

Abstract
The spike output of neural pathways can be regulated by modulating output neuron excitability and/or their synaptic inputs. Dopaminergic interneurons synapse onto cells that route signals to mammalian retinal ganglion cells, but it is unknown whether dopamine can activate receptors in these ganglion cells and, if it does, how this affects their excitability. Here, we show D1a receptor-like immunoreactivity in ganglion cells identified in adult rats by retrogradely transported dextran, and that dopamine, D1-type receptor agonists, and cAMP analogs inhibit spiking in ganglion cells dissociated from adult rats. These ligands curtailed repetitive spiking during constant current injections and reduced the number and rate of rise of spikes elicited by fluctuating current injections without significantly altering the timing of the remaining spikes. Consistent with mediation by D1-type receptors, SCH-23390 [R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine] reversed the effects of dopamine on spikes. Contrary to a recent report, spike inhibition by dopamine was not precluded by blocking I h. Consistent with the reduced rate of spike rise, dopamine reduced voltage-gated Na+ current (I Na) amplitude, and tetrodotoxin, at doses that reduced I Na as moderately as dopamine, also inhibited spiking. These results provide the first direct evidence that D1-type dopamine receptor activation can alter mammalian retinal ganglion cell excitability and demonstrate that dopamine can modulate spikes in these cells by a mechanism different from the presynaptic and postsynaptic means proposed by previous studies. To our knowledge, our results also provide the first evidence that dopamine receptor activation can reduce excitability without altering the temporal precision of spike firing.
Introduction
Dopaminergic neurons regulate the spike output of mammalian central pathways during events as diverse as working memory, goal-directed behavior, long-term potentiation, nocioception, auditory cortical reorganization, and light adaptation (Häggendal and Malmfors, 1965; Fleetwood-Walker et al., 1988; Frey et al., 1993; Williams and Goldman-Rakic, 1995; Bao et al., 2001; Bissiere et al., 2003). Anatomical and electrophysiological observations have shown that this regulation is achieved in different ways. Projection neurons synapse onto various cells in cerebral cortex, hippocampus, striatum, and spinal cord (Goldman-Rakic et al., 1989; Doyle and Maxwell, 1993; Sesack et al., 1994; Carr et al., 1999), and, in these structures, dopamine modulates neurotransmitter release, neurotransmitter responses, and/or excitability (Pirot et al., 1992; Schiffmann et al., 1995; Bamford et al., 2004). In contrast, presynaptic dopamine receptors alone modulate signal transmission at afferent fiber terminals in olfactory bulb (Hsia et al., 1999; Ennis et al., 2001) and at spinal cord inputs to nucleus tractus solitarius (Kline et al., 2002). Likewise, dopaminergic amacrine and interplexiform cells might regulate retinal ganglion cell spiking by effects upstream to these output neurons (e.g., by shifting the balance of excitatory and inhibitory inputs) (Thier and Alder, 1984). Observations suggesting that dopamine affects retinal ganglion cells indirectly include synapses of tyrosine hydroxylase-immunopositive interneurons onto bipolar and amacrine cells but not ganglion cells (Pourcho, 1982; Hokoç and Mariani, 1987; Gustincich et al., 1997), insensitivity of isolated rat retinal ganglion cells to dopamine (Guenther et al., 1994), formation of a neuromodulator by dopamine receptor activation in glial cells (Newman, 2003), and absence of dopamine receptor ligand and antibody binding, in some studies, to the ganglion cell layer (Ariano et al., 1991; Behrens and Wagner, 1995).
A few studies have found that anti-D1-type dopamine receptor antibodies bind to somata in the ganglion cell layer of rat retina (Bjelke et al., 1996; Nguyen-Legros et al., 1997), and a recent study of spikes recorded from ganglion cell layer somata in rat retinal slices attributed effects of bath-applied dopamine to ganglion cell dopamine receptors (Chen and Yang, 2007). However, these studies did not show that the somata examined were ganglion cells rather than displaced amacrine cells (Perry, 1981), did not compare effects of dopamine on ion currents commonly targeted in central neurons (Cantrell and Catterall, 2001; Poolos et al., 2002), and provided little information about which spike properties are dopamine sensitive (Tang et al., 1997; Gulledge and Jaffe, 1998). The present study therefore addresses three questions: Are D1 receptors present in cells identified anatomically as ganglion cells in rat retina? If so, what spike properties are altered by activating these receptors, and how do the changes in spikes compare with effects on voltage-gated Na+ current and on I h? Our results indicate that dopamine can regulate spiking in mammalian retinal ganglion cells by feedforward inhibition and that this can reduce spike number without altering the timing of the remaining spikes.
Materials and Methods
Animals.
Adult rat retinas were used for the experiments reported here because a wide variety of studies have indicated that dopamine is used as a neurotransmitter in this tissue (Brown and Makman, 1972; Voigt and Wässle, 1987; Bjelke et al., 1996; Puopolo et al., 2001; Partida et al., 2004; Witkovsky et al., 2005, 2008; Chen and Yang, 2007; Mills et al., 2007). Long–Evans rats [female; postnatal day 60 (P60) to P120; 150–250 g] were obtained from a commercial supplier (Harlan Bioproducts) and housed in standard cages at room temperature (∼23°C) on a 12 h light/dark cycle. Before enucleation, rats were killed by a lethal dose of sodium pentobarbital (75 mg/kg, i.p.). All animal care and experimental protocols were approved by the Animal Use and Care Administrative Advisory Committee of the University of California, Davis.
Protein isolation.
Protein was extracted from rat retinas as described previously (Partida et al., 2004). Freshly isolated retinas were frozen individually in Eppendorf tubes by dropping into liquid nitrogen. Twenty of these retinas were then transferred to a tissue grinder and homogenized in ice-cold homogenization buffer containing 0.3 m sucrose, 50 mm HEPES, 1 mm EDTA, and a protease and phosphatase inhibitor mixture [50 mm NaF, 50 mm β-glycerol phosphate, pH 8.0, 0.2 mm sodium orthovanadate, 0.1 mm l -1-chloro-3-(4-tosylamido)-4-phenyl-2-butanone (TPCK), 10 μg/ml leupeptin and pepstatin A, 1 μg/ml aprotinin, 0.1 mg/ml benzamidine, and 8 μg/ml calpain I and II inhibitors] (see below for the source of all chemicals used in this study). After centrifuging the homogenate at 13,000 × g for 15 min at 4°C, the supernatant was collected and the pellet resuspended in homogenization buffer; these two steps were repeated twice. The final pellet was discarded and the supernatant from all three spins was centrifuged at 45,000 × g for 1 h at 4°C in an ultracentrifuge. The membrane-enriched pellet from this final spin was resuspended in 500 μl of homogenization buffer and assayed by the Bradford method for total protein. This suspension was loaded at 50–100 μg of protein per lane onto a 4–12% Bis-Tris polyacrylamide gradient gel (NuPage; Invitrogen) and run with 3-(N-morpholino)-propanesulfonic acid (MOPS) running buffer. Protein standards (Magic Mark and See Blue) were run in lanes adjacent to the samples.
Western blot.
After electrophoretic separation, proteins were transferred from the polyacrylamide gradient gel to a nitrocellulose membrane (0.2 μm pore diameter). The membrane was blocked in TBST (0.1% Tween 20 in Tris-buffered saline) containing 3% dry nonfat milk for 1 h at room temperature, incubated overnight at 4°C with anti-D1a dopamine receptor antibody (see below for a list of all antibodies used in this study), rinsed in TBST, and then incubated in secondary antibody conjugated to HRP for 1 h at room temperature. After rinsing again in TBST, protein bands were visualized using ECL detection with SuperSignal West Pico Chemiluminescent Substrate. As negative control experiments for staining by the anti-D1a dopamine receptor antibody, pairs of membrane lanes from the same electrophoretic separations were processed identically and simultaneously, except for the primary antibody. One lane of each pair was incubated in anti-D1a dopamine receptor antibody, whereas the other was incubated in anti-D1a dopamine receptor antibody that had been mixed overnight at 4°C with a fourfold higher concentration of the peptide immunogen (see below).
Retrograde labeling.
Retinal ganglion cells were filled with fluorophore-coupled dextran by retrograde transport as described previously (Oi et al., 2008). Rats were anesthetized with intraperitoneally injected sodium pentobarbital (25 mg/kg). The conjunctiva was cut and the globe retracted to expose the optic nerve. After nicking the optic nerve with scissors, ∼2 mg of fixable dextran was deposited against the retinal end of the cut optic nerve fibers. In most flat-mount experiments, the dextran was 3 kDa and coupled to fluorescein; 10 kDa dextran coupled to Alexa Fluor 488 was used for vertical sections. Each rat was allowed to survive under anesthesia for up to 8 h to allow dextran to reach the retina. During this time, the animals were placed on a warming pad, turned every one-half hour, and supplied with oxygen via a funnel positioned over the nose and mouth. These rats were then killed with an overdose of sodium pentobarbital (150 mg/kg), and the retinas isolated and processed for imaging of retrogradely transported dextran, and immunohistochemistry, as described below.
Immunohistochemistry.
Transretinal (“vertical”) sections and retinal “flat mounts” were prepared and processed as described previously (Partida et al., 2004). To form vertical sections, eyes were slit along the ora serrata, fixed by immersion in 4% paraformaldehyde (dissolved in PBS, pH 7.4) for 1 h at room temperature, rinsed in PBS, equilibrated overnight at 4°C in PBS supplemented with 30% sucrose, and hemisected. After dissecting away the anterior portion, lens, and vitreous, the retina (in the remaining eyecup) was embedded in a small volume of OCT compound, and frozen in hexane, which was cooled to just above freezing in a liquid nitrogen bath. Sections were cut at a thickness of 14 μm on a cryostat, collected onto glass slides, stored at 4°C until use, rinsed with PBS, covered with blocking solution [5% normal goat serum (NGS) and 0.5% Triton X-100 in PBS] for 1 h at room temperature, and then incubated in primary antibody overnight at 4°C. After several washes in PBS, the sections were incubated with dye-conjugated secondary antibody for 1 h at room temperature. After several more washes in PBS, the sections were mounted in FluorSave reagent, overlaid with a coverslip, and imaged (see below).
As control experiments for staining by the anti-D1a dopamine receptor antibody, sets of sections from the same retina were processed identically and simultaneously, except that some of these sections were incubated in polyclonal anti-D1a dopamine receptor antibody, whereas others were incubated in anti-D1a dopamine receptor antibody that had been mixed with a twofold higher concentration of the peptide immunogen overnight at 4°C.
To form flat mounts, freshly dissected eyecups were incubated overnight in 4% paraformaldehyde at 4°C, and rinsed in PBS. The retinas were isolated from these eyecups and placed vitreous side up on a nitrocellulose filter disc. To help the retinas flatten, radial incisions were made from the retinal periphery toward the optic nerve head, the filter and retina were placed on top of the filter support of a Swinnex adapter (Millipore), and vacuum was applied via a syringe attached to the opposite side while PBS was dropped over the retina to prevent drying. Vitreous was removed with strips of filter paper, and the inner retina was then sliced from the outer retina with a scalpel. By reducing the thickness of our flat mounts, this step facilitated flattening and probably reduced the time needed for antibodies to reach the ganglion cells. The tangential sections containing the ganglion cell layer were stored in blocking solution (0.1% Triton X-100 and either 5% NGS or 5% normal donkey serum, in PBS) for 4 h at room temperature, incubated in primary antibodies for 24–48 h at 4°C, rinsed for a total of 1 h in PBS (replacing the PBS every 5 min) on a rocker, and incubated in secondary antibodies for 2 h at room temperature or 18 h at 4°C. After a final rinse, the retinas were laid ganglion cell-side-up on Superfrost/Plus slides, then covered with FluorSave reagent and a glass coverslip (either no. 1 or 1.5). To stain with more than one antibody, sections were incubated in mixtures of primary antibodies, followed by mixtures of secondary antibodies, with the same intervening and subsequent steps as for single-antibody incubations.
Antibodies.
We used the following primary antibodies: anti-D1a dopamine receptor [rabbit polyclonal AB1765P (Millipore Bioscience Research Reagents) and mouse monoclonal NB110-60017, clone SG2-D1a (Novus Biologicals)]; anti-Thy 1.1 [mouse monoclonal MAB1406 (Millipore Bioscience Research Reagents)]; and anti-Brn3a [goat polyclonal sc-31984 (Santa Cruz Biotechnology)]. Secondary antibodies for the vertical sections and flat mounts were species-specific anti-IgGs conjugated to either Alexa Fluor 488, Alexa Fluor 555, or Alexa Fluor 568 (goat anti-rabbit A-21428 or A-11036; goat anti-mouse A-11029; Invitrogen) or DyLight 543 and DyLight 649 (donkey anti-goat 705-505-147 and donkey anti-mouse 715-495-151; Jackson ImmunoResearch). The secondary antibody for the Western blot was donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase (NA934; GE Healthcare), and the secondary used for panning was goat anti-mouse IgM (115-005-020; Jackson ImmunoResearch). The immunogen used to test for the specificity of the polyclonal anti-D1a dopamine receptor antibody binding was the synthetic peptide (AG259; Millipore Bioscience Research Reagents) used to generate the antibody.
For Western blots, primary and secondary antibodies were diluted 1:1000 and 1:5000, respectively, in TBST containing 3% bovine serum albumin (BSA) (w/v). For retinal sections, primary antibodies (rabbit anti-D1aR, 1:100; mouse anti-D1aR, 1:300; and goat anti-Brn3a, 1:1000) and secondary antibodies (1:200–1:300) were diluted in blocking solution. In each control experiment, the immunogen and immunogen-blocked primary antibody were diluted in identical solutions.
Imaging.
Confocal images were obtained by excitation of the fluorescent dyes conjugated to the secondary antibodies and introduced dextran. Images of vertical sections were collected on a Bio-Rad Radiance 2100 Confocal System interfaced to an Olympus BX50WI upright microscope, whereas flat-mount images were acquired on an Olympus FluoView 300 Confocal System interfaced to an Olympus IX70 inverted microscope, as described previously (Partida et al., 2004), or an Olympus FluoView 1000 Confocal System interfaced to an Olympus IX81 inverted microscope. Excitation was provided by Ar (488 nm) and HeNe (543 nm) lasers on the Bio-Rad Radiance, by Ar (488 nm) and Kr (568 nm) lasers on the Olympus FluoView 300, and by HeNe (543 nm) and HeNe (633 nm) lasers on the Olympus FluoView 1000.
Generally, images of vertical sections were acquired as single optical sections. Most data from flat-mounted preparations were obtained as a series of optical sections through the ganglion cell layer at 1 μm intervals. When acquiring images of doubly stained tissue, we alternated between excitation wavelengths and adjusted detector settings to minimize signal arising from the other dye (Partida et al., 2004). Images attributable to each dye were separated and cropped, and changes in color space, if needed, were applied using the public domain program ImageJ (version 1.33; developed at the National Institutes of Health and available on the internet at http://rsb.info.nih.gov/ij/). Any subsequent adjustments to brightness or contrast were performed, and overlay images were generated, in Photoshop (version 8.0; Adobe Systems). Composite figures were assembled in Illustrator (version 11.0; Adobe Systems).
Cell dissociation and panning.
The dissociation of retinal ganglion cells used in this study was based on a dissociation protocol developed in our laboratory (Hayashida et al., 2004; Lee and Ishida, 2007), and, except where noted, all solutions mentioned here are the same as those presented in that protocol. Briefly, retinas were isolated from two freshly enucleated eyes. The remaining retinal tissue was incubated in an EDTA-supplemented low-Ca2+ solution for 5 min at room temperature. The retinas were then transferred to a 5 ml plastic tube containing a papain solution (16 U/ml in low-Ca2+ solution, mixed 1:1 with L-15 culture medium) and incubated for 5–15 min at 25°C. The papain solution was replaced with ovomucoid solution (0.5 mg/ml ovomucoid in low-Ca2+ solution mixed 1:1 with L-15) and incubated for 5 min at room temperature, to inhibit the enzyme activity. The retinal tissue was rinsed a few times with fresh L-15 medium [supplemented with 1 μm tetrodotoxin (TTX) and 0.025 mg/ml DNase I, pH 7.2–7.3], and triturated. Supernatant was layered over fresh L-15 medium in a 5 ml plastic tube [10 mm inner diameter (i.d.)] and allowed to sit for 20 min. The top 1–2 cm of this solution was then discarded, and the remaining solution, except for undissociated retinal pieces at the tube bottom, was transferred to an empty 5 ml plastic tube.
We isolated retinal ganglion cells from the final cell suspension by a “panning” method based on the expression of Thy1 (Barres et al., 1988). We prepared panning dishes by cutting a 13 mm hole in the bottom of 35 mm plastic tissue culture dishes and attaching a glass coverslip with Sylgard 184 (Dow Corning). The upper side of each coverslip was coated with goat anti-mouse IgM (diluted 1:200 in 0.1 m Tris, pH 9.5) for 2 h at room temperature, and then anti-Thy1 antibody for an additional 2 h at room temperature. After rinsing with PBS, each dish was filled with 2 ml of L-15 medium.
Retinal ganglion cells were panned by placing several drops of the final cell suspension onto the prepared glass area of these culture dishes. After allowing cells to settle down for 30 min at 30°C, nonadherent cells were removed from the dishes by rinsing each dish three times with L-15 medium. The dishes were then filled with culture medium (1:1 mixture of HEPES-buffered Hanks solution and L-15 medium; supplemented with 0.5 mg/ml cholesterol and 1% B-27; pH adjusted to 7.2–7.3 with HCl). After an additional 2 h at 30°C, the culture medium was replaced with fresh aliquots of the same medium. The cells were stored at 30°C for 12–16 h and the culture medium replaced once more before electrophysiological recordings.
Recording configuration and solutions.
To guard against the possibility that our recording methods hindered our ability to detect dopamine responses, we used three different patch-clamp configurations. Because we previously found that fish retinal ganglion cells respond to dopamine receptor agonists in perforated-patch (but not ruptured-patch) mode (Vaquero et al., 2001), some recordings were made in perforated-patch mode using amphotericin B as the perforating agent (100–250 μg/ml). The recording electrode solution contained the following (in mm): 110 K-d-gluconic acid, 15 KCl, 15 NaOH, 2.6 MgCl2, 0.34 CaCl2, 1 EGTA, and 10 HEPES. The pH of this solution was adjusted with MSA (methanesulfonic acid) to 7.4. The extracellular solution contained the following (in mm): 140 NaCl, 3.5 KCl, 10 d-glucose, 5 HEPES, 2.5 CaCl2, and 1.0 MgCl2; the pH of this solution was adjusted with NaOH to 7.4. These solutions were designed to contain physiological Na+, K+, and Ca2+ concentrations. In some instances, an extracellular solution with lowered Ca2+ (0.1 mm) and elevated Mg2+ (3.4 mm) concentrations was used to block voltage-gated Ca2+ current (Vaquero et al., 2001) and thus test whether the dopamine response entailed changes in Ca2+ influx (Liu and Lasater, 1994).
Even less invasively, we recorded ganglion cell responses to dopamine in cell-attached mode, as has been used to measure dopamine responses of hippocampal neurons (Surmeier and Kitai, 1997) and retinal ganglion cells spikes in situ (Diamond and Copenhagen, 1993). The recording electrode and extracellular solutions were identical, containing the following (in mm): 140 NaCl, 3.5 KCl, 10 d-glucose, 5 HEPES, 0.1 CaCl2, and 3.4 MgCl2; the pH was adjusted with NaOH to 7.4. The spikes activated by depolarization in this configuration (see Fig. 4 A) could be blocked by addition of 1 μm TTX to the superfusate flowing over the cell surface (traces not shown).
Figure 4.

Inhibition by dopamine and by SKF-38393, and reversal of these effects by SCH-23390. A, B, Cell-attached, voltage-clamp mode at 33°C; currents appear as vertical lines because of slow time base, with downward deflections occurring at peak depolarization of each spike. Spikes elicited by stepwise changes in patch electrode voltage (5 mV in A; 10 mV in B). Solution continuously superfused over the cell by U-tube microperfusion. Spikes in control solution (left, “control”) are blocked by inclusion of 10 μm dopamine (middle). Loss of spikes is complete within 2.5 min after onset of dopamine application at lower stimulus step size, but only partial at the higher step size. Spikes are completely blocked by 5 min after dopamine first reached the recording bath. Addition of SCH-23390 (so that the superfusate contains 10 μm dopamine and 10 μm SCH) blocks the response to dopamine at both stimulus strengths. C, D, Inhibition by SKF-38393. Ruptured-patch, current-clamp mode at 34°C; 200 ms injections of constant current (10 pA in C; 30 pA in D). Solution continuously superfused over the cell by U-tube microperfusion. As in cell-attached recordings, spike firing is continuous during both stimulus pulses in control solution (left, “control”) and is inhibited during same stimulus pulses by 10 μm SKF-38393 (middle, “SKF 10 μm”). Spikes are first lost at low stimulus strength and subsequently lost at higher stimulus strength, too. SCH-23390 (i.e., perfusate containing 10 μm SKF and 10 μm SCH) blocks the response to SKF. The triangles at left show reference level for all traces in each row (zero current in A, B; zero voltage in C, D).
We further tested the responses of dissociated ganglion cells to dopamine receptor ligands in ruptured-patch mode, as has been used to measure dopamine responses of other dissociated neurons (Schiffmann et al., 1995; Cantrell and Catterall, 2001) and to record dopamine responses from somata in the ganglion cell layer of rat retinal slices (Chen and Yang, 2007). The electrode-filling solution was designed to impede the loss of second-messenger and G-protein-mediated responses, and to minimize drifts in current voltage sensitivity; it contained the following (in mm): 110 K-d-gluconic acid, 15 KCl, 15 NaOH, 2.6 MgCl2, 0.34 CaCl2, 1 EGTA, 10 HEPES, 2 ATP, 0.5 GTP, and 3 reduced glutathione. The pH of this solution was adjusted with methanesulfonic acid to 7.4. The extracellular solution used during these experiments contained the following (in mm): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 0.1 CaCl2, 3.9 MgSO4, 10 d-glucose, and 0.05 sodium metabisulfite; the pH of this solution was adjusted to 7.4 by bubbling with carbogen (95% O2, 5% CO2). In some experiments, the extracellular CaCl2 and MgSO4 concentrations were both set to 2 mm.
The cell-attached and ruptured-patch recordings were performed with a patch-clamp amplifier (Axopatch 1D and Axopatch 200B; Molecular Devices). Cell-attached configuration was used to elicit and record currents corresponding to spikes in voltage-clamp mode (Perkins, 2006). Ruptured-patch configuration was used to elicit and record spikes in current-clamp mode, and to elicit and record voltage-gated Na+ current and I h in voltage-clamp mode. Because previous studies have shown that spikes are distorted by patch-clamp amplifiers (Magistretti et al., 1998), membrane voltage changes in response to exogenous current injections were measured with a discontinuous single-electrode current-/voltage-clamp amplifier (SEC-05LX; npi electronic), especially when examining spike amplitude and shape in perforated-patch mode.
The osmolality of the extracellular and recording electrode solutions were 280–290 and 260 mmol/kg, respectively. The extracellular solution was grounded via an agar bridge, and the membrane potentials reported here were corrected for liquid junction potentials attributable to differences between the extracellular and recording electrode solutions. Recordings were made either at room temperature (21–23°C) or more physiological temperatures (33–35°C). Pharmacological agents were either superfused over cells with a U-tube or added to the recording bath. For U-tube superfusion, a hole (∼500 μm i.d.) at the bottom of a U-shaped Teflon tube was positioned so that control and test solutions could be fed alternately into the tube, and passed as a continuous stream over each cell recorded from. For bulk additions, recordings were made in a shallow bath of known volume (1–1.5 ml), and, to minimize changes in the recording quality attributable to changes in bath depth, test compounds were applied manually in a small bolus (10–100 μl) of concentrated stock solution. We previously found that these methods of drug application produced similar effects (Hayashida and Ishida, 2004). Moreover, for comparison with the results shown in Figure 5, we confirmed that low doses of D1-type receptor agonists [e.g., 10 μm 2,3,4,5-tetrahydro-7,8-dihydroxy-1-phenyl-1H-3-benazepine HCl (SKF-38393)] reduced spiking when it was applied to cells by U-tube microperfusion at room temperature and that this effect was countered by R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH-23390) (10 μm) (traces not shown).
Figure 5.
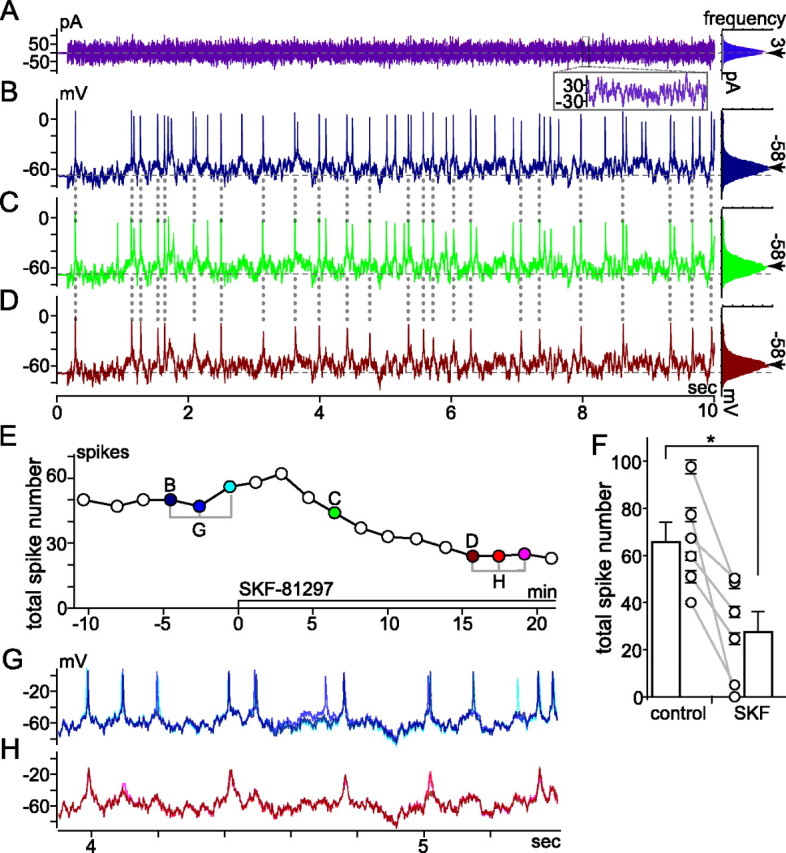
Effect of SKF-81297 on spikes elicited by fluctuating current injection; VCcCC mode recording at room temperature; perforated-patch configuration in low Ca2+ bath solution; spiking elicited by injecting fluctuating current at intervals of at least 20 s. A, Waveform of current injected (left) and histogram of current amplitude (far right). Traces of current measured at moments labeled “B” and “D” in E, and histograms constructed from these currents, are superimposed in A. Inset superimposes current recorded between 8.0 and 8.1 s of each 10 s trace. Traces from B and D are plotted in blue and red, respectively. Histograms fit to a Gaussian distribution; mean and SD are ∼3 pA (indicated by arrow) and 24 pA, respectively. B–D, Spiking and subthreshold membrane voltage changes induced by current in A, ∼4.5 min before (B), ∼6.5 min after (C), and ∼16 min after (D) SKF-81297 application began (10 μl of a 1 mm stock solution added to 0.9 ml recording bath) (see Materials and Methods). Histograms of voltages traversed during each fluctuating current injection are plotted to right. Mean voltages between and during the fluctuating current injections were set to −68 mV (dashed horizontal line through voltage traces) and −58 mV (at arrow next to each histogram), respectively. E, Time course of SKF-81297 effect on total spike number. Each point plots total number of spikes elicited by 10 s injection of fluctuating current (e.g., those in B–D are plotted in E at times labeled B–D, respectively). F, Mean ± SEM of number of spikes recorded during three injections of the current shown in A before and during the response to SFK-81297 in all cells tested (n = 6). Lines join the control and SKF values for individual cells. Bars plot the mean ± SEM of the values from all cells. The means differed significantly (*p < 0.009, paired t test). G, H, Membrane voltage changes on expanded timescale. The three traces recorded at times bracketed before SKF-81297 in E are superimposed in G; those recorded at times bracketed during SKF-81297 in E are superimposed in H. The trace and dot colors show when each recording was made and highlight the similarity in spike timing.
Current injection protocols.
Spikes were elicited by current injections that were either constant (“stepwise”) or fluctuating over time. Stepwise current injections were used to assess the capacity to spike repetitively and to facilitate comparison with results of previous studies (Liu and Lasater, 1994; Vaquero et al., 2001). During ruptured-patch recordings, the resting potential of some cells was around −65 to −70 mV, whereas in other cells, the resting potential was initially less negative. To avoid the possibility that dopamine responses differed among cells because of differences in resting potential (cf. Cantrell and Catterall, 2001), a small negative current was injected into cells, if necessary, to hold the resting membrane potential near −70 mV. Once data collection from a given cell began, no additional adjustment of the holding current was made. Current steps of various sizes were applied from the holding current to delineate both the threshold for spiking and higher spiking rates under control conditions, and to assess effects of pharmacological treatments.
Fluctuating current injections were used to examine the timing of spikes elicited by membrane potential fluctuations (cf. Mainen and Sejnowski, 1995). Membrane voltage changes in response to these current injections were measured with the SEC-05LX amplifier in discontinuous current-clamp mode. The fluctuating waveform was generated off-line by using the NEURON simulation environment [version 5.2 by J. W. Moore (Department of Neurobiology, Duke University Medical Center, Durham, NC), M. Hines (Department of Computer Science, Yale University, New Haven, CT), and T. Carnevale (Department of Neurobiology, Yale University School of Medicine, New Haven, CT)] (Hines and Carnevale, 1997), based on the Ornstein-Uhlenbeck (OU) stochastic process (Uhlenbeck and Ornstein, 1930). Numerical simulation of the OU process was given by an exact update rule (Destexhe et al., 2001) as follows: 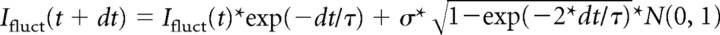 *N(0, 1), whereI
fluct is the fluctuating current, dt is the integration time step, τ is the correlation time constant, σ is SD, and N(0, 1) are normal random numbers (zero mean, unit SD). In the present study, dt, τ, and ς were 0.1 ms, 1–5 ms, and 5–100 pA, respectively. A Gaussian distribution of the current values injected was confirmed, as shown at the right edge of Figures 5
A and 7A (indicated by “frequency”). The amplitudes of these current fluctuations were adjusted for each cell so that the membrane voltage fluctuations traversed a physiological range (e.g., between −45 and −90 mV). Average membrane potentials during the fluctuating current injection, as well as at the resting state, were controlled slowly by the “voltage-clamp-controlled current-clamp (VCcCC)” technique (Sutor et al., 2003). This allowed us to elicit spikes with precise current injections and to separate effects of pharmacological agents on these spikes from effects, if any, on other properties (e.g., basal membrane potential). Before starting to collect data under the VCcCC, the electrode time constant was counterbalanced in the discontinuous voltage-clamp mode, with the supercharging and feedback capacitance neutralization circuits in the amplifier (Richter et al., 1996). To reduce electrode capacitance and its drift during the course of recordings, the patch electrode was coated with Sigmacote and the depth of the solution in the recording chamber was reduced to a minimum (∼1 mm). The switching frequency, duty cycle, and VCcCC time constant of the amplifier were set to 20–40 kHz, 1/4 (current injection/potential recording), and 100–1000 s, respectively (cf. Hayashida et al., 2004). The membrane voltage and injected current were both recorded in the VCcCC mode, and, with those amplifier settings, no distortion was discerned in the recorded traces of the current (see Fig. 5
A).
*N(0, 1), whereI
fluct is the fluctuating current, dt is the integration time step, τ is the correlation time constant, σ is SD, and N(0, 1) are normal random numbers (zero mean, unit SD). In the present study, dt, τ, and ς were 0.1 ms, 1–5 ms, and 5–100 pA, respectively. A Gaussian distribution of the current values injected was confirmed, as shown at the right edge of Figures 5
A and 7A (indicated by “frequency”). The amplitudes of these current fluctuations were adjusted for each cell so that the membrane voltage fluctuations traversed a physiological range (e.g., between −45 and −90 mV). Average membrane potentials during the fluctuating current injection, as well as at the resting state, were controlled slowly by the “voltage-clamp-controlled current-clamp (VCcCC)” technique (Sutor et al., 2003). This allowed us to elicit spikes with precise current injections and to separate effects of pharmacological agents on these spikes from effects, if any, on other properties (e.g., basal membrane potential). Before starting to collect data under the VCcCC, the electrode time constant was counterbalanced in the discontinuous voltage-clamp mode, with the supercharging and feedback capacitance neutralization circuits in the amplifier (Richter et al., 1996). To reduce electrode capacitance and its drift during the course of recordings, the patch electrode was coated with Sigmacote and the depth of the solution in the recording chamber was reduced to a minimum (∼1 mm). The switching frequency, duty cycle, and VCcCC time constant of the amplifier were set to 20–40 kHz, 1/4 (current injection/potential recording), and 100–1000 s, respectively (cf. Hayashida et al., 2004). The membrane voltage and injected current were both recorded in the VCcCC mode, and, with those amplifier settings, no distortion was discerned in the recorded traces of the current (see Fig. 5
A).
The output signals from the amplifiers were analog-filtered (2 kHz, single-pole, for the Axopatch 1D and Axopatch 200B; 5–20 kHz, two-pole Bessel for the SEC-05LX) and digitally sampled (10–50 kHz). pCLAMP software (versions 8.1.01, 8.2.0.235, and 9.2.1.9; Molecular Devices) was used for current protocol generation and data acquisition. SigmaPlot (versions 5.0.5, 8.02; SPSS) and Matlab (version 6.5.1.199709 release 13; Mathworks) were used for data analyses.
Reagents.
Reagents were obtained from the following sources: Abbott: sodium pentobarbital (0074-378-05); GE Healthcare Life Science: glycerol (56-81-5); Bio-Rad: Bradford reagent (500-0006), SDS (161-0300); BDH Laboratory Supplies: CaCl2; Calbiochem: FluorSave (B34539), 8-bromo-cAMP (203800), 8-cpt-cAMP (116812), tetrodotoxin (584411); Thermo Fisher Scientific: Triton X-100 (BP151-100); Invitrogen: B-27 (17504-044), PBS, Ca2+- and Mg2+-free, pH 7.4 (70011-044), See Blue Plus2 prestained standard (LC5925), Magic Mark protein standard (LC5600), sample buffer (NP0007), reducing agent (NP0004), MOPS running buffer (NP0001), transfer buffer (NP0006), 3 kDa dextran coupled to fluorescein (D3306), 10 kDa dextran coupled to Alexa Fluor 488 (D22910); Jackson ImmunoResearch: normal goat serum (005000121), normal donkey serum (017000121), goat anti-mouse IgM (115-005-020); Pierce: SuperSignal West Pico Chemiluminescent Substrate (34080); Roche: TPCK (874507), leupeptin (1017101), pepstatin A (253286), calpain I inhibitor (1086090), calpain II inhibitor (1086103); Sigma-Aldrich: BSA (A8806), DNase I (D4527), EDTA (E6758), NaF (S6521), β-glycerol phosphate (G6376), Sigmacote (SL2), sodium orthovanadate (S6508), aprotinin (A6279), benzamidine (B6506), Tween 20 (P9416), Ponceau S (P7767), protein A-Sepharose (P9424), dopamine hydrochloride (H8502), sodium metabisulfite (255556), SKF-38393 (D047), R-(+)-6-chloro-7,8-dihydroxy-l-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (SKF-81297) (S179), SCH-23390 (D054); VWR: OCT compound (4583). The salts (NaCl, etc.) used for electrophysiological recordings and buffers were all reagent grade and obtained from Sigma-Aldrich, unless otherwise specified. Stock solutions of dopamine (10 mm in water supplemented with Na metabisulfite) were prepared on each day of use, and then diluted by at least 1000-fold in the external superfusate solution immediately before application.
Results
Three sets of observations on adult rat retinal ganglion cells are presented here. The first visualizes D1-type dopamine receptor-like immunoreactivity in situ. The second uses three recording configurations and two current injection protocols to assess the effect of dopamine, D1-type receptor agonists, and membrane-permeant cAMP analogs, on spike generation and timing. The third compares effects of dopamine on spikes, voltage-gated Na+ current, and I h.
D1a dopamine receptor protein is present in rat retina
Although several laboratories have attempted to localize D1-type dopamine receptors in rat retina, the protein bound by the ligands used in most of these studies was neither isolated nor characterized (Tran and Dickman, 1992; Bjelke et al., 1996). We therefore estimated the molecular weight of protein bound by the polyclonal anti-D1a dopamine receptor antibody (AB1765P; Millipore Bioscience Research Reagents) used in our localizations, and tested whether this binding is inhibited by the peptide (AG259; Millipore Bioscience Research Reagents) that the antibody was directed against. On nitrocellulose blots of polyacrylamide gels in which the solubilized proteins from retinal homogenates were electrophoretically separated, this antibody stained a well focused protein band (Fig. 1 B). The molecular weight of this band was estimated to be 54 kDa by comparison of its position with molecular weight standards in an adjacent lane (Fig. 1 A), and, on all of the blots we ran, the mean estimated molecular weight was 54 ± 1 kDa (mean ± SEM; n = 3).
Figure 1.

Western blots of D1a dopamine receptor. Homogenate of snap-frozen retinas, and protein standards, separated by SDS-PAGE and transferred to nitrocellulose membranes. A, D, Molecular weight (MW) standard proteins, with MW of each indicated in kilodaltons by superimposed number. B, Retina proteins run alongside standard proteins in A and probed with anti-D1a receptor antibody. A well focused protein band is seen at migration distance corresponding to an estimated MW of 54 kDa. No other proteins are stained over the MW range shown (20–100 kDa). C, E, In a different experiment, retina proteins run alongside standard proteins in D. Lane C probed with anti-D1a receptor antibody that had been preincubated overnight with immunogen. Probing of lane E with anti-D1a receptor antibody shows a well focused protein band in E at an estimated MW of 54 kDa. A faint band is also seen within the MW range reported for glycosylated D1a receptors (here between 55 and 60 kDa). Staining of both bands (dark and faint) was blocked completely by immunogen (C).
Binding of the anti-D1a receptor antibody to this band was reduced below detectable levels by the immunogen (Fig. 1 C). These results agree with the molecular weight estimate of a rat brain protein bound by the same antibody (Huang et al., 1992) and with that of a rat brain protein bound by a different anti-D1 receptor antibody (Caillé et al., 1995) that stains the ganglion cell layer of various mammalian retinas (Nguyen-Legros et al., 1997). In some blots, a faint band was also seen within the molecular weight range reported for glycosylated D1a receptors (Karpa et al., 1999) (e.g., between 55 and 60 kDa in Fig. 1 E). Staining of this band was also blocked by immunogen (Fig. 1 C).
D1a dopamine receptor is present in rat retinal ganglion cells
Having found that anti-D1a dopamine receptor antibody binds protein solubilized from rat retina (Fig. 1), we attempted to localize this protein in situ by indirect immunofluorescence methods. In transretinal (vertical) sections, we consistently observed bright immunoreactivity in somata located in the ganglion cell layer (Fig. 2 A,B, D1a). In contrast, we never observed D1a dopamine receptor-like immunoreactivity in the outer nuclear layer (Fig. 2 A,B, D1a). Staining in the inner plexiform layer was typically diffuse and moderate in intensity, except for some large caliber dendrites (see below). The outer plexiform layer stained faintly (Fig. 2), as did some somata in the inner nuclear layer. Some of these somata may be horizontal and/or bipolar cells (Veruki and Wässle, 1996; He et al., 2000; Müller et al., 2003), but we leave this to be resolved by use of antibodies that stain inner nuclear layer somata more vividly. In any event, the staining described here (including bright and faint) appeared to be specific because it was reduced below detectable levels by preincubating the primary antibody with immunogen (Fig. 2 C).
Figure 2.
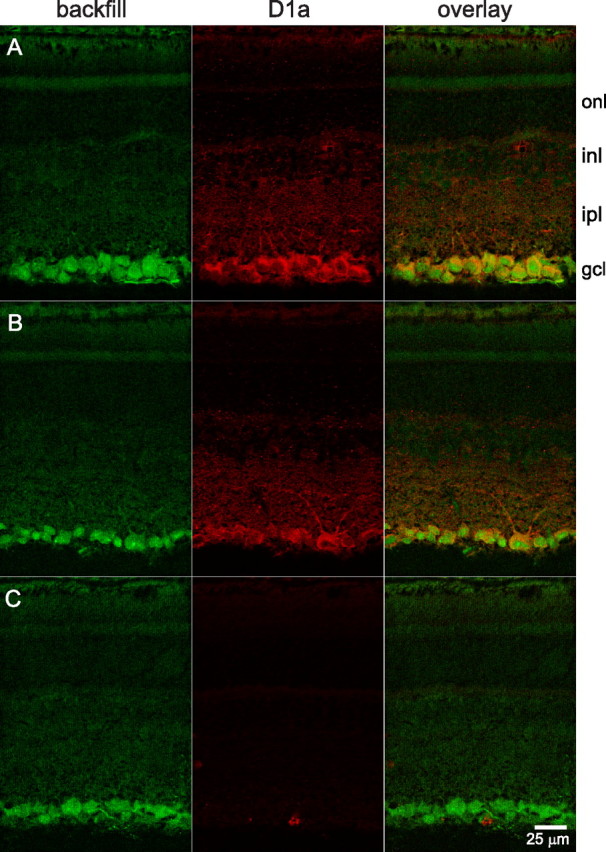
D1a receptor-like immunoreactivity in ganglion cells in vertical sections. Ganglion cells identified by retrograde transport of Alexa Fluor 488-coupled dextran introduced into the optic nerve. Labeling with anti-D1a receptor antibody visualized with Alexa Fluor 555-conjugated secondary antibody. Images are single confocal optical sections obtained with a 40× oil-immersion objective. Fluorescence attributable to each dye obtained individually. A, Direct overlay of fluorescence signals from retrogradely transported dextran (backfill, in green) and anti-D1a receptor antibody (D1a, in red) indicate ganglion cells exhibiting D1a receptor-like immunoreactivity (orange–yellow). Labels along right side indicate position of outer nuclear layer (onl), inner nuclear layer (inl), inner plexiform layer (ipl), and ganglion cell layer (gcl). B, Example of same labeling from a different retina. Immunopositive dendrites emerge from a large soma in the right half of the panel and project into the distal half of ipl. C, Images of a section obtained under the exact same conditions as B, except that the anti-D1a receptor antibody was preincubated with immunogen before application to section. Scale bar, 25 μm (applies to all panels).
We used various preparations to test the possibility that immunopositive somata in the ganglion cell layer were ganglion cells. The most direct approach was to stain for D1a receptors after filling the ganglion cell somata, via their axons in the optic nerve, with a retrogradely transportable marker. We used dextran (3 or 10 kDa) coupled to an intense fluorophore (fluorescein or Alexa Fluor 488) for this purpose, and collected confocal images from vertical sections and flat-mounted retinas. The dextran filled filamentous structures in the optic fiber layer and somata in the ganglion cell layer. We saw no evidence of marker leakage from these structures into other cell types, in that we never observed brightly fluorescing somata in the distal half of the inner nuclear layer, or in the outer nuclear layer, in vertical sections (Fig. 2) or flat-mounted retinas (not illustrated). We therefore interpret the stained somata and fibers to be ganglion cell somata and intraretinal portions of their axons, respectively (Oi et al., 2008).
The nuclei of these somata were brightly labeled, as in other studies (Dacey et al., 2003), presumably by influx through nuclear pores (Keminer and Peters, 1999). After sectioning these retinas and incubating them with anti-D1a receptor primary antibody and a secondary antibody coupled to Alexa Fluor 555, we collected confocal images of the Alexa Fluor 488 and Alexa Fluor 555 fluorescence at laser settings that produced negligible cross-signal contamination (Partida et al., 2004). Overlays of images in which Alexa Fluor 488 fluorescence was rendered green, and Alexa Fluor 555 fluorescence was rendered red, showed D1a receptor-like immunoreactivity in ganglion cells as a yellowish orange color (Fig. 2 A,B). In cells presenting both signals, the infiltration of cytoplasm and nuclei by dextran produced a green spot circumscribed by a yellow-orange belt—never vice versa. Moreover, the green region was often off-center within the cell profile, as are nuclei in rat retinal ganglion cells observed by other methods.
We obtained similar data in flat-mounted retinas. In overlays of the projected confocal images of retrogradely transported dextran (fluorescein) (Fig. 3 A) and anti-D1a receptor-like immunoreactivity (Alexa Fluor 568) (Fig. 3 B), yellow-to-orange areas indicate ganglion cells with both signals (Fig. 3 C). The most vivid, D1a-immunopositive cells showed a ring or band of D1a-like immunoreactivity. In most other cells, the D1a labeling was diffuse or confined to a less prominent ring along the periphery of the cell body. Figure 3 D is a masked version of Figure 3 C highlighting the cells in this field with significant dextran fill. A few cells (arrows) appeared to be only green and thus did not display noticeable D1a immunoreactivity. At the same time, a few cells presented labeling for D1a without a conspicuous dextran fill (not illustrated). However, these “green-only” and “red-only” cells constituted, at most, a small fraction of the cells backfilled with dextran. In a total of 5 retinal fields we examined in detail, D1a-immunoreactivity was found in 538 (i.e., 94%) of the 572 somata that displayed dextran fill, and only a total of 30 red-only cells were seen.
Figure 3.
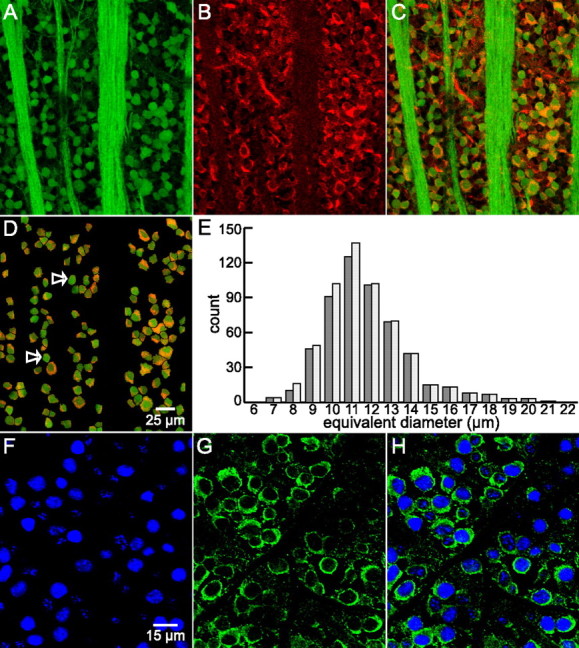
D1a receptor-like immunoreactivity in ganglion cells in flat-mounted retina. A, As in Figure 2, ganglion cell somata identified by retrogradely transported, fluorescein-coupled dextran (green in A, C, D). The same fluorescence identifies fibers in this image as intraretinal segments of ganglion cell axons, extending as fascicles between the top and bottom edges of A and C. B, Binding of polyclonal (Millipore Bioscience Research Reagents) anti-D1a receptor antibody visualized with Alexa Fluor 568-conjugated secondary antibody. C, Overlay of A and B. As in Figure 2, yellow/orange indicates regions of overlapping red (Alexa Fluor 568) and green (fluorescein) signal, signifying D1a receptor-like immunoreactivity in ganglion cells. D, Same field as C masked to highlight dextran-containing somata only. The arrows show some ganglion cells without noticeable D1a-like immunoreactivity. Fluorescent images A–D are maximum intensity z-projection of five consecutive optical sections obtained at 1 μm z-intervals with three-frame Kalman averaging. Scale bar: (in D) A–D, 25 μm. E, Side-by-side histograms of apparent size of ganglion cells identified by fluorescent dextran incorporation via retrograde transport (light bars) and the subset of these cells that also showed D1a-like receptor immunoreactivity (dark bars). Cells were masked, selected, and analyzed in ImageJ from five fields similar to (and including) D from three different retinas. Each ImageJ-reported cross-sectional area converted to diameter of an equivalent circle. Diameters are placed into 1 μm bins centered about the indicated values. F–H, Binding of an anti-D1a receptor antibody and an antibody directed against ganglion cell marker Brn3a. F and G are sequentially collected single optical sections of the ganglion cell layer of a retina incubated in anti-Brn3a and anti-goat DL549-conjugated secondary, and monoclonal (Novus) anti-D1a primary antibody and anti-mouse DL649-conjugated secondary, respectively. Fluorescence from the fluorophores is pseudocolored blue and green. H merges the images in F and G. The crisp green outline of each blue cell profile shows that the monoclonal anti-D1a antibody binds to many of the somata identified as ganglion cells in this field. Scale bar: (in F) F–H, 15 μm.
Based on recent studies of rat and mouse retina (Raymond et al., 2008; Nadal-Nicolás et al., 2009), we also identified retinal ganglion cells by the binding of antibodies against Brn3a and, for comparison with the fields in Figures 2 and 3, A–D, tested the possibility that they bind a monoclonal anti-D1 receptor antibody (NB110-60017; Novus). These were visualized by secondary antibodies conjugated to DyLight 543 and DyLight 649, and found to bind exclusively to cell nuclei (Fig. 3 F) and the rest of the somatic profile (cytoplasm and cell membrane) (Fig. 3 G), respectively. The staining pattern in overlays of the sequentially imaged fields (Fig. 3 H) corroborate those obtained with dextran fills and the polyclonal anti-D1a receptor antibody.
Our vertical sections and flat mounts of backfilled retinas revealed three properties of ganglion cell somata presenting D1a dopamine receptor-like immunoreactivity. First, somata were of the size expected for ganglion cells. In vertical sections, the measured equivalent diameter of D1a-immunopositive ganglion cells was 10 ± 2.5 μm (mean ± SD; n = 95; median, 10.2 μm). In projected optical sections from flat-mounted preparations, the average equivalent diameter of the D1a-immunopositive ganglion cells (Fig. 3 E, dark bars) was 12 ± 2 μm (mean ± SD; n = 538; median, 11.5 μm). The average size for all ganglion cells identified by retrograde transport (Fig. 3 E, light bars) was slightly (but not significantly) smaller (11.7 ± 2 μm; n = 572; median, 11.4 μm). The range of somal diameters we observed (Fig. 3 E) thus overlap with sizes found in previous studies of rat retinal ganglion cells (Huxlin and Goodchild, 1997; Sun et al., 2002). In the five fields used to generate Figure 3 E, we counted an average of 1921 ± 361 ganglion cells/mm2 (mean ± SEM). The population density of retrogradely filled cells, counted in areas that were not obscured by optic fiber tracts and blood vessels, ranged from 1038 to 2890 cells/mm2. These values overlap with the density of ganglion cells identified by retrograde fill with other markers (horseradish peroxidase, DiI, fast blue, and FluoroGold) in various rat strains (Oi et al., 2008).
Second, it was not uncommon to find immunopositive somata adjacent to one another in the ganglion cell layer (Fig. 2). These could include neighboring large and smaller somata (Figs. 2 B, 3) or rows of smaller somata (Fig. 2 A). This is consistent with our overall observation that a large fraction of the ganglion cell somata are D1a-immunopositive.
Last, some sections fortuitously captured dendrites that were large enough in caliber to follow as they emerged from somata and extended into the inner plexiform layer. Primary dendrites in Figure 2 B, for example, can be seen projecting from a large soma and beginning to reach the distal half of the inner plexiform layer. Similarly large primary dendrites also arborized in the proximal half of the inner plexiform layer.
D1-type dopamine receptor agonists modulate spike firing in dissociated rat retinal ganglion cells
Having localized D1a-type dopamine receptor-like immunoreactivity to ganglion cells, we next asked whether dopamine receptor agonists can activate receptors in these cells. Based on effects observed in retina and brain (Jensen and Daw, 1986; Schiffmann et al., 1995; Cantrell and Catterall, 2001), we tested whether dopamine, SKF-81297 (S179; Sigma-Aldrich), and SKF-38393 (D047; Sigma-Aldrich) alter spiking, and whether these effects were reversed by D1-type receptor antagonists (e.g., SCH-23390; D054; Sigma-Aldrich). To ensure that we detect responses of ganglion cells rather than effects mediated via other cells, we isolated ganglion cells by dissociating retinas and, in most cases, selected for ganglion cells by panning with anti-Thy1 antibody (Barres et al., 1988). All retinas were from 60- to 120-d-old rats, so that both our recordings and immunohistochemistry yielded observations about adult mammalian retinal ganglion cells, and because dopamine responses of juvenile and adult rat central neurons have been found to differ (Salgado et al., 2005).
To avoid disruption of intracellular signaling mechanisms as much as possible, spikes were recorded from some cells as pulsatile inward currents in cell-attached, voltage-clamp mode (Perkins, 2006). Dopamine (10 μm) first abolished spiking elicited by small current injections (Fig. 4 A, compare first three traces) and subsequently inhibited spikes elicited by larger current injections (Fig. 4 B, compare first three traces). At 3 μm, dopamine blocked spikes elicited by the small current injections and reduced the number of spikes elicited by the larger current injections (traces not shown). All of these effects were reversed by either applying a D1-type receptor antagonist (SCH-23390; 10 μm) together with dopamine (Fig. 4 A,B, far right side) or washing away the dopamine with control solution (traces not shown). Results similar to those in Figure 4, A and B, were obtained in all cells tested (n = 3 at 34°C), using dopamine at 10 μm and SCH-23390 at 10 μm.
Because the membrane potential is hardly known in cell-attached mode, we used whole-cell modes to further characterize ganglion cell responses to dopamine. Figure 4 C shows spikes elicited by 200 ms constant current steps. While control solution was microperfused over this cell, these current injections elicited continuous (i.e., “sustained”) firing of action potentials, and increases in injected current amplitude (e.g., from 10 to 30 pA in left side of Fig. 4 C,D, respectively) elicited a progressive increase in spike number (i.e., mean spike frequency when divided by the duration of each current step). These spikes showed no marked changes in amplitude, duration, or interspike interval during individual steps. The D1-specific agonist SKF-38393 (10 μm) reduced spike number and decreased instantaneous spike frequency during injection of the same current steps (Fig. 4 C,D, middle pairs of traces). Addition of the D1-type dopamine receptor antagonist SCH-23390 (10 μm, so that the perfusate contained SKF-38393 and SCH-23390) restored sustained spiking (Fig. 4 C,D, far right side). Results similar to those in Figure 4, C and D, were obtained in all cells tested (n = 5 at 34°C; n = 2 at 24°C) in experiments using SKF-38393 at 6–10 μm and SCH-23390 at 6–10 μm. Because the recordings exemplified in Figure 4 show that ligands elicited responses at concentrations equal to, or lower than, those found to be effective in other preparations (e.g., 3–10 μm dopamine and SKF-38393) (Jensen and Daw, 1986; He et al., 2000; Cantrell and Catterall, 2001; Chen and Yang, 2007; Zhang et al., 2007), we did not attempt to define dose–response relationships in any detail. Moreover, because SCH-23390 counteracted the effects of dopamine, and because SKF-38393 produced effects that were indistinguishable from those of dopamine, we did not test for effects of D2-type receptor ligands (Schorderet and Nowak, 1990).
In some, if not most, cells, the resting potential showed little or no concomitant change during responses to D1-type receptor agonists (Fig. 4 C,D). The loss of spikes without changes in resting potential differ from the combination of spike loss and depolarization induced by dopamine in ganglion cell layer somata of rat retinal slices (Chen and Yang, 2007). Instead, the response pattern observed here resembles the “silent inhibition” produced by dopamine in various preparations [e.g., rat hippocampal neurons (Stanzione et al., 1984), dissociated striatal neurons (Schiffmann et al., 1995), fish retinal ganglion cells (Vaquero et al., 2001), and putative ganglion cells from turtle retina (Liu and Lasater, 1994)].
Last, to retain the advantage of measuring whole-cell voltage while avoiding the response deterioration often seen during ruptured-patch recordings, some recordings were performed in perforated-patch mode with a discontinuous voltage-clamp amplifier. In these experiments, we tested the effect of specific D1 receptor agonists and used fluctuating current injections to examine the capacity to spike repetitively when the membrane potential is dynamically shifting. The current amplitude distribution was Gaussian (see Materials and Methods), the duration of each injection was routinely set to 10 s, and the mean value of the membrane potential during each recording epoch (i.e., before, during, and after the fluctuating current-injection) was shifted by a voltage-clamp-controlled current-clamp technique (Sutor et al., 2003). Examples of injected currents, spikes elicited under these conditions, and effects of SKF-81297, are illustrated in Figure 5. The current injected is plotted against time in Figure 5 A. The plot to the right of this trace is a histogram of the current amplitudes, showing the peak of the distribution near 3 pA. Figure 5 B plots the membrane potential during a single injection of this current while the cell was bathed in control solution, and shows that the cell generated ∼50 spikes. Spikes occurred throughout this 10 s epoch, and as shown by the six data points plotted before the zero time point in Figure 5 E, repeated injections of the same current waveform elicited approximately the same number of spikes per 10 s injection. Superimposing the responses to multiple current injections, and displaying them on an expanded timescale, shows that the spikes often occurred at the same time points during successive runs, that the spikes and the intervening membrane potential fluctuations were reproducible in shape, and that there was little temporal variability (“jitter”) in the spikes (Fig. 5 G, compare the differently colored traces; see also Fig. 5 A, inset).
During the application of SKF-81297 (Fig. 5 C,D), some of the spikes seen in the control solution were abolished, resulting in a lower spike number. As shown by the data points after t = 15 min in Figure 5 E, the spike number eventually declined by ∼50% (i.e., to a steady-state value of ∼25 spikes in this cell). As a measure of the average response of all of the cells examined this way (n = 6), we compared the spikes elicited by current injections that produced voltage fluctuations with a SD of 9 ± 2 mV (mean ± SEM of the deviation) at a controlled mean membrane potential of −59 ± 0.4 mV (mean ± SEM), similar to values observed in situ (Bloomfield and Xin, 1997). On average, this was achieved with a mean holding current of 10 ± 2 pA (mean ± SEM), and currents that fluctuated with a SD of 30 ± 5 pA (mean ± SEM) around this mean. Under these conditions, the spike number started from a mean of 66 ± 8 per 10 s epoch in control solutions, and fell by 59 ± 12% (mean ± SEM) in the presence of SKF-81297. SKF-38393 produced a similar reduction of spikes elicited by similar depolarizations (n = 2) (traces not shown). Increasing the mean amplitude of the holding current injected in the presence of D1 agonists increased the tendency of cells to spike less often during fluctuating current injections (traces not shown). In some cells, >87% of the spikes observed during the fluctuating current injections in control solutions were abolished (n = 2 with SKF-81297; n = 1 with SKF-38393). Figure 5 F plots the mean spike number before and during the response to SFK-81297 in each of the cells (n = 6) tested this way, together with the average of these spike numbers. The mean value of the current injected into these cells was adjusted to set the mean voltage at around −59 mV; the measured means ranged between −58 and −60 mV. Because of differences in input resistance from cell to cell, the mean current producing these voltages ranged between 3.2 and 13.8 pA. Likewise, to produce approximately similar numbers of control spikes, the SD of the injected current was adjusted, from cell to cell, to between 16.5 and 48.9 pA. This yielded between 40 and 98 spikes (Fig. 5 F). The inhibition by D1-type agonists developed over a course of 10–15 min, and the spike number and peak amplitude stayed at reduced levels as long as SKF-81297 was present.
In addition to reducing spike number, and as seen during the response to stepwise current injections (Fig. 4), SKF-81297 induced a decrease in spike amplitude, and a slowing of the rising and repolarizing sides of each spike (Fig. 5 H). Despite this change in spike size, shape, and number, and despite the passage of several minutes before the response reached steady state, aligning control and test recordings shows that the spikes which fired in the presence of SKF did so at almost the same moments during the 10 s current injections as spikes in the control (e.g., at the times marked by the dotted vertical lines connecting Fig. 5 B–D; compare also Fig. 5 G,H). This alignment is illustrated in more detail by histograms of the difference between (1) the time of each spike elicited by the fluctuating current and (2) the mean time of all of the temporally coaligned spikes elicited by repeated current injections before (Fig. 6 A) and after (Fig. 6 B) exposure to SKF-81297. The difference (i.e., “spike timing deviation”) between individual spikes and the average could thus be positive, negative, or zero (Fig. 6 A,B). Figure 6 shows that the control and test histograms were indistinguishable in width, measured at half-height (Fig. 6, compare A, 0.5 ms; B, 0.7 ms), and overall shape (e.g., when the histograms were superimposed after plotting the data as percentage of spike number rather than raw spike numbers) (Fig. 6 C), and that the spike time deviation did not exceed 10 ms in either the control or test solutions. Similar results were obtained from a total of four cells [three cells before and during application of SKF-81297; one cell before and during application of 8-cpt-cAMP (see below) (Fig. 7)]. The insets to A and B illustrate the jitter of a spike elicited by three current injections before and during SKF and show almost identical differences in spike timing for the spikes labeled “a” and “c” in the control and test condition, despite an increase in the deviation between the spikes labeled “a” and “b.” At the same time, these insets show a tight overlap (i.e., reproducibility) of the intervening membrane potential fluctuations (i.e., the subthreshold voltages traversed before reaching spike threshold, as well as those after spike repolarization) in each solution. These effects were recorded from ganglion cells identified either by panning or by post hoc Thy1 immunohistochemistry (not illustrated). Like the effects seen during stepwise current injections, these changes were observed in extracellular solutions containing either low (0.1 mm; n = 5) or normal (2.5 mm; n = 1) Ca2+.
Figure 6.

Spike timing deviation in absence and presence of SKF-81297. Spiking elicited and recorded using same methods as in Figure 5. Spike timing was compared moment-by-moment in three 10 s traces recorded ≤4 min before application of SKF-81297 (A), and in three 10 s traces recorded 12–16 min after application of SKF-81297 (B). For each condition, if spikes occurred at similar times in all three traces (i.e., if spikes were “temporally coaligned”), the difference between the time of each spike and the average of the times of the coaligned spikes was measured, and tallied accordingly in A or B. Time was measured from beginning of each current injection to moment of maximum change in slope (dV/dt) along rising phase of each spike. Histograms are binned at 0.2 ms, and include 165 (55 × 3) and 81 (27 × 3) spikes for A and B, respectively. Insets superimpose examples of spikes considered to be coaligned in traces recorded before (A) and after (B) SKF. All six traces (labeled a–c in A and in B) begin at identical times after start of each 10 s current injection. C, Percentage distribution of spike timing deviation. Spike number in A and B was normalized by respective total spike number and the two histograms were overlaid, with gray signifying overlap of the before (clear) and after SKF (filled) distributions.
Figure 7.

Effect of membrane-permeant cAMP analog (8-cpt-cAMP) on spikes elicited by fluctuating current injection. Recording mode, conditions, and figure format are as in Figure 5. A, Current injected (left) and histogram of current amplitude (right). The left and right parts of A superimpose two traces recorded at B and D in E, and their corresponding amplitude histograms, respectively. The inset superimposes current recorded between 8.0 and 8.1 s of each 10 s trace. The histograms fit to a Gaussian distribution with mean and SD of ∼5 pA (arrow) and 8 pA, respectively. B–D, Spiking and subthreshold membrane voltage changes induced by current in A, ∼5 min before (B), ∼5 min after (C), and ∼13 min after (D) application of 8-cpt-cAMP began (here, 2 μl of 50 mm stock solution was added to 0.9 ml recording bath). Histograms of voltages traversed during each fluctuating current injection are plotted to right. Mean voltages between and during fluctuating current injections were set to −72 mV (dashed horizontal line through voltage traces) and −58 mV (at arrow next to each histogram), respectively. E, Time course of 8-cpt-cAMP effect on total spike number. Each point plots total number of spikes elicited by 10 s injection of fluctuating current (e.g., those in B–D are plotted at correspondingly labeled times in E). F, G, Membrane voltage on expanded timescale. Three traces recorded at times bracketed before and during 8-cpt-cAMP in E are superimposed in F and G, respectively.
Membrane-permeant cAMP analogs modulate spike firing in dissociated rat retinal ganglion cells
Because D1-type receptor activation generally exerts its effects via increases in intracellular cAMP levels, we measured spikes before and while applying membrane-permeable forms of cAMP. In experiments in which we used constant current injections to initiate activity, 8-bromo-cAMP and 8-cpt-cAMP (n = 1 and n = 2, respectively, all in normal Ca2+) elicited decreases in spike amplitude and number comparable with that seen with SKF-81297 (not illustrated). These effects resembled those recorded from somata in the ganglion cell layer of retinal slices (Chen and Yang, 2007). In experiments in which the stimulus was provided by fluctuating current injection, 8-bromo-cAMP (n = 2; one each in normal and low Ca2+) and 8-cpt-cAMP (n = 1 in low Ca2+) produced effects on spike amplitude and spike number, and the time course of the onset of these changes, similar to those observed after SKF-81297/38393 applications (compare Figs. 5, 7).
For example, when the injected current produced a voltage bias and fluctuations similar to those described above (viz., mean, −58 mV; SD, 8 mV), the spike number started from a control mean value of 71 spikes per 10 s epoch, and then fell, on average, by 53% in the presence of 8-cpt-cAMP (n = 1) or 8-bromo-cAMP (n = 1). Moreover, the dotted vertical lines connecting Figure 7, B–D, indicate that as with activation of the dopamine receptor, directly increasing intracellular cAMP decreased spike number and peak amplitude without significantly impacting the temporal fidelity of the response to the injected current. Results consistent with these were obtained from one other cell but are not included in this average because the mean membrane potential was shifted only to −67 mV during the fluctuating current injection.
Even after blockade of I h, dopamine inhibits spikes and reduces voltage-gated Na+ current
Previous studies have proposed that dopaminergic modulation of retinal ganglion cell spiking entails changes in either voltage-gated Ca2+ current (Liu and Lasater, 1994), voltage-gated Na+ current (Vaquero et al., 2001; Hayashida and Ishida, 2004), or the hyperpolarization-activated cation current I h (Chen and Yang, 2007). However, the extent to which dopamine modulates spiking by different mechanisms in the species examined (turtle, goldfish, and rat, respectively) is not known, at least in part, because ion current modulation was not tested in rat under conditions that preclude indirect effects (e.g., because of dopamine effects at chemical and/or electrical synapses), not all rat ganglion cells have I h (Reiff and Guenther, 1999; Chen et al., 2004; Lee and Ishida, 2007), and the possibility that dopamine modulates voltage-gated Na+ current in rat ganglion cells was not tested. We therefore reexamined whether dopamine reduces spiking if I h is blocked, and we tested whether dopamine reduces voltage-gated Na+ current. We did not test whether dopamine modulates voltage-gated Ca2+ current because dopamine, SKF-81297, and SKF-38393 reduced spiking in normal as well as low-Ca2+/elevated-Mg2+ external saline, indicating that these responses did not entail a change in Ca2+ current.
To begin with, we depolarized and hyperpolarized single cells in voltage-clamp mode to measure voltage-gated Na+ current and I h, and we depolarized these same cells in current-clamp mode to elicit spikes. These recordings were performed in isolated cells after the extracellular Ca2+ concentration was lowered to 0.1 mm while Mg2+ was raised to 3.9 mm (Vaquero et al., 2001). The lowest row of traces in Figure 8 A shows the activation of I h by hyperpolarizations from a holding potential of −72 mV to test potentials of −77, −92, and −107 mV. As described previously in detail (Lee and Ishida, 2007), this current was slowly gating and inward in control solution (left), and it was blocked by 3 mm Cs+ (right). The top row of current traces in Figure 8 A shows that this addition of Cs+ did not affect the fast inward current activated by depolarization of this same cell to −47 mV (from the same holding potential, −72 mV). Having thereby confirmed the presence and block of I h, we then tested whether dopamine inhibits spiking. The left, middle, and right columns of Figure 8 B show the spikes elicited by 200 ms constant-current injections of 12, 22, and 32 pA, respectively, as the solution microperfused over this cell was changed from 3 mm Cs+ (top two rows), to 3 mm Cs+ plus 6 μm dopamine (next nine rows, as marked), and then 3 mm Cs+ plus 6 μm dopamine plus 5 μm SCH-23390 (next eight rows, as marked). These traces and the spike counts plotted in Figure 8 C show that dopamine inhibited spiking in this cell at all three stimulus intensities, that spike number began to decline ∼2 min before disappearing altogether, and that the loss of spikes was efficaciously antagonized by SCH-23390. Switching back to voltage clamp confirmed that I h was still blocked in this cell (traces not shown). Results similar to those in Figure 8, A–C, were obtained in all cells tested (n = 4 at 34°C), using either dopamine at 6 μm and SCH-23390 at 5 μm (n = 2) or dopamine at 3 μm and SCH-23390 at 2.5 μm (n = 2).
Figure 8.

Block of I h does not preclude spike inhibition by dopamine. Voltage-gated Na+ current, I h, and spikes elicited in a single ganglion cell in ruptured-patch configuration at 34°C. A, Currents recorded in voltage-clamp mode without leak subtraction while solution superfused over the cell were changed from control (left) to 3 mm Cs+ (right). Steps above current traces show stimulus timing and polarity. Holding potential was −72 mV. Test potentials were −47 mV to activate Na+ current (top rows) and −77, −92, −107 mV to activate I h (bottom rows). Cs+ blocks I h (including portion activated at −72 mV) (Lee and Ishida, 2007) without affecting Na+ current. The triangles at left show zero current level for each row. The insets show the Na+ current on an expanded timescale. B, Spikes then recorded in current-clamp mode in response to sequence of constant current injections (12, 22, and 32 pA) while superfusate were changed (as marked by brackets) from 3 mm Cs+ (first two rows of spikes) to 3 mm Cs+ and 6 μm dopamine (next nine rows), and then 3 mm Cs+, 6 μm dopamine, and 5 μm SCH-23390 (last eight rows). Stimulus timing is shown at top of B. Voltage traces displayed in the sequence that they were collected, with each row showing responses to same current injections, and each row initiated at 30 s intervals. Tick marks along right side show ground level for each row of traces. C plots number of spikes elicited by each current injection in B, showing inhibition of spikes by dopamine and antagonism of this response by SCH-23390.
Because our results show that spike inhibition by dopamine does not require a change in I h (Fig. 8), and also because D1 receptor agonists slowed the rate of rise of spikes (Fig. 6), we tested whether dopamine reduces the amplitude of voltage-gated Na+ current (Fig. 9). To test this possibility as in a recent study that did not find spike inhibition by dopamine in the presence of I h blocker 4-(N-ethyl-N-phenylamino)-1,2-dimethyl- 6-(methylamino)pyridinum chloride (ZD-7288) (Chen and Yang, 2007), we depolarized and hyperpolarized individual cells in ruptured-patch mode. While doing so, we monitored the amplitude of inward current as cells were exposed sequentially to the following solutions: control; 3 mm Cs+; 6 μm dopamine and 3 mm Cs+; 6 μm dopamine, 3 mm Cs+, and 5 μm SCH-23390; 6 μm dopamine, 3 mm Cs+, 5 μm SCH-23390, and 1 μm TTX; and again control. These records show that Cs+ blocked I h in this cell (Fig. 9 B2), that dopamine reduced the depolarization-activated inward current (Fig. 9 A3), that SCH-23390 blocked this effect (Fig. 9 A4), that the depolarization-activated inward current was fully abolished by 1 μm TTX (Fig. 9 A5), and that the effects of Cs+, TTX, and dopamine were reversible (Fig. 9 A6,B6). Because the test depolarizations did not activate an outward current in the presence of TTX (Fig. 9 A5), the reduction of inward current by dopamine appears to have resulted from reduction of voltage-gated Na+ current and not from augmentation of an outward current. In all cells tested (n = 6), dopamine produced a 15 ± 2% (mean ± SEM) decrease in the peak of the Na+ current activated during voltage jumps from −72 to −47 mV, and SCH-23390 restored this current to 100 ± 3% (mean ± SEM) of the control amplitude. Moreover, we found no marked difference in the test voltages that elicited minimal and maximum increases in Na+ current and I h amplitude at the beginning (Fig. 9 A1) and end (Fig. 9 A6) of this recording, indicating that the voltage sensitivity of the Na+ current and I h had not significantly drifted.
Figure 9.

Reduction of voltage-gated Na+ current by D1-type dopamine receptor activation. Ruptured-patch configuration at 34°C; voltage-clamp mode with no leak subtraction. Currents activated in a single cell by 4 ms depolarizations (A) and 1 s hyperpolarizations (B). Holding potential was −72 mV. Test potentials were −57 mV and −47 mV to activate voltage-gated Na+ current (A, gray and black traces, respectively) and −77, −92, and −107 mV to activate I h (B). Stimulus timing and polarity are shown by steps above current traces. The triangles at left show zero-current level for all traces in each row. As labeled above current traces in A, solution superfused over cell was changed from control (A1, B1) to 3 mm Cs+ (A2, B2), 6 μm dopamine and 3 mm Cs+ (A3, B3), 6 μm dopamine, 3 mm Cs+, and 5 μm SCH-23390 (A4, B4), 6 μm dopamine, 3 mm Cs+, 5 μm SCH-23390, and 1 μm TTX (A5, B5), and control (A6, B6). Peak amplitude of depolarization-activated Na+ current at −47 mV in dopamine (A3, black trace) is 10% smaller than in control (A1). This reduction was reversed by SCH-23390 (A4). The depolarization-activated current was blocked by 1 μm TTX (A5), leaving small uncompensated capacitive inward and no outward current. This TTX block, and the block of I h by Cs+, were reversed by washing with control solution (A6, B6). The activation threshold and increase in Na+ current by the increment in step depolarizations, and the increase in I h by the increment in step hyperpolarizations, were similar at the beginning and end of this recording. C, Na+ current amplitudes of all cells tested (n = 6) as in A and B. Na+ current peak amplitude normalized to value in Cs+ (left) after reduction by dopamine (middle) and recovery in SCH-23390 (right) while I h was suppressed. The mean in Cs+ differed significantly from that in Cs+ plus dopamine (*p < 0.0005, paired t test).
Nanomolar tetrodotoxin reduces spiking
The degree to which dopamine reduced Na+ current amplitude in these cells resembles that reported for other preparations (Cantrell and Catterall, 2001). Determining how dopamine reduces Na+ current will require studies beyond the findings presented here, given the complexity and variety of effects found in other preparations (Cantrell and Catterall, 2001; Carr et al., 2003; Hayashida and Ishida, 2004). Nevertheless, to further examine the possibility that decreases in Na+ current of the magnitude produced by dopamine can decrease spiking, we tested the effect of TTX on spiking at concentrations that partially reduce the whole-cell Na+ current amplitude. Figure 10 shows the depolarization-activated inward current and spikes in a single cell in voltage- and current-clamp modes, respectively, during the application of control (A1, B1), 5 nm TTX (A2, B2), and control (A3, B3) solutions. These records show that 5 nm TTX reversibly inhibits approximately the same amount of inward current as we found here with dopamine (Fig. 9) with a comparable inhibition of spiking. Similar effects were obtained in all of the cells tested this way (n = 3), with 4–5 nm TTX reducing the peak of the inward current by 18 ± 3% (mean ± SEM) and reducing spikes in all cases as shown by the traces in the middle of Figure 10 B; washing with control solution restored the inward current amplitude to 100 ± 1% (mean ± SEM) of the values recorded before TTX (Fig. 10 C). The partial reduction of current by TTX at the concentration used here is consistent with results reported previously (Hidaka and Ishida, 1998).
Figure 10.

Reduction of voltage-gated Na+ current and spiking by nanomolar tetrodotoxin. Recording mode, conditions, and figure format are as in Figure 8. A, Voltage-gated Na+ current (without leak subtraction) and spikes elicited in a single ganglion cell by depolarizations in voltage- and current-clamp modes, respectively. Current activated by voltage jump from −72 to −47 mV as solution superfused over the cell is changed from control (A1) to 5 nm TTX (A2) and then control again (A3). The triangle is positioned at zero current level. The dashed horizontal line at peak of control current highlights partial reduction of current amplitude by TTX and full recovery during wash. B, Spikes then elicited in the same cell by constant current injections (10 and 30 pA) as solution superfused over the cell is changed from control (B1) to 5 nm TTX (B2) and control (B3). At this concentration, and as seen during the response to dopamine in other cells, TTX reversibly reduced peak current amplitude by 14%, raised spike threshold (viz., abolished spiking elicited by smallest current injections), and curtailed spiking elicited by larger current injections (bottom trace, middle column). The triangles are positioned at zero voltage level for all traces in each row. C plots mean (solid bar) and SEM (error bar) of peak inward current during microperfusion of control solution, TTX (4–5 nm), and after wash with control solution, for all cells tested (n = 3). The means in control and TTX differed significantly (*p < 0.0001, paired t test).
Discussion
This study provides new information about the dopamine sensitivity of rat retinal ganglion cells, how dopamine modulates excitability in these cells, and effects of D1-type receptor activation on spike number versus timing.
Anatomy
Our D1a receptor visualizations are the first to identify immunopositive somata in the ganglion cell layer as ganglion cells. Because rat retinas contain more than a dozen morphological types of ganglion cell (Huxlin and Goodchild, 1997; Sun et al., 2002), we did not attempt the large-scale combinations of immunohistochemistry, dopamine response testing, intracellular dye injections, and dendritic analyses that would be needed to specify which types bear functional dopamine receptors (Rockhill et al., 2002). However, we observed immunopositivity in large numbers of dextran-filled somata in flat mounts and in rows of adjacent somata in vertical sections. As would also be expected if ganglion cells generally express dopamine receptors, we found D1a receptor-like immunoreactivity in somata of different diameters and in dendrites extending into different halves of the inner plexiform layer. These results are consistent with previous findings that dopamine alters light responses of on- and off-center ganglion cells (Thier and Alder, 1984; Jensen and Daw, 1986), and that some anti-D1 receptor antibodies bind to multiple sublayers of the inner plexiform layer (Veruki and Wässle, 1996; Müller et al., 2003). Having identified most, if not all, of the immunopositive somata in the ganglion cell layer as ganglion cells, and having used a dextran large enough to avoid dye coupling between ganglion and amacrine cells (Vaney, 1991), our results indicate that displaced amacrine cells (Perry, 1981) do not account for the somata we stained.
Dopaminergic modulation of spikes
During constant-current injections, dopamine, SKF-81297, and SKF-38393 promoted spike accommodation, decreased spike amplitude, and increased spike width. This is consistent with inhibitory effects of dopamine in other preparations, including striatum (Schiffmann et al., 1995), hippocampus (Stanzione et al., 1984; Cantrell and Catterall, 2001), prefrontal cortex (Gulledge and Jaffe, 1998), and fish retinal ganglion cells (Vaquero et al., 2001). Because D1-type receptor agonists accelerated accommodation during constant current injections, we wondered whether they suppress spiking under other conditions. When tested with fluctuating current injections, these agonists decreased spike number, and the timing of the remaining spikes was unaltered (Figs. 5, 6). To our knowledge, this is a novel effect of dopamine receptor activation. At the same time, this is reminiscent of modulation by two other neurotransmitters: reduction of spike number by a GABAA-type conductance in somatosensory cortical neurons (Tateno and Robinson, 2006) and increases in spike number of visual cortical neurons by acetylcholine (Tang et al., 1997), both without changes in jitter. Because these are effects on excitability, they could adjust sensitivity to changes in inputs. For example, because increased ambient illumination increases intraretinal dopamine release (Witkovsky and Dearry, 1991), the ability of dopamine to reduce excitability may be one of several mechanisms for avoiding saturation of ganglion cell light responses (Vaquero et al., 2001). A second advantage of the dopamine effect we describe here is that the ability of the remaining spikes to transfer information would be preserved, especially where spike timing and interspike intervals are critical (Usrey et al., 1998; Meister and Berry, 1999). A recent study suggests that similar needs may be filled during motion adaptation, in which spike number decreases during saccades without changing jitter (Heitwerth et al., 2005).
Another reason we tested whether dopamine receptor activation alters spike jitter is that light adaptation reduces spike jitter in ganglion cells (Lennie, 1981), and dopamine contributes to retinal light adaptation (Witkovsky and Dearry, 1991). The spike timing jitter we found in isolated cells in the presence and absence of SKF-81297 is <10 ms. Because this resembles the jitter in light-adapted ganglion cells in situ (Berry et al., 1997), our results are consistent with the possibility that increases in jitter during dark adaptation reflect jitter in signals these cells receive.
Dopaminergic modulation of voltage-gated currents
Previous studies of mammalian retinal ganglion cells attributed effects of dopamine receptor activation to changes in presynaptic inputs (Thier and Alder, 1984), electrical coupling (Mills et al., 2007), and I h (Chen and Yang, 2007). Other studies have shown that dopamine modulates several voltage-gated currents in neurons [e.g., Na+ (Cantrell and Catterall, 2001); low-threshold Ca2+ (Pfeiffer-Linn and Lasater, 1993); high-threshold Ca2+ (Cardozo and Bean, 1995); slowly inactivating K+ (Dong and White, 2003); A-current (Nisenbaum et al., 1998)], that mammalian retinal ganglion cells possess all of these currents (Ishida, 2004), and that SKF-38393 decreases voltage-gated Na+ current in fish retinal ganglion cells (Hayashida and Ishida, 2004). Our observation that the rates of rise and repolarization of individual spikes slow down suggests that inward and outward currents change. Consistent with the former, and with effects on Na+ current amplitude and spiking in other preparations (Cantrell and Catterall, 2001), dopamine reduced a TTX-sensitive inward current (Fig. 9). We suspect the slowed repolarization results from decreases in outward K+ current. However, ganglion cells appear to have as many as five different types of outward K+ current (Ishida, 2004), and protocols for selectively activating these currents are not available. This prevented us from identifying which (if any) K+ currents are modulated by dopamine, as opposed to the alternative possibility the reduced spike amplitudes we observed resulted in reduced K+ current activation and therefore slower repolarization.
We do not know why dopamine did not alter spiking in the presence of ZD-7288 in a recent study (Chen and Yang, 2007), whereas we found that dopamine inhibits spikes after blocking I h with Cs+ (Fig. 8). One possibility is that dopamine inhibited spiking primarily by reducing Na+ current in cells we recorded from, and by increasing I h in cells studied by Chen and Yang (2007). One factor may be that some rats in the latter study were younger than P20 (i.e., of ages at which cAMP shifts I h voltage sensitivity in some neurons more than in older rats) (Surges et al., 2006). The significance of this is unclear, however, because before P20, rat retinal dopaminergic interneurons are not fully developed (Kato et al., 1980; Witkovsky et al., 2005) and their tyrosine hydroxylase is not as light-responsive as in adults (Cohen and Neff, 1982; Morgan and Kamp, 1982). A second possibility is that ZD-7288 may affect currents beside I h in ganglion cells, especially as ZD-7288 can block glutamate-gated postsynaptic current (Chen, 2004) and voltage-gated calcium current (Sánchez-Alonso et al., 2008). Although Cs+ is also not entirely specific for I h (Hagiwara et al., 1976), it blocked I h without affecting Na+ current amplitude and without precluding Na+ current modulation and spike inhibition by dopamine (Figs. 8, 9). Third, some cells might not respond to dopamine. Guenther et al. (1994) originally found that young rat retinal ganglion cells are unaffected by dopamine; a small fraction of the somata we backfilled with fluorophore-coupled dextran did not display D1a receptor immunoreactivity (Fig. 4), and we encountered ganglion cells that did not respond to dopamine (results not illustrated). Whether these results apply to the cells in Figure 6 of the study by Chen and Yang (2007) is unclear. Although dopamine did not alter spiking after preincubation in ZD-7288, effects of dopamine on spiking in these cells before the application of ZD-7288 were not shown.
Receptor activation
Dopamine is released in rat retina by cells arborizing primarily in the most distal sublayer of the inner plexiform layer, in distally extending processes, and in proximal, broadly meandering processes (Nguyen-Legros et al., 1981; Ehinger, 1983; Voigt and Wässle, 1987; Bjelke et al., 1996; Witkovsky et al., 2008). Previous studies have inferred that this dopamine activates receptors in cells as far from the dopamine-releasing sites (Puopolo et al., 2001) as the pigmented epithelium and retinal ganglion cells (Piccolino et al., 1987; Dearry and Burnside, 1989; Witkovsky et al., 1993; Veruki and Wässle, 1996). Although this diffuse release makes it unlikely that dopamine encodes fine spatial detail in light falling on retinas (Ehinger, 1983), it could facilitate feedforward control of the relatively large number of ganglion cells in which we have visualized dopamine receptors by the relatively small number of dopamine-releasing cells found in mammalian retinas (Masland, 1988). This is in addition to modulatory effects known to be exerted by dopamine on photoreceptor, bipolar, horizontal, and amacrine cells (Hampson et al., 1992; Feigenspan and Bormann, 1994; He et al., 2000; Xia and Mills, 2004; Zhang et al., 2007; Ribelayga et al., 2008). For that matter, comparison with responses in other species (Piccolino et al., 1984; Witkovsky et al., 1988; Dowling, 1991; Heidelberger and Matthews, 1994; Stella and Thoreson, 2000; Vaquero et al., 2001; Ribelayga and Mangel, 2003; Ichinose and Lukasiewicz, 2007) suggests that dopaminergic modulation of these cell types is a general feature of retinas.
The results we have presented here provide direct evidence that D1-type receptors are available to mediate effects of dopamine on mammalian ganglion cells and imply that, under conditions of illumination known to alter signal transmission and network properties via dopamine (Hampson et al., 1992; Krizaj et al., 1998; Manglapus et al., 1999; Ichinose and Lukasiewicz, 2007; Mills et al., 2007; Zhang et al., 2007; Ribelayga et al., 2008), dopamine-induced changes in ganglion cell excitability will concomitantly occur. Accounting for dopamine effects on the spike output of retinas in additional detail will therefore require weighing and integrating the relative contribution of the large number of effects now known.
Footnotes
This work was supported by National Institutes of Health Grant EY08120 (A.T.I.), National Eye Institute Core Grant P30 EY12576, and a Research Award from the Plum Foundation, as well as by a departmental grant to the Department of Ophthalmology from Research to Prevent Blindness, Inc. (New York, NY). C.V.R. was partially supported by a postdoctoral fellowship of the Spanish Ministry of Education and Science EX2005-0745 (BOE14-10-2004). T.W.S. was supported by a National Institutes of Health Research Supplement to Promote Diversity in Health-Related Research.
References
- Ariano MA, Kang HC, Haugland RP, Sibley DR. Multiple fluorescent ligands for dopamine receptors. II. Visualization in neural tissues. Brain Res. 1991;547:208–222. doi: 10.1016/0006-8993(91)90964-w. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Bao S, Chan VT, Merzenich MM. Cortical remodelling induced by activity of ventral tegmental dopamine neurons. Nature. 2001;412:79–83. doi: 10.1038/35083586. [DOI] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Behrens UD, Wagner HJ. Localization of dopamine D1-receptors in vertebrate retinae. Neurochem Int. 1995;27:497–507. doi: 10.1016/0197-0186(95)00044-9. [DOI] [PubMed] [Google Scholar]
- Berry MJ, Warland DK, Meister M. The structure and precision of retinal spike trains. Proc Natl Acad Sci U S A. 1997;94:5411–5416. doi: 10.1073/pnas.94.10.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissière S, Humeau Y, Lüthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Bjelke B, Goldstein M, Tinner B, Andersson C, Sesack SR, Steinbusch HW, Lew JY, He X, Watson S, Tengroth B, Fuxe K. Dopaminergic transmission in the rat retina: evidence for volume transmission. J Chem Neuroanat. 1996;12:37–50. doi: 10.1016/s0891-0618(96)00176-7. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA, Xin D. A comparison of receptive-field and tracer-coupling size of amacrine and ganglion cells in the rabbit retina. Vis Neurosci. 1997;14:1153–1165. doi: 10.1017/s0952523800011846. [DOI] [PubMed] [Google Scholar]
- Brown JH, Makman MH. Stimulation by dopamine of adenylate cyclase in retinal homogenates and of adenosine-3′:5′-cyclic monophosphate formation in intact retina. Proc Natl Acad Sci U S A. 1972;69:539–543. doi: 10.1073/pnas.69.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé I, Dumartin B, Le Moine C, Begueret J, Bloch B. Ontogeny of the D1 dopamine receptor in the rat striatonigral system: an immunohistochemical study. Eur J Neurosci. 1995;7:714–722. doi: 10.1111/j.1460-9568.1995.tb00675.x. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol. 1995;74:1137–1148. doi: 10.1152/jn.1995.74.3.1137. [DOI] [PubMed] [Google Scholar]
- Carr DB, O'Donnell P, Card JP, Sesack SR. Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci. 1999;19:11049–11060. doi: 10.1523/JNEUROSCI.19-24-11049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA, Surmeier DJ. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- Chen C. ZD7288 inhibits postsynaptic glutamate receptor-mediated responses at hippocampal perforant path-granule cell synapses. Eur J Neurosci. 2004;19:643–649. doi: 10.1111/j.0953-816x.2003.03174.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Yang XL. Hyperpolarization-activated cation current is involved in modulation of the excitability of rat retinal ganglion cells by dopamine. Neuroscience. 2007;150:299–308. doi: 10.1016/j.neuroscience.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Chen L, Yu YC, Zhao JW, Yang XL. Inwardly rectifying potassium channels in rat retinal ganglion cells. Eur J Neurosci. 2004;20:956–964. doi: 10.1111/j.1460-9568.2004.03553.x. [DOI] [PubMed] [Google Scholar]
- Cohen J, Neff NH. Retinal amacrine cell system tyrosine hydroxylase: the development of responsiveness to light and neuroleptic drugs. Dev Brain Res. 1982;3:160–163. doi: 10.1016/0165-3806(82)90086-4. [DOI] [PubMed] [Google Scholar]
- Dacey DM, Peterson BB, Robinson FR, Gamlin PD. Fireworks in the primate retina: in vitro photodynamics reveals diverse LGN-projecting ganglion cell types. Neuron. 2003;37:15–27. doi: 10.1016/s0896-6273(02)01143-1. [DOI] [PubMed] [Google Scholar]
- Dearry A, Burnside B. Light-induced dopamine release from teleost retinas acts as a light-adaptive signal to the retinal pigment epithelium. J Neurochem. 1989;53:870–878. doi: 10.1111/j.1471-4159.1989.tb11785.x. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Rudolph M, Fellous JM, Sejnowski TJ. Fluctuating synaptic conductances recreate in vivo-like activity in neocortical neurons. Neuroscience. 2001;107:13–24. doi: 10.1016/s0306-4522(01)00344-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Copenhagen DR. The contribution of NMDA and non-NMDA receptors to the light-evoked input-output characteristics of retinal ganglion cells. Neuron. 1993;11:725–738. doi: 10.1016/0896-6273(93)90082-3. [DOI] [PubMed] [Google Scholar]
- Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci. 2003;23:2686–2695. doi: 10.1523/JNEUROSCI.23-07-02686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling JE. Retinal neuromodulation: the role of dopamine. Vis Neurosci. 1991;7:87–97. doi: 10.1017/s0952523800010968. [DOI] [PubMed] [Google Scholar]
- Doyle CA, Maxwell DJ. Direct catecholaminergic innervation of spinal dorsal horn neurons with axons ascending the dorsal columns in cat. J Comp Neurol. 1993;331:434–444. doi: 10.1002/cne.903310312. [DOI] [PubMed] [Google Scholar]
- Ehinger B. Functional role of dopamine in the retina. Prog Retin Res. 1983;2:213–232. [Google Scholar]
- Ennis M, Zhou FM, Ciombor KJ, Aroniadou-Anderjaska V, Hayar A, Borrelli E, Zimmer LA, Margolis F, Shipley MT. Dopamine D2 receptor-mediated presynaptic inhibition of olfactory nerve terminals. J Neurophysiol. 2001;86:2986–2997. doi: 10.1152/jn.2001.86.6.2986. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Bormann J. Facilitation of GABAergic signaling in the retina by receptors stimulating adenylate cyclase. Proc Natl Acad Sci U S A. 1994;91:10893–10897. doi: 10.1073/pnas.91.23.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood-Walker SM, Hope PJ, Mitchell R. Antinociceptive actions of descending dopaminergic tracts on cat and rat dorsal horn somatosensory neurones. J Physiol. 1988;399:335–348. doi: 10.1113/jphysiol.1988.sp017084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Huang Y-Y, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:9015–9019. doi: 10.1073/pnas.86.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther E, Wilsch V, Zrenner E. Inhibitory action of haloperidol, spiperone and SCH23390 on calcium currents in rat retinal ganglion cells. Neuroreport. 1994;5:1373–1376. [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB. Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci. 1998;18:9139–9151. doi: 10.1523/JNEUROSCI.18-21-09139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustincich S, Feigenspan A, Wu DK, Koopman LJ, Raviola E. Control of dopamine release in the retina: a transgenic approach to neural networks. Neuron. 1997;18:723–736. doi: 10.1016/s0896-6273(00)80313-x. [DOI] [PubMed] [Google Scholar]
- Häggendal J, Malmfors T. Identification and cellular localization of the catecholamines in the retina and the choroid of the rabbit. Acta Physiol Scand. 1965;64:58–66. doi: 10.1111/j.1748-1716.1965.tb04153.x. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Miyazaki S, Rosenthal NP. Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish. J Gen Physiol. 1976;67:621–638. doi: 10.1085/jgp.67.6.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. J Neurosci. 1992;12:4911–4922. doi: 10.1523/JNEUROSCI.12-12-04911.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida Y, Ishida AT. Dopamine receptor activation can reduce voltage-gated Na+ current by modulating both entry into and recovery from inactivation. J Neurophysiol. 2004;92:3134–3141. doi: 10.1152/jn.00526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida Y, Partida GJ, Ishida AT. Dissociation of retinal ganglion cells without enzymes. J Neurosci Methods. 2004;137:25–35. doi: 10.1016/j.jneumeth.2004.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Weiler R, Vaney DI. Endogenous dopaminergic regulation of horizontal cell coupling in the mammalian retina. J Comp Neurol. 2000;418:33–40. doi: 10.1002/(sici)1096-9861(20000228)418:1<33::aid-cne3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Dopamine enhances Ca2+ responses in synaptic terminals of retinal bipolar neurons. Neuroreport. 1994;5:729–732. doi: 10.1097/00001756-199402000-00018. [DOI] [PubMed] [Google Scholar]
- Heitwerth J, Kern R, van Hateren JH, Egelhaaf M. Motion adaptation leads to parsimonious encoding of natural optic flow by blowfly motion vision system. J Neurophysiol. 2005;94:1761–1769. doi: 10.1152/jn.00308.2005. [DOI] [PubMed] [Google Scholar]
- Hidaka S, Ishida AT. Voltage-gated Na+ current availability after step- and spike-shaped conditioning depolarizations of retinal ganglion cells. Pflugers Arch. 1998;436:497–508. doi: 10.1007/s004240050664. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Hokoç JN, Mariani AP. Tyrosine hydroxylase immunoreactivity in the rhesus monkey retina reveals synapses from bipolar cells to dopaminergic amacrine cells. J Neurosci. 1987;7:2785–2793. doi: 10.1523/JNEUROSCI.07-09-02785.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Vincent JD, Lledo PM. Dopamine depresses synaptic inputs into the olfactory bulb. J Neurophysiol. 1999;82:1082–1085. doi: 10.1152/jn.1999.82.2.1082. [DOI] [PubMed] [Google Scholar]
- Huang Q, Zhou D, Chase K, Gusella JF, Aronin N, DiFiglia M. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc Natl Acad Sci U S A. 1992;89:11988–11992. doi: 10.1073/pnas.89.24.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxlin KR, Goodchild AK. Retinal ganglion cells in the albino rat: revised morphological classification. J Comp Neurol. 1997;385:309–323. [PubMed] [Google Scholar]
- Ichinose T, Lukasiewicz PD. Ambient light regulates sodium channel activity to dynamically control retinal signaling. J Neurosci. 2007;27:4756–4764. doi: 10.1523/JNEUROSCI.0183-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida AT. Retinal ganglion cell excitability. In: Chalupa LM, Werner JS, editors. The visual neurosciences. Cambridge, MA: MIT; 2004. pp. 422–450. [Google Scholar]
- Jensen RJ, Daw NW. Effects of dopamine and its agonists and antagonists on the receptive field properties of ganglion cells in the rabbit retina. Neuroscience. 1986;17:837–855. doi: 10.1016/0306-4522(86)90049-7. [DOI] [PubMed] [Google Scholar]
- Karpa KD, Lidow MS, Pickering MT, Levenson R, Bergson C. N-linked glycosylation is required for plasma membrane localization of D5, but not D1, dopamine receptors in transfected mammalian cells. Mol Pharmacol. 1999;56:1071–1078. doi: 10.1124/mol.56.5.1071. [DOI] [PubMed] [Google Scholar]
- Kato S, Nakamura T, Negishi K. Postnatal development of dopaminergic cells in the rat retina. J Comp Neurol. 1980;191:227–236. doi: 10.1002/cne.901910206. [DOI] [PubMed] [Google Scholar]
- Keminer O, Peters R. Permeability of single nuclear pores. Biophys J. 1999;77:217–228. doi: 10.1016/S0006-3495(99)76883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Takacs KN, Ficker E, Kunze DL. Dopamine modulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol. 2002;88:2736–2744. doi: 10.1152/jn.00224.2002. [DOI] [PubMed] [Google Scholar]
- Krizaj D, Gábriel R, Owen WG, Witkovsky P. Dopamine D2 receptor-mediated modulation of rod-cone coupling in the Xenopus retina. J Comp Neurol. 1998;398:529–538. [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Ishida AT. Ih without Kir in adult rat retinal ganglion cells. J Neurophysiol. 2007;97:3790–3799. doi: 10.1152/jn.01241.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennie P. The physiological basis of variations in visual latency. Vision Res. 1981;21:815–824. doi: 10.1016/0042-6989(81)90180-2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lasater EM. Calcium currents in turtle retinal ganglion cells. II. Dopamine modulation via a cyclic AMP-dependent mechanism. J Neurophysiol. 1994;71:743–752. doi: 10.1152/jn.1994.71.2.743. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, de Curtis M, Wanke E. Modalities of distortion of physiological voltage signals by patch-clamp amplifiers: a modeling study. Biophys J. 1998;74:831–842. doi: 10.1016/S0006-3495(98)74007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Reliability of spike timing in neocortical neurons. Science. 1995;268:1503–1506. doi: 10.1126/science.7770778. [DOI] [PubMed] [Google Scholar]
- Manglapus MK, Iuvone PM, Underwood H, Pierce ME, Barlow RB. Dopamine mediates circadian rhythms of rod-cone dominance in the Japanese quail retina. J Neurosci. 1999;19:4132–4141. doi: 10.1523/JNEUROSCI.19-10-04132.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masland RH. Amacrine cells. Trends Neurosci. 1988;11:405–410. doi: 10.1016/0166-2236(88)90078-1. [DOI] [PubMed] [Google Scholar]
- Meister M, Berry MJ., 2nd The neural code of the retina. Neuron. 1999;22:435–450. doi: 10.1016/s0896-6273(00)80700-x. [DOI] [PubMed] [Google Scholar]
- Mills SL, Xia XB, Hoshi H, Firth SI, Rice ME, Frishman LJ, Marshak DW. Dopaminergic modulation of tracer coupling in a ganglion-amacrine cell network. Vis Neurosci. 2007;24:593–608. doi: 10.1017/S0952523807070575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan WW, Kamp CW. Postnatal development of the light response of the dopaminergic neurons in the rat retina. J Neurochem. 1982;39:283–285. doi: 10.1111/j.1471-4159.1982.tb04737.x. [DOI] [PubMed] [Google Scholar]
- Müller F, Scholten A, Ivanova E, Haverkamp S, Kremmer E, Kaupp UB. HCN channels are expressed differentially in retinal bipolar cells and concentrated at synaptic terminals. Eur J Neurosci. 2003;17:2084–2096. doi: 10.1046/j.1460-9568.2003.02634.x. [DOI] [PubMed] [Google Scholar]
- Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naïve and optic nerve injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Legros J, Berger B, Vigny A, Alvarez C. Tyrosine hydroxylase-like immunoreactive interplexiform cells in the rat retina. Neurosci Lett. 1981;27:255–259. doi: 10.1016/0304-3940(81)90439-0. [DOI] [PubMed] [Google Scholar]
- Nguyen-Legros J, Simon A, Caillé I, Bloch B. Immunocytochemical localization of dopamine D1 receptors in the retina of mammals. Vis Neurosci. 1997;14:545–551. doi: 10.1017/s0952523800012207. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Mermelstein PG, Wilson CJ, Surmeier DJ. Related selective blockade of a slowly inactivating potassium current in striatal neurons by (+/−) 6-chloro-APB hydrobromide (SKF82958) Synapse. 1998;29:213–224. doi: 10.1002/(SICI)1098-2396(199807)29:3<213::AID-SYN3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Oi H, Partida GJ, Lee SC, Ishida AT. HCN4-like immunoreactivity in rat retinal ganglion cells. Vis Neurosci. 2008;25:95–102. doi: 10.1017/S095252380808005X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partida GJ, Lee SC, Haft-Candell L, Nichols GS, Ishida AT. DARPP-32-like immunoreactivity in AII amacrine cells of rat retina. J Comp Neurol. 2004;480:251–263. doi: 10.1002/cne.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH. Evidence for an amacrine cell system in the ganglion cell layer of the rat retina. Neuroscience. 1981;6:931–944. doi: 10.1016/0306-4522(81)90174-3. [DOI] [PubMed] [Google Scholar]
- Pfeiffer-Linn C, Lasater EM. Dopamine modulates in a differential fashion T- and L-type calcium currents in bass retinal horizontal cells. J Gen Physiol. 1993;102:277–294. doi: 10.1085/jgp.102.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolino M, Neyton J, Gerschenfeld HM. Decrease of gap junction permeability induced by dopamine and cyclic adenosine 3′:5′-monophosphate in horizontal cells of turtle retina. J Neurosci. 1984;4:2477–2488. doi: 10.1523/JNEUROSCI.04-10-02477.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolino M, Witkovsky P, Trimarchi C. Dopaminergic mechanisms underlying the reduction of electrical coupling between horizontal cells of the turtle retina induced by d-amphetamine, bicuculline, and veratridine. J Neurosci. 1987;7:2273–2284. [PMC free article] [PubMed] [Google Scholar]
- Pirot S, Godbout R, Mantz J, Tassin JP, Glowinski J, Thierry AM. Inhibitory effects of ventral tegmental area stimulation on the activity of prefrontal cortical neurons: evidence for the involvement of both dopaminergic and GABAergic components. Neuroscience. 1992;49:857–865. doi: 10.1016/0306-4522(92)90362-6. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Pourcho RG. Dopaminergic amacrine cells in the cat retina. Brain Res. 1982;252:101–109. doi: 10.1016/0006-8993(82)90982-9. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Hochstetler SE, Gustincich S, Wightman RM, Raviola E. Extrasynaptic release of dopamine in a retinal neuron: activity dependence and transmitter modulation. Neuron. 2001;30:211–225. doi: 10.1016/s0896-6273(01)00274-4. [DOI] [PubMed] [Google Scholar]
- Raymond ID, Vila A, Huynh UC, Brecha NC. Cyan fluorescent protein expression in ganglion and amacrine cells in a thy1-CFP transgenic mouse retina. Mol Vis. 2008;25:1559–1574. [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Guenther E. Developmental changes in voltage-activated potassium currents of rat retinal ganglion cells. Neuroscience. 1999;92:1103–1117. doi: 10.1016/s0306-4522(99)00044-5. [DOI] [PubMed] [Google Scholar]
- Ribelayga C, Mangel SC. Absence of circadian clock regulation of horizontal cell gap junctional coupling reveals two dopamine systems in the goldfish retina. J Comp Neurol. 2003;467:243–253. doi: 10.1002/cne.10927. [DOI] [PubMed] [Google Scholar]
- Ribelayga C, Cao Y, Mangel SC. The circadian clock in the retina controls rod-cone coupling. Neuron. 2008;59:790–801. doi: 10.1016/j.neuron.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DW, Pierrefiche O, Lalley PM, Polder HR. Voltage-clamp analysis of neurons within deep layers of the brain. J Neurosci Methods. 1996;67:121–123. [PubMed] [Google Scholar]
- Rockhill RL, Daly FJ, MacNeil MA, Brown SP, Masland RH. The diversity of ganglion cells in a mammalian retina. J Neurosci. 2002;22:3831–3843. doi: 10.1523/JNEUROSCI.22-09-03831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado H, Tecuapetla F, Perez-Rosello T, Perez-Burgos A, Perez-Garci E, Galarraga E, Bargas J. A reconfiguration of CaV2 Ca2+ channel current and its dopaminergic D2 modulation in developing neostriatal neurons. J Neurophysiol. 2005;94:3771–3787. doi: 10.1152/jn.00455.2005. [DOI] [PubMed] [Google Scholar]
- Sánchez-Alonso JL, Halliwell JV, Colino A. ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci Lett. 2008;439:275–280. doi: 10.1016/j.neulet.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltage-gated sodium current in rat striatal neurones through a protein kinase A. J Physiol. 1995;483:95–107. doi: 10.1113/jphysiol.1995.sp020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorderet M, Nowak JZ. Retinal dopamine D1 and D2 receptors: characterization by binding or pharmacological studies and physiological functions. Cell Mol Neurobiol. 1990;10:303–325. doi: 10.1007/BF00711177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM. Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci. 1994;14:88–106. doi: 10.1523/JNEUROSCI.14-01-00088.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzione P, Calabresi P, Mercuri N, Bernardi G. Dopamine modulates CA1 hippocampal neurons by elevating the threshold for spike generation: an in vitro study. Neuroscience. 1984;13:1105–1116. doi: 10.1016/0306-4522(84)90291-4. [DOI] [PubMed] [Google Scholar]
- Stella SL, Jr, Thoreson WB. Differential modulation of rod and cone calcium currents in tiger salamander retina by D2 dopamine receptors and cAMP. Eur J Neurosci. 2000;12:3537–3548. doi: 10.1046/j.1460-9568.2000.00235.x. [DOI] [PubMed] [Google Scholar]
- Sun W, Li N, He S. Large-scale morphological survey of rat retinal ganglion cells. Vis Neurosci. 2002;19:483–493. doi: 10.1017/s0952523802194107. [DOI] [PubMed] [Google Scholar]
- Surges R, Brewster AL, Bender RA, Beck H, Feuerstein TJ, Baram TZ. Regulated expression of HCN channels and cAMP levels shape the properties of the h current in developing rat hippocampus. Eur J Neurosci. 2006;24:94–104. doi: 10.1111/j.1460-9568.2006.04880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Kitai ST. State-dependent regulation of neuronal excitability by dopamine. Jpn J Psychopharmacol. 1997;17:105–110. [PubMed] [Google Scholar]
- Sutor B, Grimm C, Polder HR. Voltage-clamp-controlled current-clamp recordings from neurons: an electrophysiological technique enabling the detection of fast potential changes at preset holding potentials. Pflugers Arch. 2003;446:133–141. doi: 10.1007/s00424-003-1008-0. [DOI] [PubMed] [Google Scholar]
- Tang AC, Bartels AM, Sejnowski TJ. Effects of cholinergic modulation on responses of neocortical neurons to fluctuating input. Cereb Cortex. 1997;7:502–509. doi: 10.1093/cercor/7.6.502. [DOI] [PubMed] [Google Scholar]
- Tateno T, Robinson HP. Rate coding and spike-time variability in cortical neurons with two types of threshold dynamics. J Neurophysiol. 2006;95:2650–2663. doi: 10.1152/jn.00683.2005. [DOI] [PubMed] [Google Scholar]
- Thier P, Alder V. Action of iontophoretically applied dopamine on cat retinal ganglion cells. Brain Res. 1984;292:109–121. doi: 10.1016/0006-8993(84)90895-3. [DOI] [PubMed] [Google Scholar]
- Tran VT, Dickman M. Differential localization of dopamine D1 and D2 receptors in rat retina. Invest Ophthalmol Vis Sci. 1992;33:1620–1626. [PubMed] [Google Scholar]
- Uhlenbeck GE, Ornstein LS. On the theory of the Brownian motion. Phys Rev. 1930;36:823–841. [Google Scholar]
- Usrey WM, Reppas JB, Reid RC. Paired-spike interactions and synaptic efficacy of retinal inputs to the thalamus. Nature. 1998;395:384–387. doi: 10.1038/26487. [DOI] [PubMed] [Google Scholar]
- Vaney DI. Many diverse types of retinal neurons show tracer coupling when injected with biocytin or Neurobiotin. Neurosci Lett. 1991;125:187–190. doi: 10.1016/0304-3940(91)90024-n. [DOI] [PubMed] [Google Scholar]
- Vaquero CF, Pignatelli A, Partida GJ, Ishida AT. A dopamine- and protein kinase A-dependent mechanism for network adaptation in retinal ganglion cells. J Neurosci. 2001;21:8624–8635. doi: 10.1523/JNEUROSCI.21-21-08624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Wässle H. Immunohistochemistry localization of dopamine D1 receptors in rat retina. Eur J Neurosci. 1996;8:2286–2297. doi: 10.1111/j.1460-9568.1996.tb01192.x. [DOI] [PubMed] [Google Scholar]
- Voigt T, Wässle H. Dopaminergic innervation of AII amacrine cells in mammalian retina. J Neurosci. 1987;7:4115–4128. doi: 10.1523/JNEUROSCI.07-12-04115.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Dearry A. Functional roles of dopamine in the vertebrate retina. Prog Retin Res. 1991;11:247–292. [Google Scholar]
- Witkovsky P, Stone S, Besharse JC. Dopamine modifies the balance of rod and cone inputs to horizontal cells of the Xenopus retina. Brain Res. 1988;449:332–336. doi: 10.1016/0006-8993(88)91048-7. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Nicholson C, Rice ME, Bohmaker K, Meller E. Extracellular dopamine concentration in the retina of the clawed frog, Xenopus laevis . Proc Natl Acad Sci U S A. 1993;90:5667–5671. doi: 10.1073/pnas.90.12.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky P, Arango-Gonzalez B, Haycock JW, Kohler K. Rat retinal dopaminergic neurons: differential maturation of somatodendritic and axonal compartments. J Comp Neurol. 2005;481:352–362. doi: 10.1002/cne.20389. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Gábriel R, Krizaj D. Anatomical and neurochemical characterization of dopaminergic interplexiform processes in mouse and rat retinas. J Comp Neurol. 2008;510:158–174. doi: 10.1002/cne.21784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XB, Mills SL. Gap junctional regulatory mechanisms in the AII amacrine cell of the rabbit retina. Vis Neurosci. 2004;21:791–805. doi: 10.1017/S0952523804045122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Zhou TR, McMahon DG. Functional heterogeneity of retinal dopaminergic neurons underlying their multiple roles in vision. J Neurosci. 2007;27:692–699. doi: 10.1523/JNEUROSCI.4478-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]