

Abstract
Juvenile Polyposis (JP) is an autosomal dominant hamartomatous polyposis syndrome that carries a significant risk for the development of colorectal cancer. Microdeletions of one of the two predisposing genes to JP, BMPR1A, have been associated with a severe form of JP called juvenile polyposis of infancy. Many of these deletions have also been found to contiguously include PTEN, which is the gene responsible for the development of Cowden syndrome. The advent of molecular techniques that localize genomic copy number variations and others that target specific genes such as multiplex-ligation probe analysis has allowed researchers to explore this area further for deletions. Here, we review the literature for microdeletions described on chromosome 10q22-23 in patients with JP and other intestinal polyposis syndromes.
Introduction
Copy number variation (CNV) is the term used to describe genomic structural variations ranging in size from kilobases (kb) to Megabases (Mb). The recent extensive utilization of genome-wide study tools such as comparative genomic hybridization (CGH) has led to the rapid expansion of these studies (1–3). Altering the copy number of dosage-sensitive regions has been well established as a mechanism leading to clinically relevant phenotypes (4), and since the early 1980s, a number microscopic deletion syndromes have been described. Microdeletion of chromosome 15q11.2q12 was identified as a cause of Prader-Willi syndrome, and reports of other disorders that contain variable deletion break-points followed, such as α-thalassemia and Duchenne muscular dystrophy (5–8). Due to the lack of accurate molecular tools that screened the whole genome for CNVs 1–50 kb in size (3, 9), investigators focused their search to previously identified regions of disease-predisposing genes, which enabled localization of smaller CNVs. This approach proved fruitful in identifying CNVs as the cause of a number of common complex traits such as Parkinson’s disease, Alzheimer’s disease and epilepsy (10–13).
Juvenile Polyposis (JP) is an autosomal dominant disorder characterized by the development of multiple hamartomatous polyps of the GI tract (14, 15). JP carries an increased risk of GI malignancy, with a lifetime risk of approximately 50% (16). In 1998, a JP gene was localized to chromosome 18q21 by linkage analysis and germline mutations were found in the tumor suppressor gene SMAD4 in several families (17, 18). In 2001, another JP locus was found in four kindreds mapping to chromosome 10q22-23. Germline mutations were identified in the bone morphogenetic protein receptor type 1A (BMPR1A), a receptor involved in the BMP signaling pathway, of which SMAD4 is the intracellular mediator (19). Prior to this, mutations in the tumor suppressor gene PTEN, also located on 10q22-23, about 1Mb telomeric to BMPR1A, had been found in the germline of patients with Cowden Syndrome (CS), in which some patients also develop hamartomatous intestinal polyps (20, 21). Prior to the discovery of BMPR1A as a JP gene, there were 2 reports of JP patients with PTEN germline mutations (22, 23). However, further review of the clinical data suggested these patients might have CS rather than JP, and Eng and Ji concluded that patients with inherited hamartoma syndromes and PTEN mutations should be considered to have CS or Bannayan-Riley-Ruvalcaba syndrome (BRRS) (24). Beside BRRS, it later became evident that PTEN mutations were present in the other distinct hamartomatous syndromes Proteus syndrome and Proteus-like syndrome ((25, 26) and reviewed in (27)).
Due to the phenotypic diversity exhibited by patients with PTEN mutations and the fact that germline single base substitutions and small deletion/insertions in the JP genes BMPR1A and SMAD4 only account for less than half of JP cases (28, 29), alternative means of gene inactivation have been explored. This was largely facilitated by the improvement and increased availability of molecular tools that identify smaller-scale CNVs. Multiplex ligation-dependent probe amplification (MLPA) is one method by which many genomic deletions/duplications have been found in target genes (30–33), and this technique has been particularly useful in the study of JP related CNVs. Here we review the literature for studies that describe chromosome 10q22-23 microdeletions in JP and other intestinal polyposis syndromes.
JP, CS and BRRS
Three subtypes of JP were defined by Sachatello et al. in 1975: 1-Juvenile polyposis coli (JPC); 2-Generalized juvenile polyposis (GJP); and 3-Juvenile polyposis of infancy (JPI)(34). The first two subtypes are distinguished by the location and extent of polyps along the GI tract with JPC having polyps in the colon only and GJP in the colon and upper GI tract. These are thought to be variable manifestations of the same condition since both subtypes can be present in the same families and commonly present in the first two decades of life (35). In contrast, JPI is characterized by a very early age of onset (often before 2 years of age) and a substantially more severe clinical course due to recurrent gastrointestinal bleeding, diarrhea and exudative enteropathy. JPI has rarely been reported in the literature, and the infrequency of this condition and its high rate of mortality have limited the study of its molecular basis in the past.
CS was first described in 1963 by Lloyd et al. (36). It is autosomal dominant and is characterized by the formation of multiple disorganized benign growths (hamartomas) and an increased risk of benign and malignant tumor development. More than 90% of affected patients manifest the phenotype before 20 years of age and almost all have CS-mucocutaneous involvement (37). Of note, thyroid abnormalities occur in over 50% of cases, 38% have macrocephaly and 40% develop GI hamartomatous polyps. Clinical diagnosis is often challenging, and can be aided by genetic testing (37). BRRS is a rare disorder that also has an autosomal dominant mode of transmission, and affected members manifest with macrocephaly, lipomatosis, angiomatosis, penile abnormalities and occasional intestinal polyps (38). It is estimated that 60% of BRRS cases have germline mutations in PTEN and some cases even overlap with CS from a clinical perspective (25). The correlation between genotype and phenotype in these two syndromes is not fully understood, and there is overlap of mutations suggesting that the two conditions are allelic (39).
Cytogenetic Studies
The first report to describe a germline chromosomal anomaly in a patient with a hamartomatous polyposis syndrome came from Jacoby et al. in 1997 (40). The authors described a 4 year-old boy referred for a 1-year history of hematochezia who had several dozen polyps of typical juvenile histology distributed along the colon on endoscopy. This patient had multiple congenital abnormalities including dysmorphic features, as well as growth and developmental delays which suggested the presence of a chromosomal abnormality. Using a standard Giemsa-banding method, metaphase analysis of peripheral leukocytes revealed an interstitial deletion of the long arm of chromosome 10, with a karyotype of 46,XY,del(10)(q22.3.24.1). This change was de novo, as cytogenetic analysis did not reveal a similar abnormality in either parent. Figure 1 illustrates the locations of the different deletions reported here and in subsequent studies.
Figure 1.
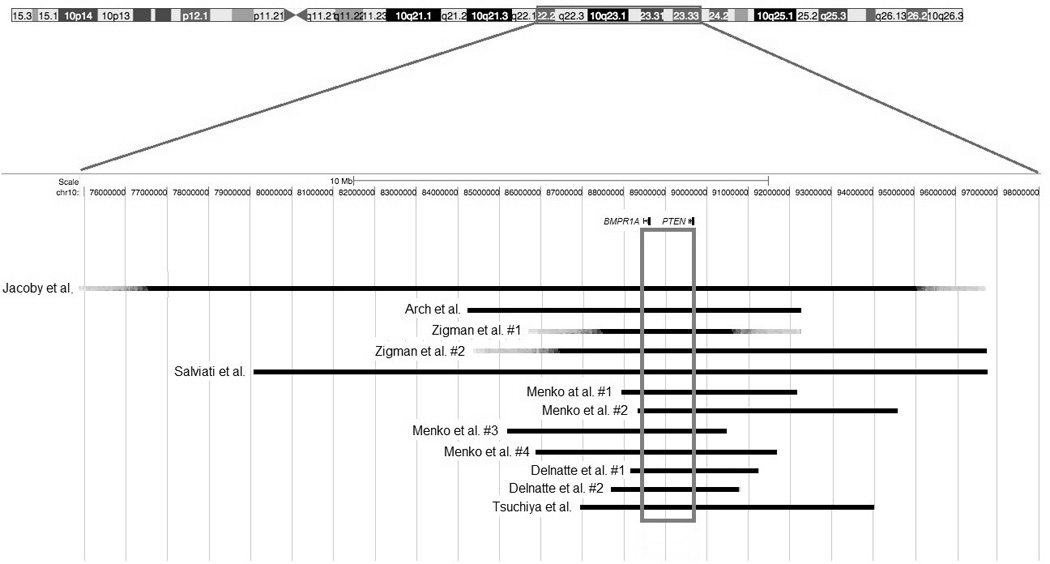
Deletions in chromosome 10q22-23 are associated with a severe form of JP. Deletions are shown in black. Deletions described by Jacoby et al. (40) and Zigman et al. (42) were studied by Giemsa banding only and the exact positions of the breakpoints were therefore not determined. BMPR1A and PTEN are within red boxes with the common area lost in all patients outlined in blue. Numbers next to the author names refer to the individual cases in the order that they were listed in their corresponding primary publications.
Later that year, Arch et al. reported a similar case (41). The authors described an 18-month old boy with a presumed diagnosis of BRRS who had multiple anomalies consistent with that diagnosis, such as remarkable macrosomia that was 7 SD above the age-adjusted mean, 2 lipomas, developmental delay and pseudopapilledema. On endoscopy, multiple hamartomatous polyps that extended throughout the duodenum and small intestine were found, and since polyps in BRRS are usually confined to the distal ileum and colon, the authors proposed that features of Cowden’s syndrome (CS) might overlap in this patient. Standard Giemsa-banding was used for the karyotyping. which revealed a hemizygous deletion of chromosome 10q23.2-q24.1. The authors further demonstrated the absence of PTEN by performing FISH using a bacterial artificial chromosome (BAC) clone specific for that gene. Examination of the parental chromosomes revealed no abnormalities.
Zigman et al. reported 2 additional patients soon afterwards (42). The first patient they described was a male who presented with developmental delay, macrocephaly, a subcutaneous hamartoma and multiple juvenile polyps in the intestine, features thought to be typical of BRRS. The second patient was a girl who additionally had complex congenital cardiac anomalies, as well as clubfoot. Both patients had abnormal karyotypes by Geimsa-staining. The deletion in the first patient was part of an unbalanced translocation with the distal short arm of chromosome 9 (46,XY,der(9)t(9;10)(p24.1;q24.1),der(10)del(10)(q23.2q24.1)t(9;10)(p24.1;q23.2)), and was unbalanced in the second patient (46,XX,del(10)(q23.1q24.2)). Chromatin loss was confirmed in both patients, however the deletion was substantially larger in the second patient (11.6 cM) compared to the first (~1cM). Since the diagnosis of JP was highly suspected in the second patient, the authors concluded that genes responsible for JP and BRRS were within that chromosomal segment. In both patients, the parental karyotypes were normal.
In 1998, Tsuchiya et al. reported another case of a 6-year old boy with rectal bleeding since the age of 2(43). He was found to have multiple juvenile polyps that extended from the duodenum to the rectum. Other clinical features included developmental delay, prominent dysmorphic facies, a large head circumference (>90th percentile) and hyperpigmentation of the mouth and penis. Cytogenetic analysis of Giemsa/Trypsin/Leishman-banded chromosomes revealed a karyotype of 46,XY,del(10)(q23.2q23.33), and the estimated size of the deletion was between 11 and 15 cM by FISH. BRRS was thought to be unlikely in this patient as there was no evidence of lipomas or hemangiomas. Moreover, CS was not confirmed either due to the lack of characteristic findings such as papillomatous skin lesions, thyroid or genitourinary abnormalities. Other family members reportedly had no such signs, and the mother’s karyotype was found to be normal.
In 2005, Delnatte et al. described 4 unrelated cases of JPI who had deletions of both BMPR1A and PTEN (44). All patients presented at an early age (between 1 and 18 months) and had multiple juvenile polyps extending throughout the upper and lower GI tracts. Furthermore, they all had macrocephaly, but hemangiomas were only found in 2 out of the 4. Facial dysmorphism was present in 3 patients and none showed evidence of mental retardation (other features are summarized in table 1). FISH was performed using BAC clones encompassing PTEN and BMPR1A in the first 3 patients, which confirmed loss of this region. In patient 4, quantitative PCR of all BMPR1A and PTEN exons demonstrated the complete loss of both genes. Interestingly, one patient was mosaic for this deletion, exhibiting it in only 17% of examined peripheral lymphocytes. Using CGH and serial microsatellite typings, the deletions in patients 1, 2 and 4 were further characterized and found to be between 1.2 and 2 Mb in length. In two of these patients, genetic testing revealed no abnormalities in either parent (data was not available from the other two patients).
Table 1.
10q deletions in patients with childhood-onset JP. A summary of the studies that describe patients presenting in childhood with JP.
| Author (year) |
Onset | Juvenile Polyps U/L |
Macrocephaly | Additional abnormalities |
Mental/ Developmental retardation |
Karyotype | Further characterization |
|---|---|---|---|---|---|---|---|
| (40) Jacoby et al. (1997) | 3 years | ?/+ | No (2nd percentile, proportionate to size) | Club foot, Tricuspid insufficiency | Yes | 46,XY,del(10)(q22.3q24.1) | |
| (41) Arch et al. (1997) | 18 months | +/+ | Yes (+7 SD) | Lipoma, hemangioma, | Yes | 46,XY,del(10)(q23.2q24.1) | 5.73 Mb, confirmed by Balciuniene et al. (51) |
| (42) Zigman et al. (1997) | Not reported | ?/+ | Yes | Subcutaneous hamartomas | Yes | 46,XY,del(10)(q23.2q24.1) | 1 cM, part of an unbalanced translocation |
| Not reported | ?/+ | Not reported | ASD, VSD, club feet | Yes | 46,XX,del(10)(q23.1q24.2) | 11.6 cM | |
| (43) Tsuchiya et al. (1998) | 2 years | +/+ | Yes (>90th percentile) | Hyperpigmentation multiple areas | Yes | 46,XY,del(10)(q23.2q23.33) | 10–15 cM |
| (44) Delnatte et al. (2006) | 1 month | +/+ | Yes (+2 SD) | Lipoma, hemangioma | No | 46,XX.ish del(10)(q23.2q23.3) | 2Mb by CGH and PCR |
| 2.5 months | +/+ | Yes (+2 SD) | None reported | No | 46,XX.ish del(10)(q23.2q23.3) | 2Mb by CGH and PCR | |
| 3 months | +/+ | Yes (+2.5 SD) | Hemangioma, speckled penis | No | 46,XY.ish del(10)(q23.2q23.3) | Detected in 17% of lymphocytes | |
| 18 months | +/+ | Yes (+4 SD) | ASD VSD | No | 46,XX,t(2;10)(q31;p15) | 1.2 Mb by microsatellite typings and PCR | |
| (45) Salviati et al. (2006) | 5 years | −/+ | No | Hemangioma, ASD, café-au-lait | Yes | 46,XX,del(10)(q22.3q23.33)dn | 12.3 Mb by FISH |
| (46) Menko et al. (2008) | 2 years | +/+ | Yes (+3 SD) | Cleft palate, VSD | Yes | 46,XY.ish del(10)(q23.2q23.31) | 2.88 Mb (SNP array) |
| 18 months | +/+ | Yes (+2 SD) | Kyphosis | Yes | 46,XY.ish del(10)(q23.2q24.31) | 4.26 Mb (SNP array) | |
| 4 years | +/+ | Yes (+2 SD) | VSD | Yes | 46,XX.ish del(10)(q23.2q23.31) | 3.55 Mb (SNP array) | |
| Not reported | Yes (98th percentile) | Kyphosis | Yes | 46,XY.ish del(10)(q23.2q23.31) | 4.01 Mb (SNP array) |
(ASD: Atrial Septal Defect; CGH: Comparative Genomic Hybridization; cM: centimorgan; dn: de novo; ish: in situ hybridization; Mb: Mega-base; SD: Standard deviation; STRP: Single tandem repeat polymorphism; U/L: upper/lower GI tract; VSD: Ventricular Septal Defect.)
These reports gave rise to the hypothesis that the more aggressive JPI is caused by deletion of the 2 contiguous genes known to cause JP, CS and BRRS (BMPR1A and PTEN). However, clinical and molecular data were not always consistent. Features associated with BRRS, such as macrocephaly, facial dysmorphy, speckled penis, and lipomas were not found in all subjects thought to have JPI, and furthermore, Zigman et al. did not find a deletion of BMPR1A in one patient (42). A later report by Salviati et al. (45) argued the notion that a more complex mechanism underlies the development of JPI, and that the phenotypic expression of contiguous BMPR1A and PTEN deletions is variable. The authors reported one patient who had a notably milder phenotype, yet was found to have an interstitial deletion of chromosome 10 that included both genes. Delnatte et al. previously suggested that this was the molecular defect underlying the severity of JPI (44). Salviati’s patient was first evaluated at 3 years of age, had no macrocephaly, only mild developmental delay, mild dysmorphic features, and a small atrial septal defect. Interestingly, high-resolution karyotyping and FISH using BAC clones spanning from10q22 to 10q23 revealed that the deletion was in fact considerably larger than those that had been previously reported (12 Mb versus 2 Mb). This added another layer of complexity, as the size of the deletion did not seem to correlate with the severity of the phenotype.
The most recent study on JPI came in 2008 by Menko et al. (46) in 2008, and provided valuable insight on the phenotypic outcome of these deletions. The authors described 4 new patients with JPI who had 10q23 microdeletions that involved both BMPR1A and PTEN. All 4 cases had macrocephaly, dysmorphic features and other congenital abnormalities. This study was unique in that it combined data from MLPA with FISH and then further characterized the deletions by SNP analysis using the Affymetrix 250k NspI array. The amount of heterozygous loss of DNA ranged between 2.88 and 4.26 Mb, with larger deletions not predicting a more severe course of disease. The authors concluded that the clinical consequences of these molecular abnormalities were fairly heterogeneous.
The Frequency of Large Deletions in JP
The first study to address the frequency of CNV in the causative genes for JP was performed by Aretz et al. in 2007 (29). The authors examined 80 unrelated cases of JP where patient DNA was first subjected to direct sequencing of BMPR1A, SMAD4, PTEN and CDH1. For MLPA analysis, 60 patients were examined (50 did not have any point mutations and another 10 had missense mutations or unspecified variants). DNA from these 60 probands was analyzed by MLPA kit P158 (MRC Holland, Amsterdam, the Netherlands). A total of 6/80 (7.5%) were found to have deletions in SMAD4 and 3/80 (3.8%) had deletions in BMPR1A. One of these latter patients had decreased amplification in the probes for the two first noncoding exons and first exon and another only had the first exon deleted. The third patient had a deletion of the entire genes of BMPR1A and PTEN. Further review of this patient’s history revealed the age at diagnosis was 3, (possibly consistent with JPI), but additional information regarding the patient’s phenotype was not reported.
A similar study by van Hattem et al. examined a well-documented group of 27 JP patients and utilized the same MLPA kit as Aretz et al. (47). To check first for point mutations and small deletions/insertions, direct sequencing was performed and mutations in SMAD4 and BMPR1A were only found in 9 (33.3%) patients. MLPA was subsequently used on the remaining 18 patients, and 4 large hemizygous deletions were found (one including all exons of SMAD4, one in exons 10 and 11 of BMPR1A, and 2 involving all exons of PTEN and BMPR1A). Both patients that had the contiguous deletion of BMPR1A and PTEN at 10q22-23 had typical multiple juvenile polyps, however one of them was reported to have thyroid carcinoma making CS also a potential diagnosis. Other pertinent features and age of onset were not mentioned.
The most recent study searching for large deletions in JP probands came from our lab in 2009 (48). A total of 102 subjects were analyzed, all meeting the clinical criteria set by Jass et al.(49). Direct sequencing identified SMAD4 and BMPR1A mutations in (42/102) 41.2% of patients. The remaining 60 patients were examined by MLPA, and two patients were found to have deletions involving parts or all of SMAD4. Two patients were found to have large deletions involving BMPR1A. One involved the promoter and 1st non-coding exon (50) and the other a contiguous deletion of BMPR1A and PTEN. This patient did not appear to have JPI based on age of diagnosis (20 years), but had intriguing manifestations suggestive of BRRS such as macrocephaly (95th percentile head circumference), a dermatofibroma, trichoepithelioma and recurrent hernias (48). A summary of the deletions found by MLPA in these studies is shown in Figure 2.
Figure 2.

Three studies used MLPA to localize deletions in JP patients. Top: Chromosome 10 with area expanded within the red square. Reference genes are shown in the middle, with MLPA probes corresponding to different coding and non-coding exons below them. (1) Calva et al. (48); (2) Aretz et al. (29); (3) Van Hattem et al. (47).
Contiguous Deletions Must Include PTEN
In this review, we found that all patients with chromosome 10 microdeletions who presented with signs of polyposis were found to have a hemizygous deletion which included both BMPR1A and PTEN. Further insight on that could be derived by examining two individual studies by Balciuniene (51) and Alliman (52). Balciuniene et al. reported three unrelated probands who had deletions in the 10q22.3-23.3 region. The first two probands had a deletion ~7.2Mb in size that included BMPR1A, but not PTEN. Both patients manifested with developmental delay and macrocephaly, and had no evidence of polyposis. The third proband was the same patient described by Arch et al. (41) in 1997. The authors confirmed the loss of both BMPR1A and PTEN in this patient and again confirmed the presence of hamartomatous polyps at that age. Alliman et al. reported an additional 6 patients with deletions similar to the first two probands reported by Balciuniene et al. (52). Likewise, none had any signs of polyposis, which suggested the requirement of a contiguous deletion of both genes for polyposis to take place. Nevertheless, due to the rarity of this condition, and the fact that point mutations and smaller deletions (detected by MLPA) in BMPR1A cause JP, the risk of polyposis in these patients with microdeletions cannot be ruled out entirely.
Summary
Mutations in BMPR1A have been found in patients with JP, whereas PTEN mutations have been described in CS and BRRS. These two genes map to chromosome 10q22-23 and contiguous deletions of both genes have been reported. We reviewed the literature for studies that describe large deletions in JP-predisposing genes and further focused on the phenotype of patients with contiguous deletions of BMR1A and PTEN.
Point mutations and small insertions/deletions in SMAD4 and BMPR1A have been identified through direct sequencing in 21.6% and 18.5% of JP cases, respectively. That suggested CNV as an alternative mechanism of inactivation of these genes. Three major independent studies utilizing MLPA have been conducted to date to screen for CNVs in patients with JP (29, 47, 48). These collectively reported a total of 6.3% of JP patients to have large germline deletions of one of the two causative genes (BMPR1A and SMAD4). Despite thorough evaluation of these two genes, a significant portion of patients with the clinical diagnosis of JP remain genetically unaccounted for, which suggests that other genes might be involved in the pathogenesis of JP.
JP usually presents in older children or young adults, but when it presents at a very early age, it is associated with a more severe clinical course and more pronounced extraintestinal manifestations, often leading to a significantly reduced life expectancy. Recently, 2 studies attempted to correlate the extent of DNA loss with the severity of the symptoms (suggesting that the hemizygous deletion of more genes could potentially lead to more pronounced manifestations), and found no clear association (45, 46). However, it is plausible that large deletions involving both BMPR1A and PTEN lead to a clinical picture that is more severe than JP. Additionally, part of the controversy may be due to the lack of a unified diagnostic definition for JPI. Salviati et al. argued that since their patient did not present prior to 2 years of age with GI bleeding, diarrhea and rectal prolapse, JPI was a questionable diagnosis (45) (referring to the criteria proposed by Sachatello in 1974 (53)). In response, Sanalville et al. argued that an age of diagnosis at 6 years points towards an onset of the disease process in infancy and that the diagnosis of JPI appropriate (54). The rarity and variability of these deletions has made the correlation between phenotype and genotype difficult. Nevertheless, the accumulation of these data can point researchers to study this area more closely and perhaps examine other genes in 10q22-23 in patients with features of these disorders. Finally, while the range of phenotypes is variable, we conclude that JPI is a unique entity that often combines features of both CS and BRRS with JP, and requires the loss of both BMPR1A and PTEN.
Acknowledgements
This work was supported by NIH Grant 1 RO1 CA136884-01, the Susser Family, and Roy J. Carver Charitable Trust.
Footnotes
Authors declare no conflict of interest.
References
- 1.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 2.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 3.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lupski JR, Wise CA, Kuwano A, et al. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- 5.Schmickel RD. Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr. 1986;109:231–241. doi: 10.1016/s0022-3476(86)80377-8. [DOI] [PubMed] [Google Scholar]
- 6.Ledbetter DH, Riccardi VM, Airhart SD, et al. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- 7.Flint J, Hill AV, Bowden DK, et al. High frequencies of alpha-thalassaemia are the result of natural selection by malaria. Nature. 1986;321:744–750. doi: 10.1038/321744a0. [DOI] [PubMed] [Google Scholar]
- 8.Francke U, Ochs HD, de Martinville B, et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Am J Hum Genet. 1985;37:250–267. [PMC free article] [PubMed] [Google Scholar]
- 9.Hurles ME, Dermitzakis ET, Tyler-Smith C. The functional impact of structural variation in humans. Trends Genet. 2008;24:238–245. doi: 10.1016/j.tig.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polymeropoulos MH, Higgins JJ, Golbe LI, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- 11.Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 12.Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erez A, Patel AJ, Wang X, et al. Alu-specific microhomology-mediated deletions in CDKL5 in females with early-onset seizure disorder. Neurogenetics. 2009;10:363–369. doi: 10.1007/s10048-009-0195-z. [DOI] [PubMed] [Google Scholar]
- 14.Chow E, Macrae F. A review of juvenile polyposis syndrome. J Gastroenterol Hepatol. 2005;20:1634–1640. doi: 10.1111/j.1440-1746.2005.03865.x. [DOI] [PubMed] [Google Scholar]
- 15.Haidle JL, Howe JR. Juvenile Polyposis Syndrome. Vol. 2011 2011. [Google Scholar]
- 16.Jass JR, Williams CB, Bussey HJ, et al. Juvenile polyposis--a precancerous condition. Histopathology. 1988;13:619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 17.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 18.Howe JR, Ringold JC, Summers RW, et al. A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am J Hum Genet. 1998;62:1129–1136. doi: 10.1086/301840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howe JR, Bair JL, Sayed MG, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 20.Nelen MR, Padberg GW, Peeters EA, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13:114–116. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 21.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 22.Olschwang S, Serova-Sinilnikova OM, Lenoir GM, et al. PTEN germ-line mutations in juvenile polyposis coli. Nat Genet. 1998;18:12–14. doi: 10.1038/ng0198-12. [DOI] [PubMed] [Google Scholar]
- 23.Lynch ED, Ostermeyer EA, Lee MK, et al. Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am J Hum Genet. 1997;61:1254–1260. doi: 10.1086/301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eng C, Ji H. Molecular classification of the inherited hamartoma polyposis syndromes: clearing the muddied waters. Am J Hum Genet. 1998;62:1020–1022. doi: 10.1086/301847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Hampel H, Thiele H, et al. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. 2001;358:210–211. doi: 10.1016/s0140-6736(01)05412-5. [DOI] [PubMed] [Google Scholar]
- 27.Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–694. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- 28.Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484–491. doi: 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44:702–709. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26:513–519. doi: 10.1002/humu.20253. [DOI] [PubMed] [Google Scholar]
- 32.Gille JJ, Hogervorst FB, Pals G, et al. Genomic deletions of MSH2 and MLH1 in colorectal cancer families detected by a novel mutation detection approach. Br J Cancer. 2002;87:892–897. doi: 10.1038/sj.bjc.6600565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor CF, Charlton RS, Burn J, et al. Genomic deletions in MSH2 or MLH1 are a frequent cause of hereditary non-polyposis colorectal cancer: identification of novel and recurrent deletions by MLPA. Hum Mutat. 2003;22:428–433. doi: 10.1002/humu.10291. [DOI] [PubMed] [Google Scholar]
- 34.Sachatello CR, Griffen WO., Jr. Hereditary polypoid diseases of the gastrointestinal tract: a working classification. Am J Surg. 1975;129:198–203. doi: 10.1016/0002-9610(75)90298-6. [DOI] [PubMed] [Google Scholar]
- 35.Stemper TJ, Kent TH, Summers RW. Juvenile polyposis and gastrointestinal carcinoma. A study of a kindred. Ann Intern Med. 1975;83:639–646. doi: 10.7326/0003-4819-83-5-639. [DOI] [PubMed] [Google Scholar]
- 36.Lloyd KM, 2nd, Dennis M. Cowden's disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963;58:136–142. doi: 10.7326/0003-4819-58-1-136. [DOI] [PubMed] [Google Scholar]
- 37.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37:828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorlin RJ, Cohen MM, Jr., Condon LM, et al. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44:307–314. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 39.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 40.Jacoby RF, Schlack S, Sekhon G, et al. Del(10)(q22.3q24.1) associated with juvenile polyposis. Am J Med Genet. 1997;70:361–364. doi: 10.1002/(sici)1096-8628(19970627)70:4<361::aid-ajmg6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 41.Arch EM, Goodman BK, Van Wesep RA, et al. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet. 1997;71:489–493. [PubMed] [Google Scholar]
- 42.Zigman AF, Lavine JE, Jones MC, et al. Localization of the Bannayan-Riley-Ruvalcaba syndrome gene to chromosome 10q23. Gastroenterology. 1997;113:1433–1437. doi: 10.1053/gast.1997.v113.pm9352843. [DOI] [PubMed] [Google Scholar]
- 43.Tsuchiya KD, Wiesner G, Cassidy SB, et al. Deletion 10q23.2-q23.33 in a patient with gastrointestinal juvenile polyposis and other features of a Cowden-like syndrome. Genes Chromosomes Cancer. 1998;21:113–118. doi: 10.1002/(sici)1098-2264(199802)21:2<113::aid-gcc6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 44.Delnatte C, Sanlaville D, Mougenot JF, et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet. 2006;78:1066–1074. doi: 10.1086/504301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salviati L, Patricelli M, Guariso G, et al. Deletion of PTEN and BMPR1A on chromosome 10q23 is not always associated with juvenile polyposis of infancy. Am J Hum Genet. 2006;79:593–596. doi: 10.1086/507151. author reply 596–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menko FH, Kneepkens CM, de Leeuw N, et al. Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet. 2008;74:145–154. doi: 10.1111/j.1399-0004.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- 47.van Hattem WA, Brosens LA, de Leng WW, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut. 2008;57:623–627. doi: 10.1136/gut.2007.142927. [DOI] [PubMed] [Google Scholar]
- 48.Calva-Cerqueira D, Chinnathambi S, Pechman B, et al. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet. 2009;75:79–85. doi: 10.1111/j.1399-0004.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- 49.Jass JR. Pathology of polyposis syndromes with special reference to juvenile polyposis. In: Utsunomiya JLH, editor. Hereditary colorectal cancer. Tokyo: Springer-Verlag; 1990. pp. 343–350. [Google Scholar]
- 50.Calva-Cerqueira D, Dahdaleh FS, Woodfield G, et al. Discovery of the BMPR1A promoter and germline mutations that cause juvenile polyposis. Hum Mol Genet. 2010;19:4654–4662. doi: 10.1093/hmg/ddq396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balciuniene J, Feng N, Iyadurai K, et al. Recurrent 10q22-q23 deletions: a genomic disorder on 10q associated with cognitive and behavioral abnormalities. Am J Hum Genet. 2007;80:938–947. doi: 10.1086/513607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alliman S, Coppinger J, Marcadier J, et al. Clinical and molecular characterization of individuals with recurrent genomic disorder at 10q22.3q23.2. Clin Genet. 2010;78:162–168. doi: 10.1111/j.1399-0004.2010.01373.x. [DOI] [PubMed] [Google Scholar]
- 53.Sachatello CR, Hahn IS, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75:107–114. [PubMed] [Google Scholar]
- 54.Sanlaville D, Delnatte C, Moungenot J-F, et al. Reply to Salviati et al. American Journal of Human Genetics. 2006:596–597. [Google Scholar]