

Abstract
Background
Genome-wide screening of patients with mental retardation using Array Comparative Genomic Hybridization (array-CGH) has identified several novel imbalances. With this genotype-first approach, the 2q22.3q23.3 deletion was recently described as a novel microdeletion syndrome. We report two unrelated patients with a de novo interstitial deletion mapping in this genomic region and presenting similar “pseudo-Angelman” phenotypes, including severe psychomotor retardation, speech impairment, epilepsy, microcephaly, ataxia and behavioural disabilities.
Methods
The microdeletions were identified by array-CGH using oligonucleotide and BAC-arrays, and further confirmed by Fluorescence In Situ Hybridization (FISH) and semi-quantitative PCR.
Results
The boundaries and sizes of the deletions in the two patients were different but an overlapping region of about 250 kb was defined, which mapped to 2q23.1 and included two genes: MBD5 and EPC2. The SIP1 gene associated with the Mowat Wilson syndrome was not included in the deleted genomic region.
Discussion
Haploinsufficiency of one of the deleted genes (MBD5 or EPC2) could be responsible for the common clinical features observed in the 2q23.1 microdeletion syndrome and this hypothesis needs further investigation.
Keywords: Abnormalities, Multiple; genetics; pathology; Child; Chromosome Deletion; Chromosomes, Human, Pair 2; Comparative Genomic Hybridization; DNA; chemistry; genetics; Humans; In Situ Hybridization, Fluorescence; Male; Phenotype; Polymerase Chain Reaction
INTRODUCTION
Associations between clinical description and submicroscopic chromosomal imbalances have already been established for microdeletion syndromes, but phenotypic recognition usually precedes the identification of the recurrent cryptic chromosomal rearrangement. Inversely, screening methods such as FISH based screening for subtelomeric chromosomal imbalances in patients with mental retardation have revealed microrearrangements for which phenotypic features were secondarily grouped. Another screening method is array-CGH which allows high resolution screening of the whole human genome for DNA copy number changes. This genome-wide approach, applied to cohorts of patients with mental retardation and dysmorphic features, detected microdeletions and microduplications in 10–15% of the patients, most of them scattered across the genome (1). However, a non-random involvement of several chromosomal regions has been observed (2) and some recurrent rearrangements were recently described, concerning 1q41q42 (3), 2p15p16.1 (4), 9q22.3 (5), 16p11.2p12.2 (6) and 17q21.31 (7–9) regions. For some of these regions, both deletions and duplications were found and their occurrence is probably caused by the Non Allelic Homologous Recombination (NAHR) mechanism due to flanking Low Copy Repeats (LCRs). Novel syndromes can then be described based on the clinical description of patients bearing the same subtle rearrangement.
We report here clinical and molecular cytogenetic findings, observed in two unrelated patients presenting similar “pseudo-Angelman” phenotypes and de novo microdeletion at 2q22.3q23.1. This submicroscopic deletion should enable the description of such a novel microdeletion syndrome and by comparing the probands with previous patients showing overlapping microdeletions, we expect to establish a clinical characterisation of this syndrome, based on phenotypic similarities. Our aim is to improve identification of new patients carrying this cryptic chromosomal rearrangement for more accurate genetic counselling and for improved medical care. Another important point is the delineation of the critical region containing dosage-sensitive genes, presumably involved in the aetiology of the matched phenotypic features, including mental retardation.
MATERIALS AND METHODS
Patients
Informed consent for genetic investigations on mental retardation was obtained from participating families. Detailed clinical history was obtained and physical examination was carried out for the two patients reported.
Subjects 1 and 2 were selected by clinical geneticists according to criteria derived from the checklist of De Vries et al. (10), which is linked with a higher likelihood of manifesting submicroscopic copy number changes, namely an intelligence quotient (IQ) less than 50 with at least one associated criterion (familial history of mental retardation, growth abnormalities, facial dysmorphic features, congenital malformations, seizures and behavorial or sleeping disabilities). Subject 1 belonged to a cohort of 64 patients presenting with mild to severe mental retardation and tested by oligonucleotide based array-CGH to identify novel microdeleted and microduplicated regions that could include a gene involved in mental retardation. The 64 patients were recruited at genetics clinical centres in the West of France (HUGO, Hôpitaux Universitaires du Grand Ouest) and subject 1 came from the clinical genetic department of Rennes University Hospital. Subject 2 was recruited at the clinical genetic department of Robert Debré University Hospital, Paris.
Array-CGH
For subject 1, oligonucleotide array-CGH was performed using the Agilent Human Genome CGH microarray 4×44K (Agilent Technologies, Santa Clara, CA, USA). The experiments were performed according to version 4.0 (June 2006) of the protocol provided by Agilent (Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis). For subject 2, BAC array was performed using the IntegraChips BAC microarray (IntegraGen SA, Evry, France).
DNA extraction and labelling
For subject 1, 3.5 μg of native test and reference sex-matched gDNAs were digested by AluI/RsaI (Promega Corporation, Madison, WI, USA). Reference gDNA was from unique male or female individuals. Samples were labelled by random priming using the Agilent Genomic DNA Labeling Kit PLUS to differentially label test and reference gDNAs with Cy5™-dUTP and Cy3™-dUTP respectively. A dye swap experiment was performed to minimise dye-related artifacts.
For subject 2, 500 ng of patient gDNA and reference gDNA (male DNA from Promega) were labelled with Cy3™-dUTP and Cy5™-dUTP respectively using a random primed labelling kit (Invitrogen™, Carlsbad, CA, USA).
For both patients, test and reference samples were combined in an equimolar way based on DNA concentration measured with a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA).
Array hybridization
For subject 1, pooled gDNAs were mixed with Human Cot-1 DNA (Invitrogen™, Carlsbad, CA, USA), Blocking Agent (Agilent Technologies) and Hybridization Buffer (Agilent Technologies). After denaturation and pre-annealing at 37°C for 30 minutes, the hybridization mixture was deposited on the microarray slide and hybridized in a hybridization chamber for 24 hours in a 65°C rotating oven. Then washing steps and acetonitrile rinsing were performed. For subject 2, test and reference gDNAs were hybridized to the IntegraChips™ BAC microarray (3172 clones spotted in quadruplicate with positions validated by end-sequencing) to enable a ratiometric analysis. Arrays were blocked by incubation in a solution containing Bovine Serum Albumin and SDS at 37 °C. The slide was pre-hybridized with formamide-based array hybridization buffer containing salmon sperm DNA at room temperature for 30 min. Hybridization mix was deposited on the array, covered with a Hybrislip (Grace Biolabs, Bend, OR, USA) and the array was placed in a Corning® hybridization chamber in a water bath at 42 °C for 3 days. Then the coverslip was removed and washing steps were performed.
Image and microarray data analysis
Microarrays were scanned using the Agilent scanner G2565BA.
For subject 1, images were analysed using Agilent Feature Extraction Software version 9.1 (CGH-v4_91 protocol) and data were imported into Agilent CGH analytics software version 3.4.27 for a graphical overview obtention and analysis. Identification of probes with a significant gain or loss was based on the log2 ratio plot deviation from 0 with cut-off values of 0.5 to 1 and −0.5 to −1 respectively.
For subject 2, fluorescent intensities were extracted by GenePix® Pro 6. The resulting files were analyzed with the program GenoCensus™ (IntegraGen SA, Evry, France). Spots with fluorescent signals indicating partial saturation (>50,000) or signals less than 2 times the mean of the local background were excluded. Cy5/Cy3 ratios of background–corrected intensities were normalized by a block Loess procedure and median ratios were obtained from the spot replicates of each clone. Clones with less than three morphologically acceptable replicates or with a coefficient of variation of the mean of the replicates that exceeded 0.2 were removed from the analysis.
Multiplex PCR/Liquid Chromatography (MP-LC)
Multiplex PCR/Liquid chromatography was performed for subject 1 to confirm array-CGH data and to test the parents for the microdeletion identified. We designed a duplex PCR using primers for an endogenous control gene, HMBS, and for the EPC2 gene exon 7, showing a loss in patient 1. Primers were designed using Primer Premier software (Premier Biosoft International, Palo Alto, CA, USA) with EPC2 exon 7 forward primer: 5′-GGGAGCGAAACAGGTGGA-3′; EPC2 exon 7 reverse primer: 5′-CCAGGCAGAAATGGCAGTGTA-3′. Duplex PCR was performed in a 25 μl reaction mixture, containing 10 % DMSO, 0.2 mM each dNTP, 2 mM MgCl2, 1.5 U DNA polymerase, 5–10 pmol of each primer and 100 ng genomic DNA, in a Master Cycler PCR System (Eppendorf, Hamburg, Germany). PCR cycling conditions were an initial step of denaturation at 94°C for 5 min, followed by 24 cycles of denaturation at 94°C for 10 sec, annealing at 50°C for 15 sec and extension at 72°C for 20 sec, and a final step of extension at 72°C for 5 min (11). The same duplex PCR mix was applied to normal controls, patient and parental gDNAs. PCR products were then separated by ion-pair reversed-phase high-performance liquid chromatography on a semi-automated 3500 Wave HS System (Transgenomic, Omaha, NE, USA) and quantified by fluorescent detection using a post-column intercalation dye, as described by Dehainault et al. (12).
FISH
FISH was performed to confirm copy number changes identified by microarray analyses. Metaphase spreads were obtained from phytohaemaglutinin-stimulated peripheral blood lymphocyte cultures.
Clones mapping to the unbalanced chromosome 2q were obtained for subject 1 from BACPAC Resource Center (Children’s Hospital Oakland Research Institute, Oakland, CA, USA) (http://bacpac.chori.org) (RP11-659J19) and for subject 2 from the RP11 library (Ensembl Genome Browser) (RP11-95O9, RP11-107E5, RP11-548K13, RP11-515K12, RP11-341F20, RP11-72H23). Bacterial culture was performed in LB medium supplemented with chloramphenicol (12.5 μg/mL) and DNA from the clone was isolated using a Nucleobond® PC 20 kit (Macherey Nagel, Düren, Germany). DNA was labelled with SpectrumGreen™-dUTP or SpectrumRed™-dUTP Vysis® (Abbott Molecular Inc., Downers Grove, IL, USA) by random priming using the Bioprime® Array CGH Genomic Labeling System (Invitrogen™, Carlsbad, CA, USA) or by nick translation using the Nick Translation Kit Vysis® (Abbott Molecular Inc., Downers Grove, IL, USA). Probes were hybridized to metaphase slides prepared from patients or parents lymphocytes. Slides were analysed with an epifluorescent microscope Olympus BX61 and images were captured using Isis® software (MetaSystems, Altlussheim, Germany). FISH signals were examined both on metaphase chromosomes and interphase nuclei.
RESULTS
Characterisation of the 2q23.1 deletion
Subject 1
Array-CGH experiments were performed using an oligonucleotide array with an average spatial resolution of approximately 35–75 kb. The analysis identified a chromosome 2 interstitial deletion of ~250 kb (Fig. 1A and 1B) at 2q23.1, confirmed by dye-swap. The proximal breakpoint mapped within the chromosome interval 148.836 Mb (last non-deleted probe) and 149.062 Mb (deletion start) from 2p telomere and the distal one within the chromosome interval 149.319 Mb (deletion end) and 149.362 Mb (first non-deleted probe) from 2p telomere. To confirm array data, multiplex PCR/Liquid Chromatography experiments were performed for the proband and the parents. The result was also further confirmed by FISH analyses using a clone mapping to the 2q23.1 region (RP11-659J19), hybridized on metaphase slides from the patient and the parents. For both validation experiments, the deletion was confirmed in the patient, while parents showed a normal result (Fig. 2 and 3) proving the de novo occurrence of the deletion.
Figure 1A-1B. Array-CGH data of patient 1.
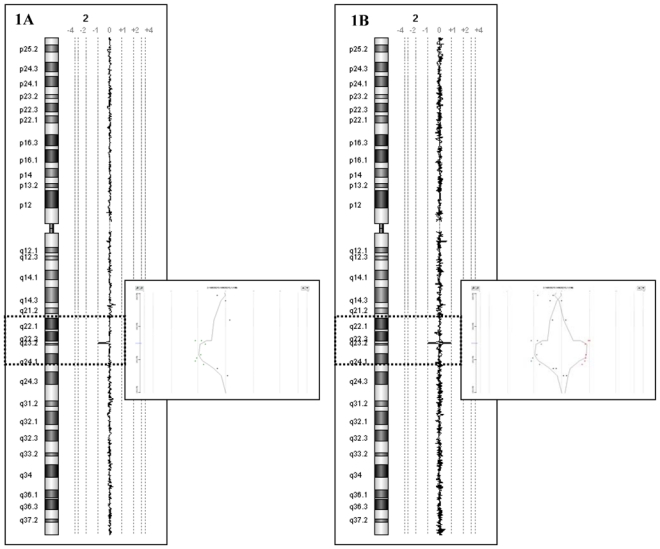
(1A) Genomic profile of chromosome 2. On the left, the chromosome 2 ideogram. On the right, the log2 ratio of chromosome 2 probes. Each dot represents a single probe spotted on the array. Dots with a value of zero represent equal fluorescence intensity ratio between sample and reference gDNAs. Copy number losses shift the ratio to the left or to the right according to the fluorescent labelling.
(1B) Dye reversal hybridization experiment shows a mirror image
Figure 2. FISH analysis of patient 1.
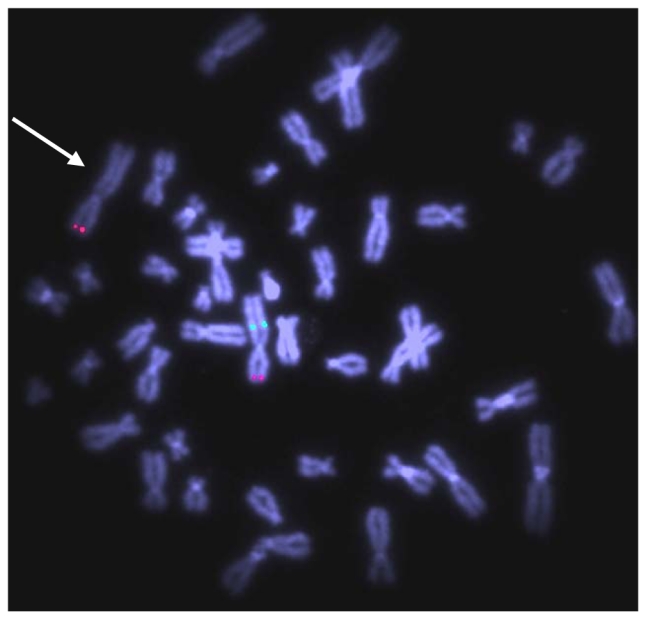
Fluorescent hybridisation with BAC clone RP11-659J19 (green) and N-Myc specific probe as a control (red) shows absence of green signal on the derivative chromosome 2 in subject 1, indicated with an arrow (20 metaphases and 25 interphase nuclei examinated).
Figure 3. Multiplex PCR/Liquid chromatography validation experiment, patient 1.
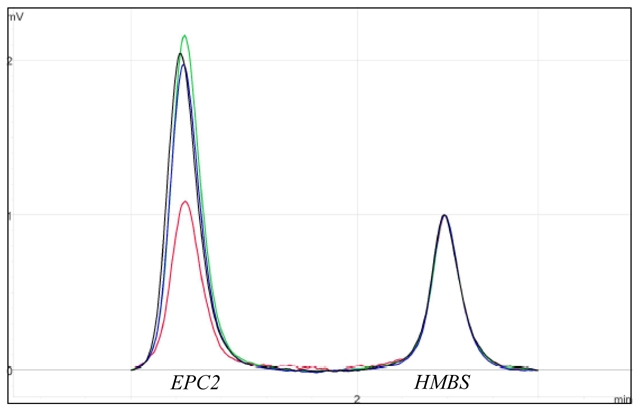
The electrophoregrams of the patient (in red) and of the parents (mother in green, father in blue) were superimposed to those of controls (in black) and normalized on HMBS2 internal control peak (148 bp) intensity. The presence of a deletion was indicated by a twofold reduction in the height of the exon 7 EPC2 amplicon peak (112 bp).
Subject 2
Array-CGH analysis was performed using BAC array 3172 clones, with an average resolution of 1 Mb. An interstitial deletion at 2q22.3-2q23.1 was revealed by decreased test/reference ratios for two adjacent clones: FE0BPADA13ZE01, FE0BPADA9ZH01 (RP11-72H23), indicating a deletion ranging from 2.4 to 5.4 Mb (Fig. 4). The deletion was confirmed by FISH analyses using clones mapping to the 2q22.3-2q23.1 region (RP11-548K13, RP11-515K12, RP11-341F20, RP11-72H23, RP11-659J19) hybridized on metaphase slides from the patient (Fig. 5). The proximal breakpoint resided between clones RP11-379G6 (non-deleted) and RP11-548K13 (deleted), and the distal breakpoint mapped between RP11-659J19 (deleted) and RP11-62P16 (non-deleted). The deletion was not present in parental gDNA samples, and is thus a de novo chromosomal aberration in the patients. Hybridizations with clones overlapping the SIP1 gene (RP11-107E5, RP11-95O9) were normal in subject 2.
Figure 4. Array-CGH data of patient 2.
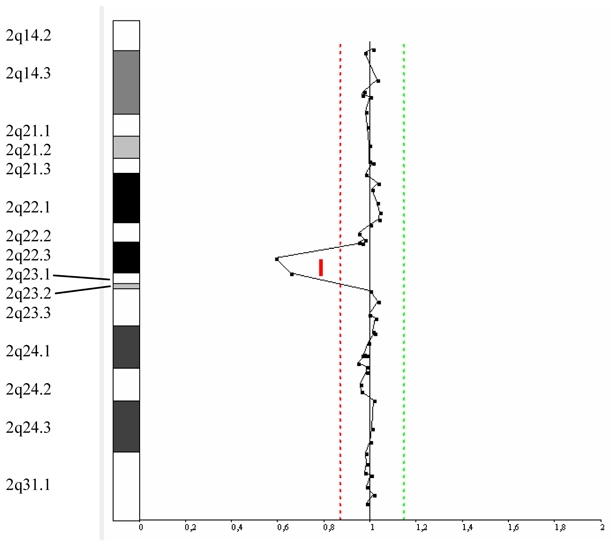
Genomic profile of chromosome 2 from 2q14.2 to 2q31.1. On the left, the chromosome 2 ideogram. On the right, the log2 ratio of chromosome 2 clones, with the thresholds for copy-number gain on the right and for copy-number loss on the left, showing deletion for 2 adjacent clones in 2q22.3q23.1.
Figure 5. FISH analysis of patient 2.
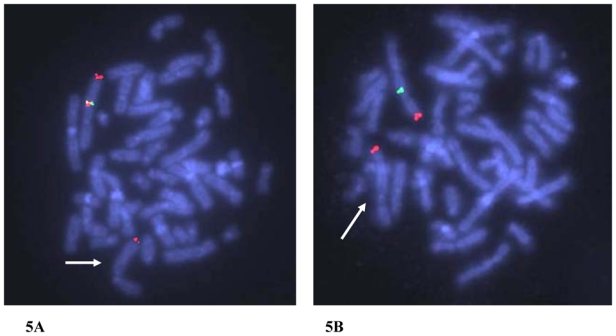
(5A) Fluorescent hybridisation with BAC clones RP11-341F20 (green) and RP11-548K13 (red) associated to a 2qter specific probe as a control (red) shows absence of signal on the derivative chromosome 2 in subject 2, indicated with an arrow (10 metaphases and 25 interphase nuclei examinated).
(5B) Fluorescent hybridisation with BAC clone RP11-659J19 (green) and 2qter specific probe as a control (red) shows absence of green signal on the derivative chromosome 2 in subject 2, indicated with an arrow (10 metaphases and 25 interphase nuclei examinated).
Clinical features of the patients with the 2q23.1 deletion
Subject 1 is a 10-year-old boy, third child of young non-consanguineous French parents (Fig. 6). He was referred to the genetics clinic for epilepsy, psychomotor retardation, gradual microcephaly and ataxia. Pregnancy and birth at 40 weeks were uneventful and birth parameters were normal: weight 3 815 g (50th percentile), height 50 cm (50th percentile), and head circumference 34 cm (50th percentile). A hip dysplasia was detected in neonatal period. Epileptic seizures were noted between the age of 3 and 9 months. Psychomotor retardation became severe: the child could not stay seated before the age of two, he could walk with ataxia at the age of four and he remained unable to speak. The secondary microcephaly became gradually more pronounced (−3 SD at the age of one year and half). At age 10 when last examined, the child could communicate via pictures, but not via language and he could understand some sentences. Behavioral problems included reduced capacity for attention and episodes of inappropriate laughter. Ataxia was still obvious but there were no longer any epileptic seizures. A moderate dysmorphia was noted (hypertelorism, flat nose, long lashes, large mouth). Other features included hypermetropia and astigmatism, scoliosis with hemivertebra of the eighth thoracic vertebra, valgus feet, severe constipation with occlusive episodes and episodes of fecal incontinence, micropenis and recurrent ear infections. At this time, the height of the child was − 2 SD, his weight −1.5 SD and his head circumference − 3 SD.
Figure 6. Dysmorphic features of the subject 1 (age 10).
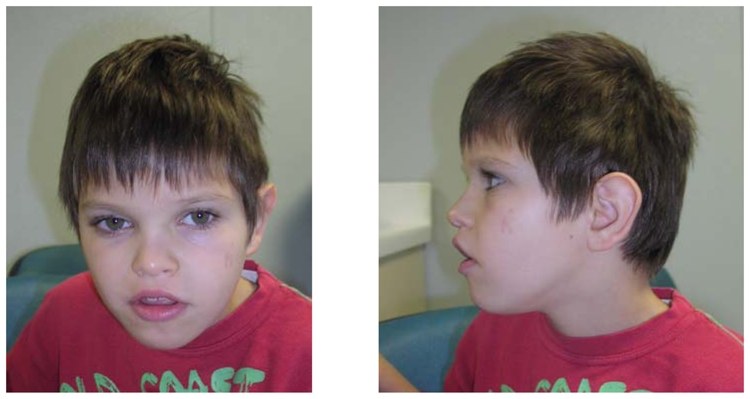
Hypertelorism, flat nose, long lashes, large mouth.
EEG was non-specific and with no Angelman syndrome pattern. Brain MRI revealed a small cerebellar vermis, with a large posterior fossa. Auditory Evoked Potentials (AEP) were normal. Standard and high resolution karyotyping, fragile X testing and analysis of ATRX, MECP2, STK9/CDK5 genes were normal. A subtelomeric rearrangement was excluded by MLPA and FISH. Angelman syndrome (study of the 15q11q12 chromosomal locus by FISH and study of the methylation pattern, complete sequencing of UBE3A) and CDG syndrome were ruled out. The search for a skew of X inactivation in the mother was negative.
Subject 2 is a 10 year-old boy, the only child of young non-consanguineous parents of French origin (Fig. 7). He was referred to the genetics clinic for mental retardation, autistic behaviour, ataxia and epilepsy. Pregnancy and birth at 39 weeks were unremarkable. Neonatal measurements were macrosomic, with a weight at 4 300g (>97th percentile), height at 52.5 cm (95th percentile) and head circumference at 36 cm (90th percentile). The neonatal period was marked by feeding difficulties, with swallowing troubles and gastro-esophageal reflux, and a baby being too calm, and who did not cry. Psychomotor retardation was progressively discovered, including retarded sitting position, unaided walking at 34 months with ataxia and severe language impairment. Epilepsy began at 3 years and was treated by valproate. Sleep patterns were perturbed, with multiple awakenings in the night. When last examined at the age of 10 years, he emitted some dissyllabic sounds but did not express any significant words. There were frequent smiles, unmotivated bursts of laughter and autistic behaviour including eye avoidance, hand flapping, bruxism and elevated anxiety in new surroundings. A tendency of stereotypies on the median line was present, amplified with stress. Auto and hetero-aggressivity were noted, with self-biting of hands and forearms. Pain sensitivity seemed decreased, with no reaction to bites and injuries. He had poor balance and ataxia, and reflexes were brisk. Facial dysmorphism included brachycephaly, low implantation of the hair on the neck, pronounced midface hypoplasia, short columella, large mouth, prognathism, macroglossia and small spaced teeth. Other features included frequent dribbling, secondarily hypodeveloped genitalia, short hands with thick striated skin and bilateral clinodactyly of the fifth, large and short thorax with pseudarthosis of the clavicle, constipation. His height was at the median (M), his weight at −1 SD and his head circumference at the median (M).
Figure 7. Dysmorphic features of subject 2 (age 10).
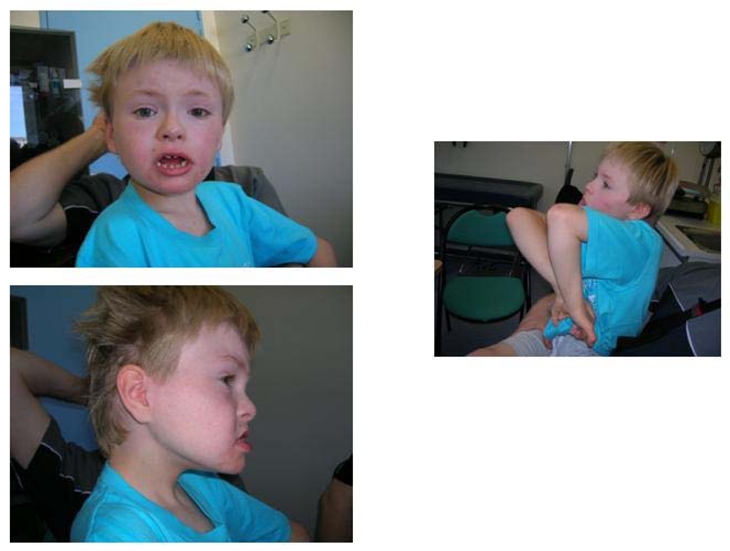
Midface hypoplasia, large mouth, prognathism, small spaced teeth, everted lower lip and spontaneous attitude with midline stereotypias.
EEG was normal, without a classical Angelman syndrome pattern. Brain MRI showed large pericerebral spaces, and a relative microcephaly with hypoplasia of frontal lobes. Visual Evoked Potentials (VEP), AEP and electroretinogram were normal. Hypoconduction was noted in the Sensitive Evoked Potential in the median, and normal on cubital and sural, performed in the wrists because of suspected hyposensitivity to pain. X-rays showed a poorly mineralized skeleton and brachymesophalangy. Standard and high resolution karyotyping, fragile X testing and ARX gene mutations screening were normal. The presence of subtelomeric rearrangements was excluded by MLPA. Angelman syndrome (study of the 15q11q12 chromosomal locus by FISH and study of the methylation pattern, complete sequencing of UBE3A), CDG syndrome and Smith-Magenis syndrome were ruled out. The maternal X-methylation pattern was normal.
DISCUSSION
We report here two unrelated patients sharing a strikingly similar association of clinical features (mental retardation, absent speech, stereotypies, happy disposition, ataxia, microcephaly, seizures) first strongly suggesting Angelman syndrome, and with a de novo interstitial deletion including 2q23.1 identified by array-CGH (Fig. 8). This 2q23.1 deletion is encompassed by three microdeletions previously reported in this chromosomal region: a 2-Mb genomic interval on 2q22.3q23.3 (13, 14), a 920-kb interval on 2q23.1q23.2 (15) and a 1.1-Mb interval on 2q22.3q23.1, corresponding to a reciprocal translocation breakpoint (16). Array-CGH also underscored at least three larger deletions (> 7-Mb) in 2q22.3q24.1, and so including the 2q23.1 region (17, patient DECIPHER 1079, patient DECIPHER 1818). These deletions never include the SIP1 gene, associated with Mowat-Wilson syndrome. Despite the heterogeneity in the size of the imbalances, common clinical findings seem to emerge including severe mental retardation, behavioural disabilities, post natal growth retardation, microcephaly and epilepsy (Table 1). The latter is present in nearly 100% of the reported cases. The overlap in clinical features between the five microdeleted patients may point to a novel microdeletion syndrome with a “pseudo-Angelman” phenotype as there are clinical features shared with Angelman syndrome, associated with frank dysmorphic features, unusual in Angelman syndrome, mainly midface hypoplasia, short upturned philtrum, and carp-shaped mouth. Neurological features seem quite consistent although non specific: severe mental retardation, delayed motor milestones, microcephaly (absolute or relative), speech impairment, behavioural disorders (happy demeanour, oral-motor behaviours) and seizures. Moreover, it is important to emphasize that EEGs did not show classical Angelman syndrome patterns. The 2q23.1 microdeletion can then be considered as a differential diagnosis of Angelman syndrome, with epilepsy as the most common finding, associated with marked dysmorphic features, unusual in Angelman syndrome. Cytogenetically visible deletions involving the 2q22q24 region have already been reported in patients sharing some of these clinical findings (18). Seizures are frequent but inconsistent in those circumstances, as opposed to seizures which are a characteristic finding in the submicroscopic cases. The pattern of clinical features has sometimes been described quite close to our “pseudo-Angelman” patients (19) but a more malformative VATER like phenotype has also been reported in a girl with a 2q22q24.2 deletion (20). However, the boundaries of the previously described 2q22q24 deletions were determined only by banding techniques and it is difficult to conclude if these deletions correspond to the highly resolved boundaries determined by array-CGH. Our assumption that deletions in the 2q23.1 region are involved in the onset of the clinical pattern described above is not supported by the existence of a copy number variant (CNV), overlapping the 2q23.1q23.2 region, reported in the Database of Genomic Variants (http://projects.tcag.ca/variation/). This CNV consisted of copy number gain and was reported once in a study of 506 healthy individuals from Northern Germany and 270 HapMap individuals. However, data from this study have not been validated (21). In addition, CNVs reported only in a single individual may correspond to a rare variant or to a false positive (22). In the course of array-CGH studies of patients with mental retardation, some chromosomal regions appeared recurrently involved, including the 2q22.3q24.2 region (2, 13, 15, 17) and a common deletion/duplication mechanism has been suggested for the 2q22.3q23.2 region by Koolen et al. (14). Analysis of this chromosomal region using the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu) and the Human Genome Segmental Duplication Database (http://projects.tcag.ca/humandup/) identified no segmental duplications. As the microdeletions reported in 2q22.3q23.2 do not share common breakpoints (Table 2) and as segmental duplications are not present in the proximal and distal breakpoint intervals, we postulate that NAHR is not likely to be the responsible mechanism, as recurrent rearrangements are typically mediated by highly homologous low-copy repeats (LCRs) flanking a chromosomal region, leading to deletion, duplication or inversion of the intervening sequence (23).
Figure 8. Map of 2q22.3-2q23.1 region.
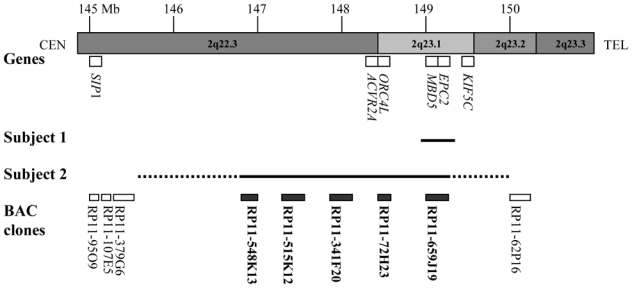
(Genome assembly, May 2004) indicating Megabase position (Mb) on chromosome 2q22.3q23.3 region, genes of interest, deleted region in subject 1 (above) and in subject 2 (under) and location of used clones for validation experiments. The dotted line represent breakpoints interval for subject 2. The deleted clones for subject 1 and/or 2 are shown in bold. CEN = centromere, TEL = telomere.
Table 1.
Clinical features of patients with microdeletion encompassing the 2q23.1 region
| Reference | Classical Angelman Syndrome | Patient 1 This study |
Patient 2 This study |
Patient 3 Vissers et al., 2003 Koolen et al., 2004 |
Patient 4 De Vries et al., 2005 |
Patient 5 De Gregori et al., 2007 |
|---|---|---|---|---|---|---|
| Sex | M | M | F | F | M | |
| Psychomotor retardation | Severe | Severe | Severe | Severe | Severe | + |
| Language impairment | Less than 6 words | Severe | Severe | Moderate | NR | + |
| Behavorial disabilities | Inappropriate laughter, happy disposition | Inappropriate laughter | Inappropriate laughter, agressivity, autistic comportment | NR | Picking of the eyes, hyperpnea, putting of full hands into the mouth | Bruxism |
| Sleeping difficulties | + | − | + | NR | NR | NR |
| Post natal growth retardation | + | + | + (weight) | + (height) | + | NR |
| Microcephaly | + | + | + (relative) | + | + | NR |
| Seizures | + | + | + | + | + | + |
| Ataxia | + | + | + | NR | NR | NR |
| Feeding difficulties | + | − | + | + | NR | NR |
| Hypotonia | + | − | − | + | NR | NR |
| Facial dysmorphy | Mild | Coarse facies | Mild | |||
| ▪ Brachycephaly | + | − | + | − | − | |
| ▪ Large mouth | + | + | + | − | + | |
| ▪ Broad chin | + | − | − | + | + | |
| ▪ Prognathism | + | − | + | − | − | |
| ▪ Teeth anomalies | + | − | + | + | − | |
| ▪ Macroglossia | + | − | + | − | − | |
| ▪ Midface hypoplasia | + | − | + | − | − | |
| ▪ Other | Hypertelorism Flat nose Long lashes |
Low implantation of the hair on the neck Short columella |
Up-slanting palpebral fissures, hypotelorism Small upturned nose Malformed ears Small-carp shaped mouth Narrow flat palate |
High forehead Full round face Light blue irises Small ears, large lobules Short philtrum Full lips |
||
| Hand/foot abnormalities | − | + | + | + | + | NR |
| ▪ Clinodactyly fifth finger | − | + | + | + | ||
| ▪ Syndactyly II-III | − | − | + | − | ||
| ▪ Sandal gap | − | − | + | − | ||
| ▪ Valgus feet | + | − | − | − | ||
| Other congenital features | Micropenis Constipation Scoliosis, hemivertebra Hypermetropia Astigmatism |
Hypogenitalism Constipation Pseudoarthrosis of the clavicle |
Hirsutism (lower extremities) | NR | ||
presence, −: absence, NR: Not Reported
Table 2. Molecular features of patients with microdeletion encompassing the 2q23.1 region.
Map positions refer to the Genome Assembly May 2004. Proximal breakpoints are localised in the region between the end of the last non-deleted BAC or probe before the deletion and the start of the first deleted BAC (start clone) or probe. Distal breakpoints are localised in the region between the end of the last deleted BAC (end clone) or probe and the start of the first non-deleted BAC or probe after the deletion. For BAC-arrays, the minimal size of copy number changes is calculated from the start of the first clone to the end of the last clone with decreased ratios.
| Patient 1 This study |
Patient 2 This study |
Patient 3 Vissers et al., 2003 Koolen et al., 2004 |
Patient 4 De Vries et al., 2005 |
Patient 5 De Gregori et al., 2007 |
|
|---|---|---|---|---|---|
| Deleted region | 2q23.1 | 2q22.3-q23.1 | 2q22.3-q23.2 | 2q23.1-q23.2 | 2q22.3-q23.1 |
| Array method | Oligo-array, Agilent 4×44K | BAC-array, 3172 clones | BAC array, 3569 clones | BAC-array, 32 447 clones | Oligo-array, Agilent 244 K |
| Breakpoints of imbalance | |||||
| -region of proximal breakpoint | RP11-379G6/RP11-548K13 | RP11-67J2/RP11-89L3 | |||
| 148.836–149.062 | 145.268–146.891 | 145.410–146.734 | 149.17 (start clone) | 148.14–148.18 | |
| -region of distal breakpoint | RP11-659J19/RP11-62P16 | RP11-72H23/RP11-79A11 | |||
| 149.319–149.362 | 149.268–150.720 | 148.694–151.147 | 150.09 (end clone) | 149.32–149.33 | |
| Deletion minimal size (kb) | ~250 | ~2 400 | ~2 000 | ~920 | ~1 100 |
| Candidate genes | ACVR2A | ACVR2A | ACVR2A | ||
| ORC4L | ORC4L | ORC4L | |||
| MBD5 | MBD5 | MBD5 | MBD5 | MBD5 | |
| EPC2 | EPC2 | EPC2 | EPC2 | EPC2 | |
| KIF5C | KIF5C | ||||
The de novo deletion may create haploinsufficiency of genes within the overlapping deletion region. Grosso et al. (24) show a strong association between chromosome 2 long arm aberrations and epilepsy, suggesting that many genes which are present in the 2q22.3-2q37 region have significant roles in epileptogenesis. The smallest region of overlap for the microdeletions in 2q22.3q23.3 is about 250 kb. It includes two genes, MBD5 and EPC2, but does not involve two other genes, KIF5C and AVCR2A, that were candidates in the first description of the microdeletion by Koolen et al. (14). The KIF5C gene encodes a neuronal kinesin; this protein is a member of the kinesin superfamily and is enriched in neurons, with a pan-neuronal distribution in mice. Disruption of KIF5C in mouse leads to smaller brain size and loss of peripheral neurons (25). ACVR2A is expressed in human trophoblast cells, ovary and brain. Knockout mice develop skeletal and facial abnormalities, suppression of the follicle stimulating hormone and reproductive failure (26, 27). Even if some clinical features could have been explained by these candidates, our study limits the focus to EPC2 and MBD5. EPC2 (Enhancer of Polycomb C-terminus homolog 2) is a member of the polycomb protein family, involved in heterochromatin formation (28). These proteins were reported to belong to an acetyltransferase complex and to have both transcription activating and repressing functions (28). MBD5 is particularly interesting as it is a member of the methyl CpG-binding-domain protein family (MBD), important in regulation of gene expression and is expressed in the brain and also in many other adult tissues (29, 30). The protein MBD5 was first identified because of its MBD sequence, shared notably by MECP2, implicated in Rett syndrome and it also harbors a PWWP motif, involved in chromatin modification. Recently, a 200 kb deletion affecting the first 7–10 exons of the MBD5 gene was described in a patient with severe mental retardation, slight retarded motor development, seizures and sandal gap, thus supporting the hypothesis that MBD5 is a strong candidate gene involved in the phenotype associated with the 2q23.1 microdeletion. However, for the moment, sequence analysis of MBD5 in 415 children with mental retardation did not reveal neither deletions nor nonsense mutations (31).
The next step to support the hypothesis that haploinsufficiency of the MBD5 gene should be a novel cause of mental retardation will be a large-scale screening of individuals with mental retardation and a “pseudo-Angelman” phenotype, to identify similar microdeletions and point mutations. Identification of additional cases of microdeletion in the 2q23.1 region will allow more accurate genotype-phenotype correlations. Nevertheless, the non-specificity of the Angelman-like phenotype may involve abnormalities in other chromosomal regions that should be tested by a targeted method like Multiplex Ligation-Dependant Probe Amplification (http://www.mrc-holland.com), or other genes like the recently described SLC9A6 gene (32).
Acknowledgments
We thank Pr Jean Mosser, who manages the transcriptomic platform of the OUEST Genopole®-IFR140 (Rennes).
We thank all our colleagues who referred cases, and the patients and their families for participating in this study. We are grateful to Hubert Journel (Génétique Médicale, CHBA, Vannes) who organizes the Berder genetics seminar which permitted us bringing together these two cases with “pseudo-Angelman” phenotype and 2q23.1 microdeletion.
This research was supported by grants from the Centre Hospitalier et Universitaire de Rennes (concours post-internat), PHRC inter-régional du Grand Ouest and from the Centre Hospitalier et Universitaire Jean Verdier, Paris.
We are grateful to Beverley Osborne (UMR 6061, Rennes, France) and to Peter Brooks (Integragen®) for revising the English text.
Footnotes
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd to permit this article (if accepted) to be published in JMG and any other BMJPGL products and sublicences such use and exploit all subsidiary rights, as set out in our licence (http://jmg.bmj.com/ifora/licence.pdf).
References
- 1.Menten B, Maas N, Thienpont B, Buysse K, Vandesompele J, Melotte C, De Ravel T, Van Vooren S, Balikova I, Backx L, Janssens S, De Paepe A, De Moor B, Moreau Y, Marynen P, Fryns JP, Mortier G, Devriendt K, Speleman F, Vermeesch JR. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan YS, Jayakar P, Zhu H, Barbouth D, Sacharow S, Morales A, Carver V, Benke P, Mundy P, Elsas LJ. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridization. Hum Mutat. 2007;28:1124–1132. doi: 10.1002/humu.20581. [DOI] [PubMed] [Google Scholar]
- 3.Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, Shaikh T. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- 4.Rajcan-Separovic E, Harvard C, Liu X, McGillivray B, Hall JG, Qiao Y, Hurlburt J, Hildebrand J, Mickelson EC, Holden JJ, Lewis ME. Clinical and molecular cytogenetic characterisation of a newly recognised microdeletion syndrome involving 2p15–16. 1. J Med Genet. 2007;44:269–276. doi: 10.1136/jmg.2006.045013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redon R, Baujat G, Sanlaville D, Le Merrer M, Vekemans M, Munnich A, Carter NP, Cormier-Daire V, Colleaux L. Interstitial 9q22. 3 microdeletion: clinical and molecular characterisation of a newly recognised overgrowth syndrome. Eur J Hum Genet. 2006;14:759–767. doi: 10.1038/sj.ejhg.5201613. [DOI] [PubMed] [Google Scholar]
- 6.Ballif BC, Hornor SA, Jenkins E, Madan-Khetarpal S, Surti U, Jackson KE, Asamoah A, Brock PL, Gowans GC, Conway RL, Graham JM, Jr, Medne L, Zackai EH, Shaikh TH, Geoghegan J, Selzer RR, Eis PS, Bejjani BA, Shaffer LG. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2–p12. 2. Nat Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 7.Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, Porter K, Prigmore E, Krepischi-Santos AC, Varela MC, Koiffmann CP, Lees AJ, Rosenberg C, Firth HV, de Silva R, Carter NP. Microdeletion encompassing MAPT at chromosome 17q21. 3 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 8.Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, de Vries BB. A new chromosome 17q21. 31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 9.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 10.de Vries BB, White SM, Knight SJ, Regan R, Homfray T, Young ID, Super M, McKeown C, Splitt M, Quarrell OW, Trainer AH, Niermeijer MF, Malcolm S, Flint J, Hurst JA, Winter RM. Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet. 2001;38:145–150. doi: 10.1136/jmg.38.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bendavid C, Dubourg C, Pasquier L, Gicquel I, Le Gallou S, Mottier S, Durou MR, Henry C, Odent S, David V. MLPA screening reveals novel subtelomeric rearrangements in holoprosencephaly. Hum Mutat. 2007;28:1189–1197. doi: 10.1002/humu.20594. [DOI] [PubMed] [Google Scholar]
- 12.Dehainault C, Lauge A, Caux-Moncoutier V, Pages-Berhouet S, Doz F, Desjardins L, Couturier J, Gauthier-Villars M, Stoppa-Lyonnet D, Houdayer C. Multiplex PCR/liquid chromatography assay for detection of gene rearrangements: application to RB1 gene. Nucleic Acids Res. 2004;32:e139. doi: 10.1093/nar/gnh137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Chy CO, Straatman H, Van Der Vliet W, Huys EH, Van Rijk A, Smeets D, Van Ravenswaaij-Arts CM, Knoers NY, Van Der Burgt I, De Jong PJ, Brunner HG, Van Kessel AG, Schoenmakers EF, Veltman JA. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koolen DA, Vissers LE, Nillesen W, Smeets D, Van Ravenswaaij CM, Sistermans EA, Veltman JA, de Vries BB. A novel microdeletion, del(2)(q22.3q23. 3) in a mentally retarded patient, detected by array-based comparative genomic hybridization. Clin Genet. 2004;65:429–432. doi: 10.1111/j.0009-9163.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 15.de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, Smeets D, Sistermans EA, Feuth T, Van Ravenswaaij-arts CM, Van Kessel AG, Schoenmakers EF, Brunner HG, Veltman JA. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Gregori M, Ciccone R, Magini P, Pramparo T, Gimelli S, Messa J, Novara F, Vetro A, Rossi E, Maraschio P, Bonaglia MC, Anichini C, Ferrero GB, Silengo M, Fazzi E, Zatterale A, Fischetto R, Previderé C, Belli S, Turci A, Calabrese G, Bernardi F, Meneghelli E, Riegel M, Rocchi M, Guerneri S, Lalatta F, Zelante L, Romano C, Fichera M, Mattina T, Arrigo G, Zollino M, Giglio S, Lonardo F, Bonfante A, Ferlini A, Cifuentes F, Van Esch H, Backx L, Schinzel A, Vermeesch JR, Zuffardi O. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: a study of 59 patients. J Med Genet. 2007;44:750–762. doi: 10.1136/jmg.2007.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid BM, Nordenskjöld M. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J Med Genet. 2005;42:699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maas SM, Hoovers JM, van Seggelen ME, Menzel DM, Hennekam RC. Interstitial deletion of the long arm of chromosome 2: a clinically recognizable microdeletion syndrome? Clin Dysmorphol. 2000;9:47–53. doi: 10.1097/00019605-200009010-00010. [DOI] [PubMed] [Google Scholar]
- 19.McMilin KD, Reiss JA, Brown MG, Black MH, Buckmaster DA, Durum CT, Gunter KA, Lawce HJ, Berry TL, Lamb OA, Olson CL, Weeks FF, Yoshitomi MJ, Jacky PB, Olson SB, Magenis RE. Clinical outcomes of four patients with microdeletion in the long arm of chromosome 2. Am J Med Genet. 1998;78:36–43. [PubMed] [Google Scholar]
- 20.Woods CG, Koehn DC, McFadden DE, Evans JA, van Allen MI. A case report of a child with an interstitial deletion of chromosome 2 (q22;q24.2) and the VATER association phenotype. Abstract. D. W. Proc Greenwood Genet Ctr. 1993;12:119–120. [Google Scholar]
- 21.Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Hum Mol Genet. 2007;16(Spec No 2):R168–73. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 22.Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- 23.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 24.Grosso S, Pucci L, Curatolo P, Coppola G, Bartalini G, Di Bartolo R, Scarinci R, Renieri A, Balestri P. Epilepsy and electroencephalographic anomalies in chromosome 2 aberrations A review. Epilepsy Res. 2008;79:63–70. doi: 10.1016/j.eplepsyres.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng C, Huang TH, Jeung EB, Donaldson CJ, Vale WW, Leung PC. Expression of the type II activin receptor gene in the human placenta. Endocrinology. 1993;133:3046–3049. doi: 10.1210/endo.133.6.8243335. [DOI] [PubMed] [Google Scholar]
- 27.Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360. doi: 10.1038/374356a0. [DOI] [PubMed] [Google Scholar]
- 28.Doyon Y, Selleck W, Lane WS, Tan S, Côté J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagase T, Kikuno R, Ishikawa K, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7:143–150. doi: 10.1093/dnares/7.2.143. [DOI] [PubMed] [Google Scholar]
- 30.Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics. 2003;4:1. doi: 10.1186/1471-2164-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagenstaller J, Spranger S, Lorenz-Depiereux B, Kazmierczak B, Nathrath M, Wahl D, Heye B, Glaser D, Liebscher V, Meitinger T, Strom TM. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, Sjøholm H, Server A, Samuelsson L, Christianson A, Tarpey P, Whibley A, Stratton MR, Futreal PA, Teague J, Edkins S, Gecz J, Turner G, Raymond FL, Schwartz C, Stevenson RE, Undlien DE, Strømme P. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet. 2008;82:1003–10. doi: 10.1016/j.ajhg.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]