panel of SOXs proteins documented for stem cell markers exhibit increased expression in tumor cells indicating expansion of putative cancer stem cells. Finally, we find growth inhibition in BE3 cells with a γ-secretase inhibitor (GSIXXI), but not in SKGT-4 cells. Unlike SKGT-4 cells, BE3 cells have activated Notch signaling with disruption of TGF-β signaling.
Conclusions
Our study demonstrates a potential therapeutic value for targeted therapy in esophageal adenocarcinoma in the setting of loss of β2SP/TGF-β with concomitant constitutively active Notch signaling.
Keywords: TGF-β, Notch, Smad4, β2SP, Oct4, Jagged1, Hes1, stem/progenitor cells, cancer stem cell, Barrett's esophageal adenocarcinoma
Introduction
Esophageal cancer is the sixth leading cause of cancer death in the world. It represents 1% of cancers diagnosed in the United States, with an estimated 16,640 new cases reported in 2010 (ACS 2010). The incidence of esophageal adenocarcinoma, a type of esophageal cancer, has risen at an alarming rate in the United States and other Western countries over the last 30 years[1,2]. Esophageal adenocarcinoma is thought to arise through multiple stages of carcinogenesis, including the replacement of the normal squamous epithelial lining with a columnar intestinal metaplasia called Barrett's esophagus[3]. Barrett's esophagus is likely to be secondary to the chronic acid and bile exposure in gastroesophageal reflux disease (GERD) [4]. Patients with Barrett's esophagus are at higher risk of developing esophageal dysplasia and subsequently, adenocarcinoma, at a rate of approximately 0.5-1% per year [5]. The prognosis for patients presenting with advanced esophageal adenocarcinoma is poor, with a 5-year survival of 0.9% [6]. The clonal/stem cell origin of esophageal cancer may present one reason for its poor prognosis. Molecular signatures, identifying the transition from normal esophageal stem cells into cancer stem/progenitor cells, are of paramount importance for developing new therapeutics.
TGF-β signaling is implicated in cell-cycle control, differentiation, and modulation of a number of cancers, particularly of the gastrointestinal tract [7-9]. TGF-β signals through activation of type I and type II transmembrane serine/threonine kinase receptors (TBRI and TBRII). These receptors then recruit intracellular molecules, Smad2 and Smad3, which further complex with Smad4. We have previously demonstrated that a β-2 spectrin, (β2SP or embryonic liver fodrin, ELF), provides the crucial adaptor functions for Smad2/3 and Smad4 [10]. The Smad2-3/4 complex then translocates to the nucleus to target downstream gene activation, such as the up-regulation of p21, p15, p16, RUNX3 and down-regulation of CDK4 and c-myc[11,12]. There is some evidence of dysfunctional TGF-β signaling in Barrett's associated adenocarcinoma. Low mRNA levels of the TGF-β Type II receptor (TBRII) have been reported in 27% of Barrett's-associated adenocarcinoma[13], while LOH of Smad4 (Ch 18q21.1) was found in 45% of cases. Smad4 mRNA expression was progressively reduced in the metaplasia-dysplasia-adenocarcinoma sequence (p<0.01) and smad4 promoter methylation was found in 70% of primary Barrett' adenocarcinoma samples [14]. Impaired TGF-β and Smad4 signaling prevents cell cycle arrest and promotes invasion in esophageal adenocarcinoma cells by increased expression of urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor 1(PAI-1) through MAPK pathways[15]. Moreover, RUNX3, a target gene of TGF-β signaling, has been shown down-regulated in Barrett's-related adenocarcinoma [16]. In addition, we have found that deletion of β-2 spectrin, the crucial adaptor for Smad2/3 and Smad4 resulted in a dramatic and spontaneous formation of liver and gastrointestinal cancers including esophageal cancer. These studies suggest that loss of TGF-β signaling is an important factor in Barrett's-related adenocarcinoma. Thus while disruption of TGF-β signaling has been observed, their modulators especially β-2 spectrin are not clearly delineated in Barrett's related adenocarcinoma.
Notch signaling pathway is implicated in stem cell self-renewal, cell-fate determination, and terminal differentiation[17,18]. Notch signaling is active in hematopoietic stem cells (HSCs) self-renewal in vivo and is down-regulated as HSCs differentiate[19]. Aberrant activation of Notch signaling has been reported in some hematologic malignancies and multiple solid tumors[19,20]. The basic molecular players of Notch signaling are its ligands Delta and Jagged and the Notch receptors (Notch 1 to Notch 4) [18]. Cells expressing Delta or Jagged bind with cells expressing the Notch receptor, which results in the release of an intracellular domain of the Notch receptor (ICN1) by a cascade of proteolytic cleavages by both alpha and gamma secretases. ICN1 then translocates to the nucleus and complexes with CBF-1 and CSL. This complex further recruits transcriptional co-factors converting it from a transcriptional repressor to activator[21]. Canonical Notch signaling may then activate the repressor Hes-1, whose function is to maintain the undifferentiated paradigm that directly opposes the differentiating signals associated with TGF-β induced p21 activation [22]. Notch signaling has been indicated in cancer development, however, has not been well studied for Barrett's-related adenocarcinoma.
We and others have recently observed multiple gastrointestinal cancers including gastric and esophageal in mouse mutants of the TGF-β pathway; the tumors potentially arise from a clonal population of dysfunctional stem cells with activation of oncogenic events. In this study, we provide evidence that Barrett's-related adenocarcinoma could result from a dysfunctional population of stem cells arising from disrupted TGF-β and subsequent activation of Notch signaling.
Materials and Methods
Materials
γ-secretase inhibitor XXI (GSIXXI) was purchased from Calbiochem (La Jolla, CA) and prepared in DMSO at 1mM stock concentration. TGF-β was obtained from Sigma Chemical (St. Louis, MO). Antibodies against cell cycle inhibitors-p21, p16 and p15 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against p-Smad3, Jagged-1, Hes-1, SOX-2 and β-catenin were purchased from Cell Signaling Technology (Beverly, MA). SOX-9 and SOX-4 were purchased from Chemicon (Billerica, MS). The antibodies agaist β2SP, Smad4, TBRII, Runx3, CDK4, and Oct3/4 were from (Santa Cruz Biotechnology, Zymed, Abcam, Chemicon, CA, USA).
Cell lines and cell cultures
The human adenocarcinoma cell lines FLO-1, SKGT-4, BE3 and OE33 were kindly provided by Dr. Uma Raja and Dr. Sushovan Guha (The University of Texas M. D. Anderson Cancer Center, Houston, TX) and have been previously described[23,24]. The h-TERT-immortalized BE cell lines CP-A and CP-C (kind gift from Dr. Peter Rabinovich, Fred Hutchinson Cancer Center, Seattle, WA and supplied by Dr. Xiao-Chun Xu, M. D. Anderson Cancer Center) were grown in MCDB-153 medium supplemented with 5% fetal calf serum, 20 ng/ml EGF (Gibco, Grand Island, NY), 140 μg/ml bovine pituitary extract, 5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml selenium (Sigma), as described previously[25]. As an untreated solvent control, cells were exposed to dimethyl sulfoxide (DMSO; Sigma) at a final concentration of <0.1%. For trypan blue exclusion analysis, cells were treated with TGF-β and GSI XXI for indicated time, mixed with 0.4% trypan blue (1:1), and examined with a light microscope for dye exclusion.
Cell proliferation assays
Cell proliferation assays were done using the CellTiter 96 aqueous nonradioactive cell proliferation assay (MTS) according to the instructions of the manufacturer (Promega Co. Madison, WI). SKGT-4 and BE3 cells were seeded onto 96-well plates (3 × 103cells/well). Twenty-four hours later, the cells were treated with TGF-β and GSIXXI at indicated dose and time in DMEM. As an untreated solvent control, cells were treated with DMSO (for GSIXXI control) or 1mg/ml BSA in 4mM HCI of PBS (for TGF-β control) (Sigma Chemical Co., St. Louis, MO). The medium and reagents were changed once at 72 hours. All assays were performed in triplicate and repeated at least three times.
Protein extraction and Western blot analysis
Total cell lysates were prepared in 2% SDS lysis buffer containing 330 mM Tris-HCl (pH 8.8), 2% SDS, 10% glycerol, and one mini tablet protease inhibitor cocktail (Roche Diagnostics Corp., Indianapolis, IN). The protein concentration of supernatant was determined using the BCA protein assay kit (Pierce, Rockford, IL). Equal amounts of protein (70 μg proteins per well) were loaded and subjected to electrophoresis on 10% or 12% Tris-glycine gels. Western blot analyses were performed as previously described, and immunoreactive bands were visualized by chemiluminescence detection [26].
Transient transfection, and luciferase reporter assays
Hes-1 luciferase reporter [27] was kindly provided by Dr. P Zhang, (U.T. M.D.Anderson Cancer Center, Houston, TX). Plasmid was prepared using the Genopure plasmid midi kit from Roche Diagnostics. Renilla Luciferase Control Reporter (pRL-TK Vector) (Amersham-Pharmacia, Arlington Heights, IL) was used to normalize transfection efficiency.
For transient transfection, Cells were seeded at a concentration of 4 × 105 cells per well in 6 well plates. After overnight culture, the cells were transfected with DNA (1μg of Hes-1 luciferase reporter and 0.2 μg of Renilla vector) mixed with 3 μl of FuGENE 6 (Roche Diagnostics) according to the manufacturer's protocol. Cells were harvested for measurement of luciferase activity by dual luciferase assay system (Promega) with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA). The values represent the mean and standard deviation of at least three independent experiments.
Tumor specimens
Archival formalin-fixed and paraffin-embedded human tissues from esophageal adenocarcinoma, Barrett's esophgus and normal esophagus were obtained from the Department of Pathology, Lombardi Cancer Center, Georgetown University Medical Center, Washington DC. Additional normal squamous esophageal tissues were obtained from the Department of Pathology, U.T.M.D. Anderson Cancer Center, Houston. The patient population included thirty-eight with esophageal adenocarcinoma with varying risk factors, representing different grades and stages of disease and and sixteen with Barrett's esophagus and nine normal esophagi. The former included patients with earlier stage (stage I) and localized disease (stage II-III) to encompass the different stage of esophageal adenocarcinoma. All of the specimens were collected after endoscopy, esophageal resection, or autopsy. Immunohistochemical labeling was performed as previously described [28]. All human tissue procedures were approved by the Institutional Review Board of Georgetown University Medical Center, Washington D.C. and U.T.M.D. Anderson Cancer Center, Houston.
Immunohistochemistry and Histology
Antibodies against β2SP (β-2 spectrin or ELF), Smad4, TBRII, Notch pathway members Jagged1, Hes1, CDK4, RUNX3, and embryonic stem cell marker Oct3/4 were used to determine the expression of these proteins by immunohistochemistry as previously described[28]. β2SP, Smad4, TBRII, and CDK4 labeling was measured in three different grades; ++, intense labeling; +, moderate labeling; and -, loss of labeling.
Statistical Analysis
Global χ2 test was used to test the hypothesis that the coefficient of each variable was equal to 0. Tissue sample sets of immunohistochemical data were compared to assess the significance. A P value of <0.05 was required for statistical significance, and all tests were two-sided. All tests were done with SPSS 10.1 software (SPSS, Inc., Chicago, IL).
Results
Loss of β2SP, Smad4 and TBRII expression in Barrett's esophagus and esophageal adenocarcinoma -- Loss of TGF-β signaling
To determine whether impaired TGF-β signaling occurs in esophageal adenocarcinoma, immunohistochemical analysis was performed on fifty-seven human esophagi specimens. 38 samples represented esophageal adenocarcinoma, 16 represented Barrett's and 9 represented normal esophagi. In normal esophageal mucosa, β2SP is highly expressed in the transit amplifying population. In these cells, which have a high proliferative potential before progressing to terminally differentiated keratinocytes, β2SP is found to be strongly expressed in both the nucleus and the cytosol (Figure 1a). β2SP expression is diminished, however, in both Barrett's and esophageal adenocarcinoma (p<0.004) (Figure 1b and c). Furthermore, 60% of Barrett's specimens and greater than 70% of esophageal adenocarcinoma specimens demonstrate no nuclear β2SP staining (Table 1). Similarly, Smad4 is universally expressed in the nucleus of transit amplifying cells of normal esophagus (Table 1 and Figure 1d). Meanwhile, 40% of Barrett's and greater than 75% of esophageal adenocarcinoma specimens demonstrate weak or absent Smad4 staining (p=0.013) (Table 1 and Figure 1 e and f). Interestingly, TBRII is expressed in 100% of normal and 57% of Barrett's esophagi specimens with decreased expression in esophageal adenocarcinoma (p=0.004) (Table 1 and Figure 1 g-i).
Figure 1.
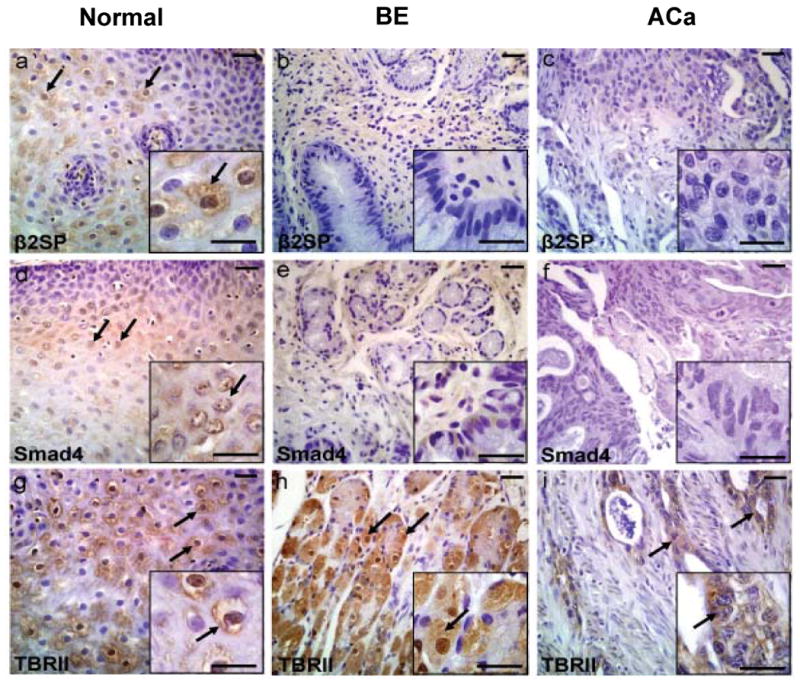
Decreased expression of TGF-β signaling components in BE and Aca tissues. Immunohistochemical analysis of TGF-β members- β2SP, Smad4 and TβRII expression were performed in human normal, Barrett's Esophagus (BE) and esophageal adenocarcinoma (ACa) tissues using β2SP, Smad4 and TβRII antibodies as described in material and methods. β2SP expression is demonstrated in normal esophagus, Barrett's esophagus and adenocarcinoma (a-c). Smad4 expression is showed in normal, Barrett's esophagus and adenocarcinoma (d-f). TBRII expression is illustrated in normal, Barrett's esophagus and adenocarcinoma (g-i). Inset shows the respective figure at higher magnification. Scale bar is 50 μM.
Table 1. IHC staining results for normal, BE and adenocarcinoma tissues.
TGF-β signaling components expression in normal, Barrett's esophagus (BE), and esophageal adenocarcinoma (Aca) tissues. TGF-β signaling components-β 2SP, Smad4 and TβRII and CDK4 were detected in tissues of normal, Barrett's esophagus (BE), and esophageal adenocarcinoma (Aca) tissues by immunohistochemistry as described in material and methods. n represents the number of samples evaluated. ++, represents intense staining; +, moderate staining; and -, no staining. Numbers represent percentage of specimens staining positive.
| Tissue type | Percentage of labeling | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β2SP *p=0.004 | Smad4 *p=0.013 | TBRII *p=0.004 | CDK4 *p=0.053 | |||||||||||||
| ++ | + | - | N | ++ | + | - | N | ++ | + | - | N | ++ | + | - | N | |
| Normal | 0 | 100 | 0 | 9 | 0 | 100 | 0 | 8 | 50 | 50 | 0 | 8 | 0 | 0 | 100 | 9 |
| BE | 24 | 14 | 62 | 11 | 15 | 45 | 40 | 10 | 19 | 38 | 44 | 16 | 14 | 21 | 64 | 14 |
| Aca* | 11 | 16 | 73 | 23 | 2 | 20 | 79 | 29 | 8 | 24 | 68 | 38 | 50 | 25 | 25 | 8 |
Note: Data obtained were statistically significant at P<0.05. ++, intense; +, moderate; -, loss of or reduced labeling. N: number of the samples for each group.
Hes1 and Jagged1 expression in Barrett's and esophageal adenocarcinoma -- Activation of Notch signaling
To evaluate the activation of Notch signaling, expression of Notch target Hes-1 was studied via immunohistochemical analysis. Hes-1 represses the transcription of tissue-specific transcription factors, thereby maintaining stem or progenitor (transit-amplifying) cells via inhibition of differentiation[20]. In normal esophageal tissue, Hes1 is strongly expressed in the basal layer (Figure 2A-a). This is consistent with previous studies indicating that cellular proliferation is limited to the basal layer and that migration to the suprabasal layers is associated with initiation of differentiation. Thereby, canonical Notch signaling is activated mainly in the basal layer to maintain the balance of stem and progenitor cells. Interestingly, in Barrett's esophagus specimens, Hes1 expression is localized to columnar cells and in adenocarcinoma, nuclear Hes1 expression is nearly ubiquitous (Figure 2A-c).
Figure 2.

Up-regulation of Notch signaling in Barrett's adenocarcinoma tissues and cell lines. (A). Hes1 and Jagged1 were detected in tissues of normal, Barrett's esophagus (BE), and esophageal adenocarcinoma (Aca) tissues by immunohistochemistry as described in material and methods. Inset shows the respective figure at higher magnification. Scale bar is 50 μM. Hes1 expression is showed in normal (a), BE (b), and in adenocarcinoma (c). Jagged1 staining is in normal tissue (d), in BE (e) and ACa (f). Inset shows the respective figures at higher magnification. Scale bar is 50 μM. (B). Increased expression of Hes-1, Jagged-1 and Notch intracellular domain (ICN1) were in Barrett's adenocarcinoma cells compared with Barrett's cells by immunoblotting. Immunoblots were performed to use cell lysate were from Barrett's cells (CP-A, CP-C) and adenocarcinoma cells (FLO-1, SKGT-4, BE3 and OE33) to analyse Notch signaling components-Hes-1, ICN1 and Jagged1 expression as described in materials and methods. (C). Increases Hes-1 transcriptional activity in Barrett's adenocarcinoma cells was determined by transient transfection of Hes-1 luciferase promoter reporter as described in Material & Methods.
The Notch ligand Jagged1 expression is used to localize canonical Notch signaling via immunohistochemical analysis. Jagged1 expression in normal esophagus is found in clusters of cells in the basal layer (Figure 2A-d). In Barrett's esophagus specimens, Jagged1 expression is localized to columnar cells, while in adenocarcinoma both nuclear and cytoplasmic labeling for Jagged1 is observed, indicating the activation of Notch signaling (Figure 2A-e,f)). To further confirm the activation of Notch signaling in Barrett' and esophageal adenocarcinoma (EA) cells, we determine the Notch signaling components by immunoblotting and found that marked increased expression of Hes-1 and slight increase of intracellular domain of Notch-1(ICN1) in all EA cells compared with Barrett's cells (CP-A, CP-C); Jagged-1 were absent in both CP-A and CP-C Barrett' cells but expressed in two out of four cell lines (50%)(Figure 3B).
Figure 3.

Evaluation of Oct4 expression in normal and esophageal adenocarcinoma (ACa). (A) Oct4+ was detected in tissues of normal (a) and esophageal adenocarcinoma (Aca) tissues (b) by immunohistochemistry as described in material and methods. Inset shows the respective figures at higher magnification. Scale bar is 50 μM. (B). Immunoblots were performed to analyze SOX-9, SOX-2 and SOX-4 and β-catenin expression using cell lysate were from Barrett's cells (CP-A, CP-C) and adenocarcinoma cells (FLO-1, SKGT-4, BE3 and OE33) as described in materials and methods.
To elucidate the transcriptional activity of Hes-1 as consequence of activation of Notch signaling, the luciferase reporter of Hes-1 has been used to characterize the transcriptional activity of Hes-1. Barrett' and EA cell lines were transfected with Hes-1 luciferase construct and then determine its activity after 48 hours. We found that increased Hes-1 transcriptional activity in EA cells compared to Barrett' cells with the most in BE3 cells (Figure 2C) which may due to dysfunctional of TGF-β signaling. This further emphasizes that esophageal adenocarcinoma overexpress the Notch signaling pathway, thereby maintaining an undifferentiated phenotype.
Oct3/4 localization indicates a continued undifferentiated pool of cells
Given the undifferentiated pool of cells seen with Hes1 and Jagged1 immunohistochemical staining, we next evaluated the potential source of these undifferentiated cells. We labeled cells for the embryonic stem cell marker Oct3/4. The Oct4 gene has been noted as being specifically expressed in embryonic stem cells and in tumor cells, but not in cells of differentiated tissues[29]. In normal esophagus, Oct3/4 expression is localized to the basal layer and confined to 2-3 cells that occupy the center of the basal layer invagination (Figure 3A-a). Oct3/4 expression in the normal esophagus specimens is consistent with previous studies localizing an esophageal stem cell niche. In esophageal adenocarcinoma, however, larger and more diffusely positive Oct3/4 cells are observed. Interestingly, the Oct4 positive cells are no longer confined to a cluster of cells (Figure 3A-b). In summary, in normal tissue Oct3/4 is localized to the basal layer in 2-3 positive cell clusters, and in adenocarcinoma it is present in more than 12% of the total cells. Moreover, the Oct3/4 expression pattern is very similar to Hes1 expression in both normal and cancer tissue. These similar expression patterns may indicate that esophageal cancer cells are a product of aberrant esophageal stem cells.
In addition, a panel of SOXs proteins including SOX-2, SOX-4 and SOX-9 has been documented for stem cell or amplified cell lineage markers and are essential for pluripotency and self-renewal of embryonic stem cells[30-33]. Correspondent to the Oct4 staining in tumor tissues, we found that SOX-9 is highly up-regulated in all adenocarcinoma (Aca) tumor cell lines compared to Barrett's cells, and SOX-4 also increased in certain extent in all Aca cells, while 50% of Aca cells express SOX-2 protein, which has been reported as a lineage-survival oncogene in lung and esophageal squamous cell carcinoma[30] (Figure 3B). Expression of β-catenin is increased in all Aca cells as well (Figure 3B). These data indicate there are expansion of aberrant stem cells named cancer stem cells in Aca tumor tissues and cell lines compared to normal tissue and Barrett' cells.
CDK4 and RUNX3 expression -- Functional consequence of disrupted TGF-β signaling
Given the tumor suppressor activity of TGF-β signaling, we decided to evaluate the functional consequence of its disruption and evaluate RUNX3 and CDK4 expression. The functional ability of β2SP to translocate Smad2 and Smad3 to the nucleus may modulate the Runt domain transcription factor RUNX3, which is involved in TGF-β mediated cell-cycle arrest by inducing the up-regulation of p21cip1/waf [34]. In normal esophagus, expression of RUNX3 is well localized to the transit amplifying population of cells. In Barrett's and adenocarcinoma specimens, however, expression of this transcription factor is absent (Figure 4A d-f). Meanwhile, CDK4, a cell-cycle marker of proliferation, is weakly expressed or absent in normal esophagus (Table 1 and Figure 2a), but strongly expressed in 35% of Barrett's and 75% of esophageal adenocarcinoma specimens (Table 1 and Figure 4A a-c). The cyclin-dependent kinase (CDK) inhibitors p15, p16, p21 are known to be regulated by TGF-β signaling[35]. We questioned the status of these CDK inhibitors in Barrett's and Aca cells as consequence of dysfunctional TGF-β signaling. As expected, P21, P15 and P16 were lost in CP-A and CP-C Barrett' cells and in most of Aca cell lines (Figure 4B).
Figure 4.
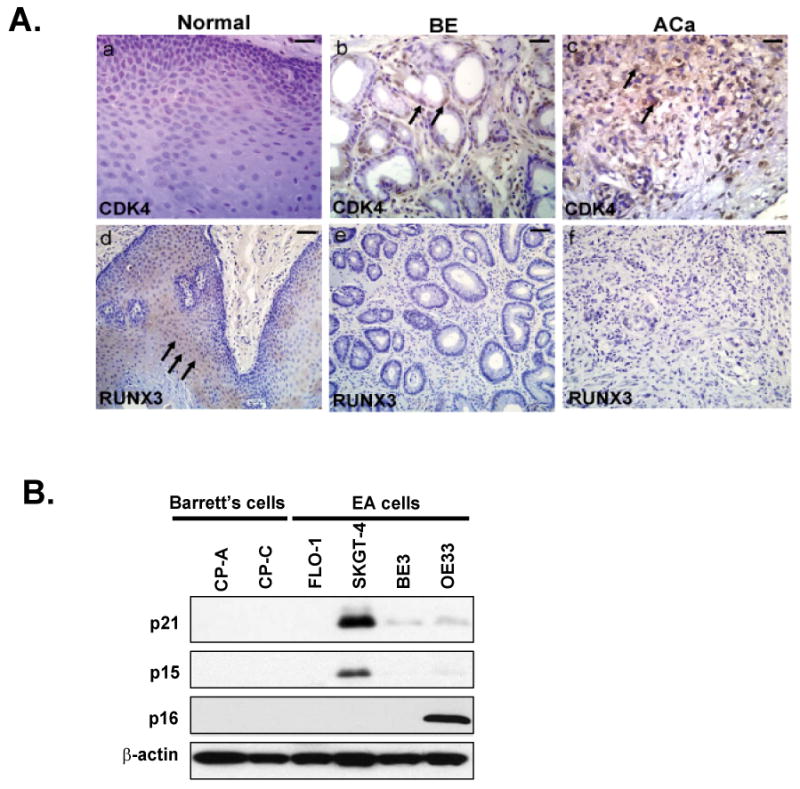
Expression of CDK4 and RUNX3 in normal, Barrett's Esophagus (BE) and esophageal adenocarcinoma (ACa). (A). CDK4 expression(a-c) and RUNX3 (d-f) expression were detected in tissues of normal, Barrett's esophagus (BE), and esophageal adenocarcinoma (Aca) tissues by immunohistochemistry as described in material and methods. Scale bar is 50 μM. (B). Immunoblots were performed to analyze cell cycle inhibitors- P21, P15 and P16 expression using cell lysate from Barrett's cells (CP-A, CP-C) and adenocarcinoma cells (FLO-1, SKGT-4, BE3 and OE33) as described in materials and methods.
Inhibition of Notch signaling by using a γ-secretase inhibitor suppresses proliferation of BE3 cells but not SKGT-4 cells
Two human esophageal adenocarcinoma cell lines, BE3 and SKGT-4 were used to assess the impact of inhibiting Notch signaling on cell proliferation using the MTS assay. The BE3 cell line is TGF-β deficient, while the SKGT-4 cell line maintains intact TGF-β signaling. After stimulation with TGF-β at 1ng/ml, neither cell line exhibits cell proliferation inhibition compared with controls (data not shown). When treating both BE3 cells and SKGT-4 cells with different dosage of γ-secretase inhibitor (GSIXXI), dose dependent inhibition was shown only in BE3 cells with high Notch signaling (Figure 2C and 5B) but not in SKGT-4 cells (Figure 5A). These results suggest that deficient TGF-β signaling in the presence of constitutively active Notch are necessary for effective treatment with a γ-secretase inhibitor.
Figure 5.

Inhibition of cell growth by γ-secretase inhibitor (GSI XXI) only in cells with activated Notch signaling by MTS Assay. The SKGT-4 and BE3 cell lines were seeded in 96-well plates and treated with γ-secretase inhibitor (GSI XXI) from 500nM to 5μM for 72 hours and a nonradioactive MTS cell proliferation assay was performed to determine the rate of proliferation. Values shown represent the mean and standard deviation of triplicate experiments. * p>0.05, **p<0.05.
Discussion
Disruption of TGF-β signaling is an important factor in Barrett's esophagus and esophageal adenocarcinoma. Loss of the tumor suppressor function of TGF-β signaling through Smad4 in esophageal cancer has been previously described as a cause of tumor progression due to the loss of the transcription factor RUNX3, loss of p16, p21 and gain of CDK4 [16,36]. Moreover, TGF-β signaling exhibits functional synergism with Notch signaling in the regulation of Hes-1, a direct target of the Notch pathway [37,38]. Both Notch and TGF-β signaling also converge to regulate the CDK4 inhibitor p21. In addition to the effects of cell-cycle regulator genes, TGF-β has regulatory roles in stem cell biology with opposing functions to Notch signaling. While the TGF-β pathway is required for stem cell differentiation, Notch maintains the undifferentiated phenotype of stem cells[18]. Disruption in TGF-β and Notch signaling could give rise to cells that are unable to differentiate or unable to maintain the differentiated state. These cells have been referred to as cancer-initiating stem cells or cancer stem cells and have been reported in cancers of the breast, prostate and colon [39]. Analogous studies are not yet to be performed in esophageal adenocarcinoma.
Notch signaling is one of key pathways constituting the stem cell signaling network[17]. Aberrant activation of Notch signaling has been reported in gastrointestinal cancers including colon cancer and pancreatic cancers [20,40]. Functionality of Notch activation in tumor initiation and progression is of more recent vintage and emerging. This study provides evidence for the first time that Notch signaling is activated in Barrett's associated esophageal adenocarcinoma tissues and cell lines. Hes-1 is an important notch signaling target and mediator. We demonstrated that Hes-1 expression is up-regulated in Barrett's associated adenocarcinoma tissues and highly up-regulated in all adenocarcinoma cell lines examined. The Hes-1 transcriptional activity was increased in EA cells as well. γ-secretase inhibitor has been shown to inhibit tumor cell growth in both colon cancer and pancreatic cancer [41]. Recent data from Hans Clevers's laboratory has showed that Notch inhibition by GSI XXI converted the proliferative Barrett's epithelial cells into terminally differentiated goblet cells[42]. We found that aberrant activation of Notch and Hes-1 could be due to the dysfunction of TGF-β signaling β2SP and Smad4. γ-secretase inhibitor GSI XXI inhibits cell proliferation only in BE3 with dysfunction of TGF-β and high notch signaling but not in SKGT-4 cells and FLO-1 and OE33 other esophageal adenocarcinoma cell lines with lower Notch signaling. Results from this study may yield important new therapeutic strategies and will be a first step toward the goal of personalized cancer treatment based on molecular characteristics in both TGF-β and Notch signaling in this lethal cancer.
Our results suggest that the presence of a dysfunctional stem cell pool is due to the loss of β2SP/TGF-β and activation of Notch signaling (Fig 6). Barrett's esophageal adecarcinoma has been thought to be a result of clonal evolution. Previously, we observed that loss of TGF-β signaling via loss of the adaptor protein β2SP and TBRII could result in dysfunctional progenitor/stem cells giving rise to hepatocellular carcinoma [43]. Similarly, in esophageal adenocarcinoma, β2SP expression was shown to be absent. TBRII, however, was expressed in greater than 30% of the adenocarcinoma specimens (Table 1). Thereby, disruption of TGF-β signaling in esophageal adenocarcinoma appears after TBRII activation and likely involves β2SP and Smad4. Down-regulation of Smad4 is due to several different mechanisms including methylation, deletions, and protein modification [14]. Meanwhile, RUNX3 expression has been evaluated in esophageal adenocarcinoma cell lines and it was found that the restoration of RUNX3 expression via transfection was able to produce robust inhibition of cell growth [44]. Moreover, β2SP/Smad4 double heterozygous mice develop multiple gastric tumors with E-Cadeherin/β-catenin complexes in gastric epithelial cells of these mutant mice[45].
Figure 6.
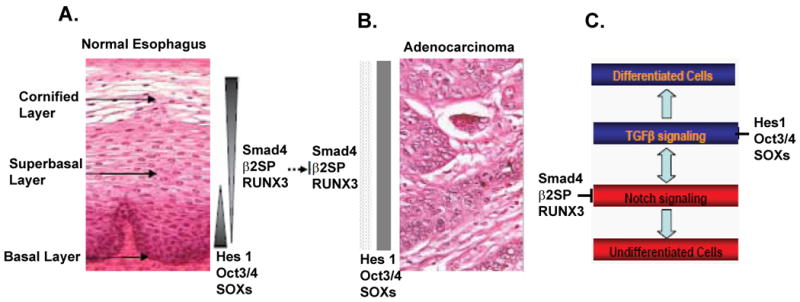
Changes of the expression patterns of TGFβ and Notch signaling components in switch from normal esophagus to adenocarcinoma. A. Different layers of normal esophagus are shown with corresponding expression patterns of Hes1, Oct3/4, Smad4, β2SP and RUNX3. B. Loss of normal hierarchy of esophageal epithelium in adenocarcinoma. Hes1, oct3/4, Smad4, β2SP and RUNX3 are not expressed in gradients as in normal tissue. While expression of Hes1, oct3/4 and SOXs proteins is increased, the expression of Smad4, β2SP and RUNX3 are decreased compared to normal tissue. C. Model of the interaction between notch and TGFβ signaling pathways in maintaining the hierarchy of esophagus epithelium.
Furthermore, the presence of larger pockets of Oct3/4 positive cells in esophageal adenocarcinoma suggests that this cancer may be stem cell driven. These cancer stem cells are likely characterized by dysregulated TGF-β and Notch signaling. Moreover, in normal esophageal tissue, TGF-β and Notch signaling components were strongly expressed in the transit amplifying region while stem cells were localized to the basal cell layer. In adenocarcinoma, however, Oct4 expression became ubiquitous. A panel of SOXs proteins has been documented for stem cell or amplified cell lineage markers[30,31]. SOX-2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinoma[30]. SOX4 is involved in murine embryogenesis and significantly up-regulated in ApcMin adenomas and human tumor cell lines[46]. SOX-9 is a target gene of Wnt/β-catenin signaling and its expression marks a subset of CD24-expressing small intestine epithelial stem cells [31,47]. We demonstrated that expression of SOX-2, SOX-4 and SOX-9 are increased in Barrett's esophageal cancer cell lines further indicating the expansion of cancer stem cells in tumor cell lines. This expansion of cancer stem cells is likely further stimulated by the loss of TGF-β mediated suppression, leaving a population of immortal cells primed for progression through the metaplasia-dysplasia-adenocarcinoma cycle of esophageal adenocarcinoma progression.
Our study, for the first time, demonstrates a role for adaptor protein β2SP in esophageal adenocarcinoma via loss of TGF-β signaling and activation of Notch signaling. Loss of β2SP/Smad4, resulting in the disruption of TGF-β signaling, could contribute to the activation of Notch signaling via Hes-1, a Notch signaling molecule (Figure 6). The interplay between the TGF-β and Notch pathway is critical in the transformation of esophageal stem cells. Moreover, rescue of TGF-β signaling by restoration of β2SP-Smad4 or Notch inhibition by γ-secretase inhibitors in the setting of dysfunctional of TGF-β signaling could hold promise for new personalized therapeutic approaches in esophageal adenocarcinoma.
Abbreviations
- TGF-β
transforming growth factor-β
- β2SP
β-2 spectrin
- ELF
Embryonic Liver fodrin
- TBRII
transforming growth factor-β receptor II
- CSC
cancer stem cell
- Oct4
Octamer-4, a homeodomain transcription factor of the POU family
- Aca
esophageal adenocarcinoma
References
- 1.Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100:1184–7. doi: 10.1093/jnci/djn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blot WJ, Devesa SS, Kneller RW, Fraumeni JF., Jr Rising incidence of adenocarcinoma of the esophagus and gastric cardia. Jama. 1991;265:1287–9. [PubMed] [Google Scholar]
- 3.Hirota WK, Loughney TM, Lazas DJ, et al. Specialized intestinal metaplasia, dysplasia, and cancer of the esophagus and esophagogastric junction: prevalence and clinical data. Gastroenterology. 1999;116:277–85. doi: 10.1016/s0016-5085(99)70123-x. [DOI] [PubMed] [Google Scholar]
- 4.Falk GW. Gastroesophageal reflux disease and Barrett's esophagus. Endoscopy. 2001;33:109–18. doi: 10.1055/s-2001-11669. [DOI] [PubMed] [Google Scholar]
- 5.Jenkins GJ, Doak SH, Parry JM, et al. Genetic pathways involved in the progression of Barrett's metaplasia to adenocarcinoma. Br J Surg. 2002;89:824–37. doi: 10.1046/j.1365-2168.2002.02107.x. [DOI] [PubMed] [Google Scholar]
- 6.Paulson TG, Reid BJ. Focus on Barrett's esophagus and esophageal adenocarcinoma. Cancer Cell. 2004;6:11–6. doi: 10.1016/j.ccr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 7.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2004;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 8.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 9.Sporn MB, Roberts AB. The transforming growth factor-betas: past, present, and future. Ann N Y Acad Sci. 1990;593:1–6. doi: 10.1111/j.1749-6632.1990.tb16095.x. [DOI] [PubMed] [Google Scholar]
- 10.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–7. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 11.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–71. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 12.Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000;12:235–43. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 13.Garrigue-Antar L, Munoz-Antonia T, Antonia SJ, et al. Missense mutations of the transforming growth factor beta type II receptor in human head and neck squamous carcinoma cells. Cancer Res. 1995;55:3982–7. [PubMed] [Google Scholar]
- 14.Onwuegbusi BA, Aitchison A, Chin SF, et al. Impaired transforming growth factor beta signalling in Barrett's carcinogenesis due to frequent SMAD4 inactivation. Gut. 2006;55:764–74. doi: 10.1136/gut.2005.076430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onwuegbusi BA, Rees JR, Lao-Sirieix P, Fitzgerald RC. Selective loss of TGFbeta Smad-dependent signalling prevents cell cycle arrest and promotes invasion in oesophageal adenocarcinoma cell lines. PLoS One. 2007;2:e177. doi: 10.1371/journal.pone.0000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulmann K, Sterian A, Berki A, et al. Inactivation of p16, RUNX3, and HPP1 occurs early in Barrett's-associated neoplastic progression and predicts progression risk. Oncogene. 2005;24:4138–48. doi: 10.1038/sj.onc.1208598. [DOI] [PubMed] [Google Scholar]
- 17.Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–6. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 18.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 19.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107:2223–33. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 20.Katoh M, Katoh M. Notch signaling in gastrointestinal tract (review) Int J Oncol. 2007;30:247–51. [PubMed] [Google Scholar]
- 21.Baron M. An overview of the Notch signalling pathway. Semin Cell Dev Biol. 2003;14:113–9. doi: 10.1016/s1084-9521(02)00179-9. [DOI] [PubMed] [Google Scholar]
- 22.Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–8. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 23.Raju U, Ariga H, Koto M, et al. Improvement of esophageal adenocarcinoma cell and xenograft responses to radiation by targeting cyclin-dependent kinases. Radiother Oncol. 2006;80:185–91. doi: 10.1016/j.radonc.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Soldes OS, Kuick RD, Thompson IA, et al. Differential expression of Hsp27 in normal oesophagus, Barrett's metaplasia and oesophageal adenocarcinomas. Br J Cancer. 1999;79:595–603. doi: 10.1038/sj.bjc.6690094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palanca-Wessels MC, Klingelhutz A, Reid BJ, et al. Extended lifespan of Barrett's esophagus epithelium transduced with the human telomerase catalytic subunit: a useful in vitro model. Carcinogenesis. 2003;24:1183–90. doi: 10.1093/carcin/bgg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Mazurek N, Liu C, et al. Galectin-3 mediates nuclear beta-catenin accumulation and Wnt signaling in human colon cancer cells by regulation of glycogen synthase kinase-3beta activity. Cancer Res. 2009;69:1343–9. doi: 10.1158/0008-5472.CAN-08-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P, Yang Y, Nolo R, Zweidler-McKay PA, Hughes DP. Regulation of NOTCH signaling by reciprocal inhibition of HES1 and Deltex 1 and its role in osteosarcoma invasiveness. Oncogene. 2010;29:2916–26. doi: 10.1038/onc.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Y, Katuri V, Srinivasan R, et al. Transforming growth factor-beta suppresses nonmetastatic colon cancer through Smad4 and adaptor protein ELF at an early stage of tumorigenesis. Cancer Res. 2005;65:4228–37. doi: 10.1158/0008-5472.CAN-04-4585. [DOI] [PubMed] [Google Scholar]
- 29.Tai MH, Chang CC, Kiupel M, et al. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26:495–502. doi: 10.1093/carcin/bgh321. [DOI] [PubMed] [Google Scholar]
- 30.Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gracz AD, Ramalingam S, Magness ST. Sox9 expression marks a subset of CD24-expressing small intestine epithelial stem cells that form organoids in vitro. Am J Physiol Gastrointest Liver Physiol. 2010;298:G590–600. doi: 10.1152/ajpgi.00470.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mfopou JK, Chen B, Mateizel I, et al. Noggin, Retinoids, and Fibroblast Growth Factor Regulate Hepatic or Pancreatic Fate of Human Embryonic Stem Cells. Gastroenterology. 2010 Mar 3; doi: 10.1053/j.gastro.2010.02.056. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Mallanna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol. 2010 May 15; doi: 10.1016/j.ydbio.2010.05.014. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasegawa K, Yazumi S, Wada M, et al. Restoration of RUNX3 enhances transforming growth factor-beta-dependent p21 expression in a biliary tract cancer cell line. Cancer Sci. 2007;98:838–43. doi: 10.1111/j.1349-7006.2007.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–32. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blokzijl A, Dahlqvist C, Reissmann E, et al. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163:723–8. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takizawa T, Ochiai W, Nakashima K, Taga T. Enhanced gene activation by Notch and BMP signaling cross-talk. Nucleic Acids Res. 2003;31:5723–31. doi: 10.1093/nar/gkg778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 40.Mysliwiec P, Boucher MJ. Targeting Notch signaling in pancreatic cancer patients--rationale for new therapy. Adv Med Sci. 2009;54:136–42. doi: 10.2478/v10039-009-0026-3. [DOI] [PubMed] [Google Scholar]
- 41.Yin L, Velazquez OC, Liu ZJ. Notch signaling: Emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010 Mar 31; doi: 10.1016/j.bcp.2010.03.026. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 42.Menke V, van Es JH, de Lau W, et al. Conversion of metaplastic Barrett's epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech. 2010;3:104–10. doi: 10.1242/dmm.003012. [DOI] [PubMed] [Google Scholar]
- 43.Tang Y, Kitisin K, Jogunoori W, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445–50. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torquati A, O'Rear L, Longobardi L, et al. RUNX3 inhibits cell proliferation and induces apoptosis by reinstating transforming growth factor beta responsiveness in esophageal adenocarcinoma cells. Surgery. 2004;136:310–6. doi: 10.1016/j.surg.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 45.Katuri V, Tang Y, Li C, et al. Critical interactions between TGF-beta signaling/ELF, and E-cadherin/beta-catenin mediated tumor suppression. Oncogene. 2006;25:1871–86. doi: 10.1038/sj.onc.1209211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reichling T, Goss KH, Carson DJ, et al. Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005;65:166–76. [PubMed] [Google Scholar]
- 47.Bastide P, Darido C, Pannequin J, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol. 2007;178:635–48. doi: 10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]