

Abstract
Gemcitabine is a deoxycytidine analog used in the treatment of various solid tumors. However, tumors often develop resistances over time, which becomes a major issue for most gemcitabine-related chemotherapies. In the present study, a previously reported stearoyl gemcitabine nanoparticle formulation (GemC18-NPs) was evaluated for its ability to overcome gemcitabine resistance. In the wild type CCRF-CEM human leukemia cells, the IC50 value of GemC18-NPs was 9.5-fold greater than that of gemcitabine hydrochloride (HCl). However, in the CCRF-CEM-AraC-8C cells that are deficient in the human equilibrative nucleoside transporter-1, the IC50 of GemC18-NPs was only 3.4-fold greater than that in the parent CCRF-CEM cells, whereas the IC50 of gemcitabine HCl was 471-fold greater than that in the parent CCRF-CEM cells. The GemC18-NPs were also more cytotoxic than gemcitabine HCl in the deoxycytidine kinase deficient (CCRF-CEM/dCK−/−) tumor cells. Similar to gemcitabine HCl, GemC18-NPs induced apoptosis through caspase activation. Another gemcitabine-resistant tumor cell line, TC-1-GR, was developed in our laboratory. In the TC-1-GR cells, the IC50 of GemC18-NPs was only 5% of that of gemcitabine HCl. Importantly, GemC18-NPs effectively controlled the growth of gemcitabine resistant TC-1-GR tumors in mice, whereas the molar equivalent dose of gemcitabine HCl did not show any activity against the growth of the TC-1-GR tumors. Proteomics analysis revealed that the TC-1-GR cells over-expressed ribonucleotide reductase M1, which was likely the cause of the acquired gemcitabine resistance in the TC-1-GR cells. To our best knowledge, this presents the first report demonstrating that a nanoparticle formulation of gemcitabine overcomes gemcitabine resistance related to ribonucleotide reductase M1 over-expression.
Keywords: Cytotoxicity, in vivo anti-tumor activity, proteomics, nucleoside transporter, deoxycytidine kinase, siRNA
1. Introduction
Gemcitabine (2′-2′-difluorodeoxycytidine, dFdC) is a deoxycytidine analog, which is used to treat various solid tumors such as ovarian cancer, non-small cell lung cancer, pancreatic cancer, and breast cancer [1, 2]. It is also an attractive candidate for combination therapy because of its favorable toxicity profile [3]. Combination therapies with cisplatin, etoposide and mitomycin are active against many other solid tumors such as bladder cancer, gastric cancer, and esophageal cancer [4, 5].
However, tumors acquire resistance over time, which becomes a major issue for most gemcitabine-related chemotherapies [6]. The resistance is related to the mechanism of action of gemcitabine. Gemcitabine is transported into cells by nucleoside transporters such as the human equilibrative nucleoside transporter-1 (hENT1) [7]. Decreased expression of hENT1 confers lower gemcitabine toxicity in cells by blocking the cellular uptake of gemcitabine [8]. After cellular uptake, gemcitabine is transformed by a deoxycytidine kinase (dCK) into gemcitabine monophospate, which is further phosphorylated to gemcitabine diphosphate (dFdCDP), and then gemcitabine triphosphate (dFdCTP) [9]. The metabolite, dFdCTP, is intercalated into DNA by DNA polymerase alpha to inhibit DNA synthesis and induce cells to undergo apoptosis [10]. The dFdCDP acts as a ribonucleotide reductase (RR) inhibitor [2, 11], which leads to increased incorporation of gemcitabine into DNA. On the other hand, gemcitabine is deaminated to its inactive form by adenosine or cytidine deaminases (CDA) [12, 13]. Thus, nucleoside transporters, dCK, deaminases, RR, and the accumulation of dFdCDP seem to be important for the development of resistance to gemcitabine. Of particular importance, the RR is believed to play a key role in resistance to gemcitabine in many tumor cells in culture [14, 15] and in vivo [16, 17], and there is evidence that the effectiveness of gemcitabine treatment is correlated to the level of ribonucleotide reductase M1 (RRM1) expression in tumor cells. For example, clinically, non-small cell lung cancer patients with a low level of RRM1 mRNA expression had a significantly longer median survival when treated with gemcitabine/cisplatin [18, 19], but patients with biliary tract cancers and a higher expression of RRM1 were resistant to gemcitabine treatment [20].
There have been extensive research efforts to overcome gemcitabine resistance. For example, amino acid ester prodrugs of gemcitabine were synthesized, and they were not as sensitive as gemcitabine to deamination by CDA [21]. In order to facilitate the uptake of gemcitabine by cells with decreased expression of nucleotide transporters, a lipophilic gemcitabine pro-drug was synthesized by esterifying gemcitabine at the 5′ position with an elaidic fatty acid [22]. Gemcitabine was also conjugated with cardiolipin to increase its uptake [23]. A phospholipid gemcitabine conjugate was shown to overcome both nucleoside transporter-deficiency and dCK-deficiency in cancer cells in culture [24], but it is unknown whether the phospholipid gemcitabine conjugate was effective in vivo.
An increasing amount of evidence had pointed to the promise of using nanoparticles to combat cancer cell resistance to chemotherapy [25], but the utilization of a gemcitabine nanoparticle formulation to overcome gemcitabine resistance is limited. Most relevantly, Reddy et al. reported that a nanoparticle formulation of gemcitabine prepared using gemcitabine covalently coupled to 1,1′,2-tris-nor-squalenic acid (4-(N)-tris-nor-squalenoyl-gemcitabine) was more cytotoxic than gemcitabine in two gemcitabine resistance cell lines, a human leukemia cell line (CCRF-CEM-AraC-8C) and a murine leukemia cell line (L1210 10K) in culture [26]. The L1210 10 K cells were dCK deficient, and the CCRF-CEM-AraC-8C cells were hENT1 deficient [26].
Previously, a novel stearoyl gemcitabine nanoparticle formulation was developed in our laboratory by incorporating a stearic acid amide derivative of gemcitabine, stearoyl gemcitabine (GemC18), into solid lipid nanoparticles engineered from lecithin/glycerol monostearate-in-water emulsions [27]. In mice with pre-established model mouse or human tumors, the stearoyl gemcitabine nanoparticles (GemC18-NPs) were significantly more effective than gemcitabine HCl in controlling tumor growth [27]. The improved anti-tumor activity of the GemC18-NPs was not simply due to the GemC18 per se because the same GemC18 dissolved in Tween 20 micelles failed to show any significant anti-tumor activity in mice [27]. In an effort to develop a strategy to overcome resistance to gemcitabine, the feasibility of overcoming tumor resistance to gemcitabine using the GemC18-NPs was evaluated. It was discovered that the GemC18-NPs can overcome gemcitabine resistance related to the over-expression of RRM1, not only in culture, but also in mice.
2. Materials and Methods
2.1. Materials and cell lines
Tween 20, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), stearic acid (C18), caspase-3 assay kit, iodoacetamide, urea, and thiourea were from Sigma-Aldrich (St. Louis, MO). Gemcitabine HCl was from U.S. Pharmacopeia (Rockville, MD). Soy lecithin was from Alfa Aesar (Ward Hill, MA). Glycerol monostearate was from Gattefosse Corp. (Paramus, NJ). N-3,4-tridhydroxy-benzamide (didox) was from Cayman Chemical (Ann Arbor, MI). Biolytes, Bio-Safe Coomassie blue staining solution, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), dithiothreitol (DTT), laemmli sample buffer, β-mercaptoethanol, nitrocellulose membrane, strip IPG (pH 3–10), Tris-HCl gel, and precision plus protein standards were from Bio-Rad (Hercules, CA). The duplex small interfering RNA (siRNA) oligonucleotides for RRM1 (UUAAUAACUGGGCUU CUGGGCUCUC and GAGAGCCCAGAAGCCCAGUUAUUAA), the negative universal control siRNA (Cat. No: 12935-300), and the Lipofectamine™ RNAiMAX were from Invitrogen (Carlsbad, CA).
Human leukemia cell line, CCRF-CEM (# CCL-119), and mouse lung cancer cell line, TC-1 (# CRL-2785), were from the American Type Culture Collection (Rockville, MD). CCRF-CEM-AraC-8C cells (hENT1 deficient), CCRF-CEM/dCK−/− cells (dCK deficient), and CCRF-CEM-AraC-8D cells (dCK deficient) were kindly provided by Dr. Buddy Ullmann (Oregon Health & Science University, Portland, OR), Dr. Margaret Black (Washington State University, Pullman, WA), and Dr. Beverly S. Mitchell (Stanford University School of Medicine, Stanford, CA), respectively. The cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (all from Invitrogen).
2.2. Preparation of stearoyl gemcitabine nanoparticles
GemC18-NPs were prepared as previously described [27]. Briefly, 3.5 mg of soy lecithin, 0.5 mg of glycerol monostearate, and 5 mg of GemC18 were placed into a 7 mL glass vial. One mL of de-ionized and filtered (0.22 μm) water was added into the mixture, which was then maintained on a 70–75 °C hot plate while stirring, with occasional water-bath sonication (Bransonic Ultrasonic Cleaner, Danbury, CT), until the formation of homogenous slurry. Tween 20 was added in a step wise manner to a final concentration of 1% (v/v). The resultant emulsions were allowed to cool to room temperature while stirring to form nanoparticles. Particle size and zeta potential were measured using a Malvern Zetasizer Nano ZS (Westborough, MA). In a short 20-day preliminary stability study, the size of the nanoparticles did not change significantly when the nanoparticles in aqueous suspension were stored in ambient condition (Supplemental Fig. S1). When stored at 37 °C in PBS (pH 6.01 or 7.4) for 72 h, no significant particle size increase, nor GemC18 degradation, was observed (Supplemental Fig. S2).
2.3. Development of TC-1-GR cell line
The gemcitabine resistant cell line, TC-1-GR, was developed by culturing TC-1 cells with gradually increasing concentration of gemcitabine HCl over a 3-month period as previously described [28]. The maximum concentration used was 1 μM. Cells were grown in 75 cm2 flasks in RPMI 1640 containing 10% FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin in 5% CO2 at 37 °C.
2.4. In vitro cytotoxicity assay
Cells (10,000/well for leukemia cells, 5,000/well for TC-1 and TC-1-GR cells) were seeded in 96-well plates. After overnight incubation, they were further incubated in the presence of various concentrations of gemcitabine HCl, the equivalent amount of GemC18-NPs (no more than 40 M of GemC18), or didox, an inhibitor of RRM1, for 48 h (TC-1 or TC-1-GR cells) or 72 h (CCRF-CEM, CCRF-CEM-AraC-8C, CCRF-CEM-AraC-8D, and CCRF-CEM/dCK−/−) at 37 °C, 5% CO2. To compare the cytotoxicities of GemC18-NPs and GemC18, TC-1-GR cells (5,000/well) were allowed to grow for 24 h and incubated with different concentrations of GemC18 in GemC18-NPs or in trace amount of dimethyl sulfoxide (DMSO) for 48 h. RPMI 1640 medium alone or medium with trace amount of DMSO were used as a control. GemC18-free nanoparticles equivalent to GemC18-NPs that contain 10 M or less GemC18 were not toxic to TC-1-GR cells (less than 50 M in the CCRF-CEM cells and derivative lines). The number of cells alive was quantified using an MTT assay. Absorbance was measured at 570 nm and 630 nm using a BioTek Synergy™ HT Multi-Mode Microplate Reader (BioTek Instruments, Winooski, VT). Penicillin and streptomycin in the cell culture medium did not significantly affect the cytotoxicity of the GemC18-NPs (Supplemental Table S1). The fraction of affected (killed) cells (Fa) and the fraction of unaffected (live) cells (Fu) at every dose were calculated, and the Log (Fa/Fu) values were plotted against the Log (concentration of gemcitabine) [29]. IC50 was the dose at Log (Fa/Fu) = 0. The experiment was repeated at least three times.
2.5. Assay of caspase-3 activity
Caspase-3 activity was determined using a Sigma caspase-3 assay kit. Briefly, CCRF-CEM cells (10,000 cells/well in 150 μl) were seeded in 96-well plates. After overnight incubation, the cells were treated for 72 h with gemcitabine HCl or GemC18-NPs at concentrations ranging from 0.22 to 314 nM. At the end of incubation, the cells were washed with PBS (10 mM, pH 7.4), centrifuged (3,000 × g for 5 min at 4 °C), and re-suspended in 20 μL of cell lysis buffer (20 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) at 4 °C for 10 min. The cell lysate was centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatant, representing cytoplasmic extracts, was transferred to a new plate and mixed with the assay substrate, acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcourmarin (Ac-DEVD-AMC). The mixture was incubated for up to 72 h for the hydrolysis of the Ac-DEVD-AMC by caspase 3 to release the fluorescent AMC, which was quantified by measuring the fluorescence intensity at 360 nm (excitation)/460 nm (emission) according to the manufacturer’s instruction using a BioTek Synergy™ HT Multi-Mode Microplate Reader. The unit of the caspase 3 activity was mol AMC/min/mL.
2.6. Animal studies
National Institutes of Health guidelines for animal use and care were followed. Animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Texas at Austin. Female nu/nu mice (18–20 g) were from Charles River (Wilmington, MA). TC-1 or TC-1-GR tumors were established in the right flank of mice by subcutaneous (s.c.) injection of 5 × 105 cells. Starting on day 4–5 after tumor cell implantation, tumors became visible (3–4 mm), and mice (n = 5–7) were randomized and injected via the tail vein with 0.56 mg gemcitabine HCl, the molar equivalent of GemC18 in nanoparticles (i.e., 1 mg GemC18), or 200 μL of sterile mannitol (5%, w/v) (as a negative control) [27]. Tumor size was measured three times a week with a caliper, and tumor volume was calculated based on the following equation: tumor volume (mm3) = 1/2 [length x (width)2].
2.7. Two-dimensional electrophoresis
To identify altered expressions of proteins in the TC-1-GR cells, a 2-dimensional sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was carried out. Protein lysates (100 μg) from TC-1 cells or TC-1-GR cells were dissolved in rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, 0.2% (v/v) biolytes, 100 mM DTT, and 0.001% bromophenol blue), sonicated for 10 min, and applied to a Ready Strip IPG (pH 3–10). The strips were then actively rehydrated at 50 V for 15 h in a protein isoelectric focusing (IEF) cell (Bio-Rad). IEF was performed at 20 °C as following: 250 V for 15 min, 8,000 V for 2.5 h rapid gradient and 60,000 V h. The strips were stored at −80 °C until a second dimension electrophoresis was performed. Prior to the second dimension separation, gel strips were equilibrated for 10 min in 375 mM Tris–HCl (pH 8.8) containing 6 M urea, 2% (w/v) SDS, 20% (v/v) glycerol, and 2% DTT. The strips were then equilibrated for 10 min in the same buffer containing 2.5% iodoacetamide in place of DTT. Eight-sixteen percent gradient criterion Tris–HCl gels (Bio-Rad) were used to perform the second dimension electrophoresis. Precision plus protein standards were run along with the sample at 130 V for 115 min. The gels were then stained in Bio-Safe Coomassie blue staining solution (Bio-Rad) and scanned using a Kodak Image Station 440CF (Rochester, NY). This experiment was repeated 3 times.
2.8. Identification of proteins using mass spectrometry
After comparing the protein expression scanned by Kodak, the gel spots with significant differential expression (p < 0.05, n =3) were excised to identify proteins. The gel pieces were digested in gel by trypsin as described by Chung et al. [30]. MALDI-MS/MS analysis for the digested peptides was performed as described by Marley et al. [31]. MALDI-MS/MS data were processed with GPS explorer 2.0 to create Mascot-searchable files as described by Chavez et al. [32]. Mascot software (Matrix Science, London, UK) was used to aid in the interpretation of tandem mass spectra. Searches were performed using the SwissProt database (Swiss Institute of Bioinformatics, Geneva, Switzerland).
2.9. Semi quantitative reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from TC-1 and TC-1-GR cells (1 × 107) using a QIAGEN RNeasy mini kit. Complementary DNA (cDNA) was generated from the RNA samples (1 μg) using reverse transcriptase reaction with oligo dT primers (Invitrogen SuperScript III™). PCR was completed utilizing cDNA from the reverse transcription and primer sets (RRM1 forward: 5′-CCCAATGAGTGTCCTGGTCT-3′, RRM1 reverse: 5′-TTCTGCTGGTTGCTCTTCC-3′, RR M2 subunit (RRM2) forward: 5′-CCTACTAACCCCAGCGTTGA-3′, RRM2 reverse: 5′-GCACTGGGAAGCTCTGAAAC-3′, CDA forward: 5′-CTCTCGTGAGGCCAAGAAGT-3′, CDA reverse: 5′-TCAGGGCTATTGCCATCTCT-3′, dCK forward: 5′-TGAGGATTGGGAAGTGGTTC-3′, dCK reverse: 5′-GAGCTTGCCATTGAGAGAGG-3′, hENT1 forward: 5′-CCAGGTACCTTTGGCTCTCA-3′, hENT1 reverse: 5′-ACTGCTCCCCTGGAATTTTT-3′, β-actin forward: 5′-TGTGATGGTGGGAATGGGTCAGAA-3′, β-actin reverse: 5′-TGCCACAGGATTCCATACCCAAGA-3′), which amplified a 201, 178, 199, 214, 202 and 695 base pair fragment of mouse RRM1, RRM2, CDA, dCK, hENT1, and β-actin genes, respectively. Reactions were conducted using an Eppendorf Mastercycler (Hauppauge, NY): an initial denaturation at 95 °C for 5 min, followed by 25 (RRM1) or 35 cycles (RRM2, hENT1, dCK and CDA) of 95 °C for 30 s, 58 °C for 60 s and 68 °C for 60 s, and a 5 min final extension at 68 °C. PCR products (20 μl) were analyzed using agarose gel electrophoresis. This experiment was repeated three times.
2.10. Western blot
TC-1 or TC-1-GR cell lysate proteins (20 μg) were mixed with Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, and 0.01% Bromophenol Blue) with 5%-mercaptoethanol. Proteins were denatured at 95 °C for 5 min. One-dimensional electrophoresis was performed with 4–15% Criterion Tris–HCl gels. Precision plus protein standards (Dual color) were run along with the samples at 130 V for 70 min. Electrophoresis was performed in buffer solution consisting of 25 mM Tris, 192 mM glycine, and 0.1% SDS (pH 8.3). The proteins were transferred to nitrocellulose membrane at 0.3 A for 2 h. The membranes were incubated in the first antibody (RRM1 polypeptide IgG, Aviva System Biology, San Diego, CA) and then in the second antibody (anti-rabbit IgG-HRP conjugate, Aviva). The membranes were incubated in SuperSignal West Pico Chemiluminescent substrate (Pierce, Rockford, IL) and then exposed to an X-ray film to visualize the protein bands. After the detection of RRM1, the membranes were stripped in buffer containing 62.5 mM Tris-HCl (pH 6.7), 2% SDS, 0.1 M β-mercaptoethanol at 50 °C for 10 min, incubated with anti-rabbit β-actin IgG (Aviva), and then with the second antibody (anti-rabbit IgG-HRP conjugate). The β-actin signals were visualized as described above.
2.11. Inhibition of RRM1 by transfection with siRNA
TC-1-GR (5 × 104) cells were plated in 96-well plates in RPMI 1640 medium supplemented with 10% heat-inactivated FBS. When cells became 80% confluent, they were transfected with the siRNA oligonucleotide (20 nM) or the negative control siRNA using 0.2 μL Lipofectamine™ RNAiMAX. Forty-eight hours after transfection, the medium was refreshed. Cells were treated with gemcitabine HCl for 48 h, and cell numbers were determined using MTT assay. For the Western blotting, TC-1-GR (1 × 105) cells were plated in 6-well plates in RPMI 1640 medium supplemented with 10% heat-inactivated FBS. When cells became 80% confluent, they were transfected with the siRNA oligonucleotide (20 nM) or the negative control siRNA using 5 μL Lipofectamine™ RNAiMAX. Forty-eight hours after transfection, cells were lysed, and Western blotting was carried out as described above.
2.12. Statistics
Statistical analyses were completed using ANOVA followed by Student-Newman Keul test. A p-value of < 0.05 (two-tail) was considered statistically significant.
3. Results
3.1. Stearoyl gemcitabine nanoparticles can overcome gemcitabine resistance in tumor cells deficient in hENT1 or dCK in culture
The size and zeta potential of the GemC18-NPs were 191 ± 5 nm and -31 ± 0.9 mV, respectively [27]. The polydispersity index of the GemC18-NPs was 0.24 ± 0.08 (n = 4) [27]. The negative zeta potential was likely due to the lecithin, which contains negatively charged phospholipids. Data from both gel permeation chromatography and photon correlation spectroscopy showed that all the GemC18 was incorporated into the nanoparticles [27]. Initially, the in vitro cytotoxicity of the GemC18-NPs was assessed and compared to that of gemcitabine HCl in human leukemia cell line, CCRF-CEM, and its derivative lines, CCRF-CEM-AraC-8C and CCRF-CEM/dCK−/−. In the wild type CCRF-CEM cells, the IC50 of GemC18-NPs was 19.0 ± 3.2 nM, 9.5 times greater than that of gemcitabine HCl (2.0 ± 0.6 nM) (Fig. 1A). In the hENT1 deficient CCRF-CEM-AraC-8C cells, the IC50 of GemC18-NPs was 64 ± 21 nM, only 3.4 times greater than that in the parent CCRF-CEM cells. However, the IC50 of the gemcitabine HCl was 942 ± 90 nM in the CCRF-CEM-AraC-8C cells, 471 times greater than that of the parent CCRF-CEM cells (Fig. 1B). In other words, in the hENT1 deficient cell line, the GemC18-NPs were 15-fold more cytotoxic than gemcitabine HCl.
Fig. 1. Cytotoxicity of GemC18-NPs and gemcitabine HCl in human leukemia cells in culture.

The cytotoxicity was measured after 72 h of incubation with CCRF-CEM cells (A), CCRF-CEM-AraC-8C cells (B), or CCRF-CEM/dCK−/− cells (C). Each point was the mean ± S.D. from at least three replicates.
In the CCRF-CEM/dCK−/− cells, the IC50 values of gemcitabine HCl and GemC18-NPs were 189,300 ± 26,100 nM and 24,200 ± 3,600 nM, respectively, which were 94,650- and 1,274-fold greater than those in the parent CCRF-CEM cells (Fig. 1C). In other words, in the CCRF-GEM/dCK−/− cells, the GemC18-NPs were 7.8-fold more cytotoxic than gemcitabine HCl. In another dCK deficient derivative, CCRF-CEM-AraC-8D cells, the IC50 values of gemcitabine HCl and GemC18-NPs were 89,800 ± 32,900 nM and 27,800 ± 4,500 nM, respectively, which were 44,900- and 1,463-fold greater than that in the parent CCRF-CEM cells.
3.2. Stearoyl gemcitabine nanoparticles induced tumor cells to undergo apoptosis
To test whether GemC18-NPs induced apoptosis through caspase activation, CCRF-CEM cells were treated with GemC18-NPs or gemcitabine HCl. In cells treated with gemcitabine HCl, the caspase-3 activity was maximized at 22 nM of gemcitabine (Fig. 2A). The IC50 of gemcitabine HCl in the CCRF-CEM cells was 2.0 ± 0.6 nM. Thus, it seemed that the maximum caspase-3 activity was detected at a concentration about 10 times of the IC50. In cells treated with GemC18-NPs, the caspase-3 activity was maximized at 314 nM of gemcitabine (Fig. 2A), which was 17 times of the IC50 value of GemC18-NPs (19.0 ± 3.2 nM). In a time dependency experiment, the capsase-3 activities in the lysates of cells treated with gemcitabine HCl or GemC18-NPs increased significantly as a function of incubation time (Fig. 2B).
Fig. 2. Caspase-3 activity in CCRF-CEM cells treated with gemcitabine HCl or GemC18-NPs.

Cells were treated for 72 h with gemcitabine HCl or GemC18-NPs. Cell lysates were incubated with assay substrates for up to 72 h to determine caspase-3 activity. (A). Dose dependence after 48 h of incubation. (B). Time dependence when incubated with 22 nM gemcitabine HCl or 314 nM GemC18 in nanoparticles. Data reported are mean ± SD (n = 3). * indicates p < 0.01 in t-test for comparison with the result at 0 nM of gemcitabine HCl or GemC18-NPs.
3.3. Development of a gemcitabine resistant TC-1 tumor cell line (TC-1-GR)
A gemcitabine resistant TC-1 cell line was developed by culturing the TC-1 cells with increasing concentrations of gemcitabine HCl. After 3 months of continuous culturing, the IC50 of gemcitabine HCl in the resultant TC-1 cells, named TC-1-GR, was increased by 3,582-fold (to 34.9 ± 15.4 M) (Fig. 3A). However, the IC50 of GemC18-NPs in the TC-1-GR cells was increased by only 41-fold (to 1.84 ± 0.25 M). In fact, in the TC-1-GR cells, the IC50 of the GemC18-NPs was only about 5% of that of the gemcitabine HCl, demonstrating that in TC-1-GR cells in culture, the GemC18-NPs were more cytotoxic than gemcitabine HCl. The cytotoxicity of the GemC18-NPs was not simply from the GemC18 alone. As shown in Fig. 3B, GemC18-NPs were more cytotoxic than GemC18 (dissolved in DMSO) to the TC-1-GR cells. There was no morphological difference between the TC-1 and TC-1-GR cells in culture. The doubling times of the TC-1 and TC-1-GR were 22.9 ± 0.9 and 20.1 ± 0.8 h, respectively (p = 0.01, n = 3).
Fig. 3. (A). Cytotoxicity of gemcitabine or GemC18-NPs in TC-1 and TC-1-GR cells in culture. (B). Comparison of the cytotoxicities of GemC18 and GemC18-NPs.

The cytotoxicity was measured after 48 h of incubation. In A, the IC50 values are the mean ± SD from at least three different determinations (*, p < 0.01 in t-test comparison of gemcitabine HCl and GemC18-NPs in the same cell line). Data shown in B are mean from six different determinations. Standard deviation was not shown for clarity.
3.4. Stearoyl gemcitabine nanoparticles, not gemcitabine HCl, can inhibit the growth of the gemcitabine resistant TC-1-GR tumors in mice
To investigate whether GemC18-NPs can inhibit the growth of the TC-1-GR tumors in vivo, TC-1 or TC-1-GR tumors were established in athymic mice, which were then treated with gemcitabine HCl or GemC18-NPs. Treatment of TC-1 tumor-bearing mice with either gemcitabine HCl or GemC18-NPs resulted in a significant delay in the growth of the TC-1 tumors (Fig. 4A). However, in mice with pre-established TC-1-GR tumors, only treatment with the GemC18-NPs significantly delayed the tumor growth, whereas treatment with gemcitabine HCl did not show any significant anti-tumor activity (Fig. 4B). GemC18-free nanoparticles were not included because data from a previous study showed that the blank nanoparticles did not have any anti-tumor activity in mice [27].
Fig. 4. The anti-tumor activity of GemC18-NPs in mice.
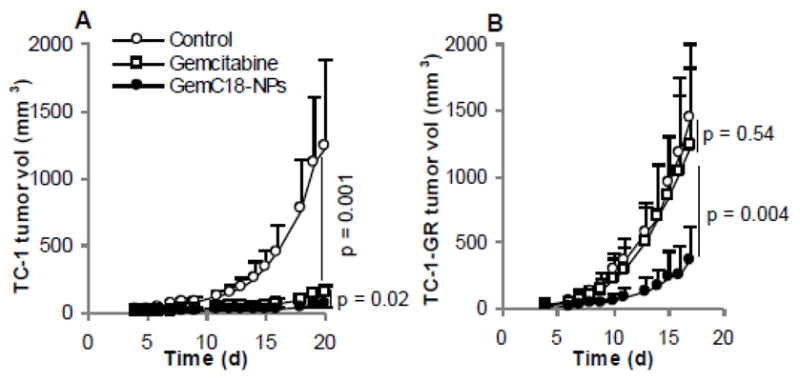
TC-1 (A) or TC-1-GR (B) tumors were established in nude mice. When tumors became 3.5–4 mm, mice were injected (i.v.) with gemcitabine HCl or GemC18-NPs. Mice were dosed on days 4 and 12 in A (4 and 11 in B). The p values shown are for the last day the tumor sizes were measured. Data reported are mean ± SD (n = 5–7).
3.5. Ribonucleotide reductase M1 over-expressed in TC-1-GR cells
To identify proteins that may be involved in the TC-1-GR cell’s ability to resist gemcitabine, the total protein expressions in TC-1 cells and TC-1-GR cells were compared using 2D SDS-PAGE, and proteins with differential expressions were identified using mass spectrometry. Based on the scanned values in SDS-PAGE gels, the expressions of six proteins were significantly decreased in the TC-1-GR cells compared to TC-1 cells (Fig. 5, Table 1). However, the expressions of vimentin and RRM1 were significantly increased in the TC-1-GR cells (9.7-fold and 16-fold, respectively) (Fig. 5, Table 1). In a semi-qualitative analysis of mRNA expression, only RRM1 expression was increased in TC-1-GR cells (7.3-fold), whereas the expressions of RR small subunit (RRM2), CDA, dCK, and hENT1 mRNA in TC-1-GR cells were not different from that in TC-1 cells (Fig. 6A). Immunoblotting confirmed the up-regulation of the expression of the RRM1 protein in TC-1-GR cells as well (Fig. 6B), and the expression of the RRM1 in TC-1-GR cells was down-regulated by transfecting the cells with RRM1-specific siRNA (Fig. 6B). Didox is known to be cytotoxic, partially by inhibiting RRM1. As shown in Fig. 6C, the IC50 of didox in TC-1-GR cells was 223 ± 19 μM, 5.4-fold greater than that in TC-1 cells, further suggesting that the RRM1 expression was up-regulated in TC-1-GR cells. Finally, to confirm that the increased RRM1 expression was related to the decreased sensitivity of TC-1-GR cells to gemcitabine, RRM1-specifc siRNA was transferred into TC-1-GR cells. In vitro cytotoxicity test showed that the siRNA-transfected TC-1-GR cells became more susceptible to gemcitabine HCl than TC-1-GR cells transfected with a control siRNA (Fig. 6D).
Fig. 5. 2-D SDS-PAGE of TC-1 (A) and TC-1-GR (B) cell lysates.
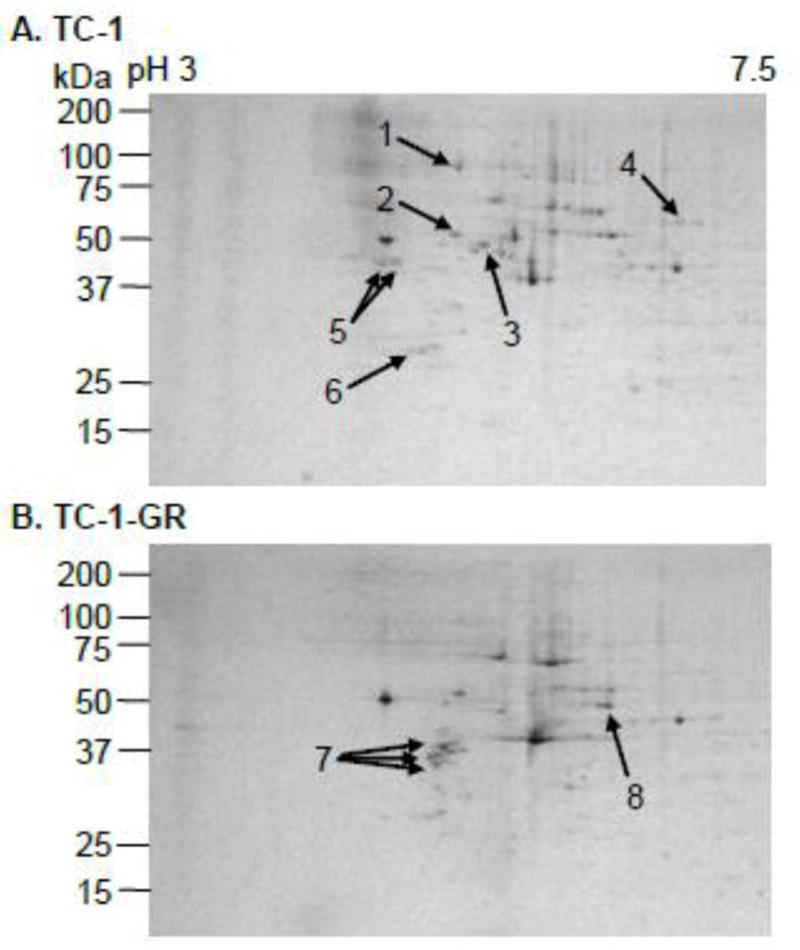
1: Endoplasmin precursor, 2: Protein disulfide-isomerase precursor, 3: Tubulin β-5 chain, 4: Lamin-A/C, 5: Calumenin precursor, 6: Elongation factor 1-β, 7: Vimentin, 8: RRM1.
Table 1.
Identification of proteins with altered expression in TC-1 and TC-1-GR cells.
| Protein No. a | Protein Name | Protein Expression b | TC-1-GR/TC-1 Ratio c | p-value d | |
|---|---|---|---|---|---|
| TC-1 | TC-1-GR | ||||
| 1 | Endoplasmin precursor e | 2023 ± 508 | 204 ± 956 | 0.10 | 0.02 |
| 2 | Protein disulfide-isomerase precursor | 1847 ± 186 | 1165 ± 250 | 0.63 | 0.02 |
| 3 | Tubulin β-5 chain | 1673 ± 277 | 212 ± 234 | 0.13 | 0.01 |
| 4 | Lamin-A/C | 461 ± 133 | 140 ± 137 | 0.30 | 0.04 |
| 5 | Calumenin precursor | 898 ± 168 | 35 ± 101 | 0.04 | 0.002 |
| 6 | Elongation factor 1-β | 548 ± 151 | 83 ± 179 | 0.15 | 0.03 |
| 7 | Vimentin | 89 ± 7 | 867 ± 308 | 9.74 | 0.01 |
| 8 | Ribonucleotide reductase large subunit (RRM1) | 139 ± 77 | 2267 ± 185 | 16.31 | 0.001 |
Protein numbers are corresponding to those in Fig. 5.
The unit of protein expression was arbitrarily compared to that of protein standard scanned in the 2D SDS-PAGE gel. Data shown are mean ± S.D. (n = 3).
TC-1-GR/TC-1 ratio was determined from the average of three separate gels scanned using a Kodak Image Station 440-CF.
Student’s t-test was used to compare the expression of proteins in TC-1 and TC-1-GR cells.
Alternative name is heat shock protein 90 or 94 kDa glucose-regulated protein.
Fig. 6.

(A). RT-PCR. Experiment was repeated three times with similar results.
(B) 1. Immunoblotting analysis of RRM1 in TC-1 and TC-1-GR cells. 2. Immunoblotting analysis of RRM1 in TC-1-GR cells transfected with RRM1-specific siRNA or control siRNA (Con). In A and B, β-Actin was used as an internal control.
(C). The IC50 values of didox in TC-1 and TC-1-GR cells. The cytotoxicity of didox was measured after 48 h of incubation. IC50 values were calculated from three different determinations.
(D). Cytotoxicity of gemcitabine in TC-1-GR cells transfected with RRM1-specific siRNA. Cytotoxicity of gemcitabine HCl at 1.6 μM and 35 μM was measured after 48 h of incubation. Con: cells transfected with negative control siRNA; siRNA: cells transfected with siRNA specific to RRM1. Data are presented as mean ± SD (n = 3) in C and D.
4. Discussion
In the present study, the feasibility of using the stearoyl gemcitabine nanoparticles previously developed in our laboratory to overcome gemcitabine resistance in cancer cells was evaluated. The GemC18-NPs were 15- and 7.8-fold more cytotoxic than gemcitabine HCl in the hENT1 deficient CCRF-CEM-AraC-8C cells and the CCRF-CEM/dCK−/− cells, respectively, although in the parent CCRF-CEM cells, gemcitabine HCl was 9.5-fold more cytotoxic than GemC18-NPs. Similar to gemcitabine HCl, GemC18-NPs induced tumor cells to undergo apoptosis through caspase activation. The anti-tumor activity of GemC18-NPs in the gemcitabine resistant TC-1-GR cells, developed in our laboratory, was also tested. In TC-1-GR cells in culture, the IC50 value of GemC18-NPs was only about 5% of that of gemcitabine HCl. In mice with pre-established TC-1-GR tumors, GemC18-NPs effectively inhibited the tumor growth, but gemcitabine HCl did not show any significant anti-tumor activity. In an effort to elucidate the mechanism of gemcitabine resistance by the TC-1-GR cells, it was discovered that the TC-1-GR cells over-expressed RRM1, which is known to play an important role in gemcitabine resistance.
Initially, the human leukemia cell line CCRF-CEM was used to study the mechanism of action of GemC18-NPs because of its well established mutant lines, CCRF-CEM-AraC-8C and CCRF-CEM/dCK−/− [26]. Gemcitabine is hydrophilic and requires membrane proteins called nucleoside transporters for cellular uptake [33]. In the wild type CCRF-CEM cells, gemcitabine HCl was more cytotoxic than GemC18-NPs (Fig. 1A). In the hENT1 deficient CCRF-CEM-AraC-8C cells, gemcitabine cannot enter the cells efficiently, but the GemC18-NPs were able to efficiently deliver the stearoyl gemcitabine into cells by endocytosis, which may explain the observation that the IC50 value of gemcitabine HCl was 15-fold greater than that of GemC18-NPs (Fig. 1B). Data from a previous study confirmed the uptake of the GemC18-NPs by tumor cells in culture [27]. The development of resistance to gemcitabine was shown to correlate strongly with deficiency of hENT1 expression in human breast cancer cells [34] and pancreatic adenocarcinoma cells [6], and the resistance to cytarabine (ara-C), another nucleoside analog, was attributed to the reduced expression of hENT1 in acute myeloid leukemia cells as well [35]. Thus, the GemC18-NPs may be useful in the treatment of gemcitabine resistant cancers. Similarly, imatinib-resistant tumor cells showed decreased activity and expression for both ENT1 and ENT2 [36], and the GemC18-NPs may be useful in the co-treatment of imatinib resistant cancers as well.
However, the GemC18-NPs were not as cytotoxic to the CCRF-CEM/dCK−/− cells as to the CCRF-CEM-AraC-8C cells, suggesting that gemcitabine in the GemC18-NPs still required the dCK to be activated to gemcitabine triphosphate before being incorporated into DNA. This is in agreement with a previous report showing that a lipophilic fatty acid ester derivative of gemcitabine transverses into tumor cells efficiently, but was inactive in dCK deficient cells [22]. The findings in the dCK deficient cells and the hENT1− cells are generally in agreement with what was reported by Reddy et al. [26].
To further evaluate the ability of GemC18-NPs to overcome gemcitabine resistance, GemC18-NPs were tested in a gemcitabine resistant TC-1 cell line, TC-1-GR, developed in our laboratory. The IC50 value of gemcitabine HCl in the TC-1-GR cells was 3,582-fold greater than that in the parent TC-1 cells. In contrast, the IC50 of GemC18-NPs in the TC-1-GR cells was only 41-fold greater than that in the TC-1 cells. In other words, in the TC-1-GR cells, GemC18-NPs were 19-fold more cytotoxic than gemcitabine HCl, which prompted us to test whether GemC18-NPs can inhibit the growth of the TC-1-GR tumors in mice. The GemC18-NPs were effective against both TC-1 and TC-1-GR tumors in mice (Fig. 4), whereas the gemcitabine HCl was effective only against TC-1 tumors in mice, not TC-1-GR tumors (Fig. 4). The anti-tumor activities of GemC18-NPs that contained polyethylene glycol (molecular weight, 2000, PEG(2000)) and gemcitabine HCl in TC-1 or TC-1-GR tumors in nude mice were compared as well. Again, both gemcitabine HCl and PEG-GemC18-NPs delayed the growth of TC-1 tumors, but for the TC-1-GR tumors in nude mice, only the PEG-GemC18-NPs significantly delayed the tumor growth (Supplemental Fig. S3). We did not directly compare the PEG-GemC18-NPs and GemC18-NPs in the TC-1-GR cells in nude mice. Data from previous studies showed that the PEG-GemC18-NPs and GemC18-NPs had similar anti-tumor activities against TC-1 tumors in C57BL/6 mice and BxPC-3 tumors in nude mice [27]. Inclusion of PEG(2000) in the nanoparticles increased the accumulation of the GemC18-NPs in tumors by more than 6-fold after intravenous injection [27], likely by prolonging the blood circulation time of GemC18-NPs and the enhanced permeation and retention (EPR) phenomenon. According to Cho et al. (2008), for nanoparticles to reach tumor tissues, their particle size should be up to 100 nm, which is large enough to prevent their leakage into blood capillaries, but small enough to escape capture in the reticuloendothelial system [37–39]. The GemC18-NPs used in this study are 191 ± 5 nm. It is expected that the EPR phenomenon contributed to their accumulation in the tumor tissues.
Proteomics tools were used to identify proteins that were expressed differently in the TC-1 and TC-1-GR cells. It was found that 6 proteins were down-regulated and 2 proteins were up-regulated in TC-1-GR cells, as compared to TC-1 cells (Table 1). Additional proteins may be identified using other techniques. At this moment it is not clear how the down-regulation of these proteins was related to gemcitabine resistance in TC-1-GR cells. The endoplasmin precursor and protein disulfide-isomerase precursor were down-regulated in TC-1-GR cells (Table 1). They are part of the protein folding machinery in endoplasmic reticulum. Their down-regulation may suggest improper folding of proteins in the TC-1-GR cells. Consequently, it may have led to the degradation of mis-folded proteins and, subsequently, the malfunction of the endoplasmic reticulum in the TC-1-GR cells. Tubulin β-5 and Lamin-A/C were also down-regulated, in agreement with Cecconi et al.’s finding that in pancreas cells treated with gemcitabine, these two proteins were down-regulated by 2.5-fold [40]. The calumenin precursor, a Ca2+-regulating protein, was down-regulated as well. Calumenin precursor was found down-regulated in metastatic UMSCC10B cells [41]. A nother down-regulated protein was the elongation factor 1-β, which is critical for protein synthesis [42].
On the other hand, vimentin was up-regulated. The emergence of the epithelial to mesenchymal transitions (EMT) was shown during tumor progression in human solid tumors [43]. EMT was also induced as part of secondary events subsequent to the acquisition of metastatic sites by cancer cells. During the acquisition of the EMT characteristics, cancer cells lose the expression of proteins that promote cell to cell contact, such as E-cadherin and γ-catenin, and gain the expression of mesenchymal markers, such as vimentin, fibronectin, and N-cadherin, leading to enhanced cancer cell migration and invasion [44]. Vimentin, one of mesenchymal markers, was increased 9.7-fold in TC-1-GR cells. This is in agreement with Shah et al.’s report that vimentin was increased in gemcitabine resistant pancreatic cells [45]. It was also found that the doubling time of TC-1-GR cells was shorter than that of TC-1 cells, and the TC-1-GR tumors seemed to grow slightly more aggressively than TC-1 tumors in mice, which may be related to the increased expression of vimentin in the TC-1-GR cells.
The other protein that was up-regulated in the TC-1-GR cells was the RRM1, which was also confirmed by RT-PCR, western blotting, and indirect evidence such as the cytotoxicity of didox in the cells. RT- PCR confirmed the over-expressing of RRM1 in TC-1-GR cells, but not other genes that are known to be related to gemcitabine resistance (e.g., RRM2, hENT1, dCK, and CDA) (Fig. 6A). In the immunoblotting in Fig. 6B, RRM1 was not detectable in TC-1 cells. Bergman et al. (2005) could not detect RRM1 protein in the wild type of colon 26-A cells using Western blotting as well [17]. RR acts as a critical enzyme in de novo DNA synthesis. It converts ribonucleotides to deoxyribonucleotides for DNA polymerization and repair [46]. RR increases the deoxynucleoside triphosphate (dNTP) pool in the cells, which may lead to decreased incorporation of dNTP analogues such as triphosphorylated gemcitabine into DNA, and thus, reduce the cytotoxicity of gemcitabine [14]. On the other hand, the diphosphorylated form of gemcitabine, dFdCDP, acts as an RR inhibitor [2, 11]. Therefore, gemcitabine and RR are mutual inhibitors. Data in the present study confirmed the increased expression of RRM1 in the TC-1-GR cells, which is consistent with findings by others using other cell lines [15, 17, 47]. For example, Rosell et al. (2001) reported that RR is an important target for gemcitabine resistance [48]. Therefore, the increased RRM1 expression was likely responsible for the resistance to gemcitabine in the TC-1-GR cells, which was further supported by the observation that transfection of RRM1 siRNA into TC-1-GR cells made the cells more sensitive to gemcitabine HCl (Fig. 6D). RRM1 is believed to play a key role for gemcitabine resistance in vitro [14, 15] and in vivo [16, 17]. Moreover, increasing evidence pointed to the inverse correlation between the expression of RRM1 in tumors and the sensitivity of the tumors to gemcitabine not only in cell lines in culture, but also in clinics [18–20].
It is clear that the stearoyl gemcitabine nanoparticles were effective against the gemcitabine resistant TC-1-GR tumors in vitro and in vivo. However, it remains unknown how the GemC18-NPs overcome gemcitabine resistance in the TC-1-GR tumors with increased expression of RRM1. Here is what is known so far. First, preliminary data in our laboratory showed that the TC-1-GR cells were still resistant to gemcitabine HCl that was physically mixed with GemC18-free nanoparticles (data not shown), demonstrating that the blank nanoparticles per se were not responsible for the GemC18-NPs’ ability to more effectively kill the TC-1-GR cells than gemcitabine HCl. The finding also indicated that it is not that the GemC18 was degraded extracellularly, and the resultant free gemcitabine was responsible for the biological effect observed. Second, our previous data showed that the GemC18-NPs formulation was critical for the potent anti-tumor activity of the GemC18 [27]. For example, intravenous injection of GemC18-NPs (150 g GemC18/mouse for 5 times) to mice with TC-1 tumors significantly delayed the tumor growth, but the same dose of GemC18 in Tween 20 micelles did not show any activity [27]. Finally, in culture, the GemC18 was not as cytotoxic to TC-1-GR cells as GemC18-NPs (Fig. 3B). Gemcitabine is rapidly metabolized and eliminated, resulting in its short half-life [49]. However, amino acid amide derivatives of gemcitabine at the 4-(N)-position on the cytidine ring were reported to be resistant to deamination [50]. GemC18-NPs have the amide structure with a stearoyl group at the 4-(N)-position on the cytidine ring. Data from our previous studies showed that the hydrolysis or release of the gemcitabine from the GemC18-NPs was slow [27]. Therefore, it is possible that the slow and continuous supply of gemcitabine from GemC18-NPs may have resulted in the steady production of dFdCDP, an inhibitor of the RRM1. More experiments are needed to be completed to elucidate the mechanism(s) responsible for the ability of the GemC18-NPs to overcome resistance against gemcitabine related to increased RRM1 expression. Nevertheless, to our best knowledge, this represents the first report demonstrating that a nanoparticle formulation of gemcitabine overcome gemcitabine resistance in tumors that over-expressed RRM1. This finding will likely have clinical relevance considering that the level of RRM1 in tumors in patients was found to be inversely correlated to the effectiveness of gemcitabine therapy.
5. Conclusion
It was found that a stearoyl gemcitabine nanoparticle formulation can overcome gemcitabine resistance related to the over-expression of RRM1, not only in culture, but also in mice. When fully developed, the stearoyl gemcitabine nanoparticles may represent a more efficacious gemcitabine formulation.
Supplementary Material
Acknowledgments
This work was supported in part by a National Cancer Institute grant (CA135274) to Z.C.
Footnotes
Author’s contribution: W-G.C, Figures 1, 2, 4, 5, 6, S3, Table 1 and manuscript preparation; M.A.S., Figures 3, S1, S2, Table S1; B.R.S., Figures 3, S1; D.S.P.L-P., GemC18 synthesis; Z.C., experimental design and manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaye SB. Gemcitabine: current status of phase I and II trials. J Clin Oncol. 1994;12:1527–1531. doi: 10.1200/JCO.1994.12.8.1527. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–572. [PubMed] [Google Scholar]
- 3.van Moorsel CJ, Veerman G, Bergman AM, Guechev A, Vermorken JB, Postmus PE, Peters GJ. Combination chemotherapy studies with gemcitabine. Semin Oncol. 1997;24:S7-17–S17-23. [PubMed] [Google Scholar]
- 4.Peters GJ, van der Wilt CL, van Moorsel CJ, Kroep JR, Bergman AM, Ackland SP. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther. 2000;87:227–253. doi: 10.1016/s0163-7258(00)00086-3. [DOI] [PubMed] [Google Scholar]
- 5.van Moorsel CJ, Peters GJ, Pinedo HM. Gemcitabine: Future Prospects of Single-Agent and Combination Studies. Oncologist. 1997;2:127–134. [PubMed] [Google Scholar]
- 6.Andersson R, Aho U, Nilsson BI, Peters GJ, Pastor-Anglada M, Rasch W, Sandvold ML. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44:782–786. doi: 10.1080/00365520902745039. [DOI] [PubMed] [Google Scholar]
- 7.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–4357. [PubMed] [Google Scholar]
- 8.Garcia-Manteiga J, Molina-Arcas M, Casado FJ, Mazo A, Pastor-Anglada M. Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2′,2′-difluorodeoxycytidine- induced cytotoxicity. Clin Cancer Res. 2003;9:5000–5008. [PubMed] [Google Scholar]
- 9.Bergman AM, Pinedo HM, Peters GJ. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine) Drug Resist Updat. 2002;5:19–33. doi: 10.1016/s1368-7646(02)00002-x. [DOI] [PubMed] [Google Scholar]
- 10.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- 11.Pereira S, Fernandes PA, Ramos MJ. Mechanism for ribonucleotide reductase inactivation by the anticancer drug gemcitabine. J Comput Chem. 2004;25:1286–1294. doi: 10.1002/jcc.20054. [DOI] [PubMed] [Google Scholar]
- 12.Neff T, Blau CA. Forced expression of cytidine deaminase confers resistance to cytosine arabinoside and gemcitabine. Exp Hematol. 1996;24:1340–1346. [PubMed] [Google Scholar]
- 13.Bouffard DY, Laliberte J, Momparler RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45:1857–1861. doi: 10.1016/0006-2952(93)90444-2. [DOI] [PubMed] [Google Scholar]
- 14.Goan YG, Zhou B, Hu E, Mi S, Yen Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res. 1999;59:4204–4207. [PubMed] [Google Scholar]
- 15.Davidson JD, Ma L, Flagella M, Geeganage S, Gelbert LM, Slapak CA. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64:3761–3766. doi: 10.1158/0008-5472.CAN-03-3363. [DOI] [PubMed] [Google Scholar]
- 16.Cao MY, Lee Y, Feng NP, Xiong K, Jin H, Wang M, Vassilakos A, Viau S, Wright JA, Young AH. Adenovirus-mediated ribonucleotide reductase R1 gene therapy of human colon adenocarcinoma. Clin Cancer Res. 2003;9:4553–4561. [PubMed] [Google Scholar]
- 17.Bergman AM, Eijk PP, van Haperen Ruiz VW, Smid K, Veerman G, Hubeek I, van den Ijssel P, Ylstra B, Peters GJ. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005;65:9510–9516. doi: 10.1158/0008-5472.CAN-05-0989. [DOI] [PubMed] [Google Scholar]
- 18.Rosell R, Danenberg KD, Alberola V, Bepler G, Sanchez JJ, Camps C, Provencio M, Isla D, Taron M, Diz P, Artal A. Ribonucleotide reductase messenger RNA expression and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res. 2004;10:1318–1325. doi: 10.1158/1078-0432.ccr-03-0156. [DOI] [PubMed] [Google Scholar]
- 19.Ceppi P, Volante M, Novello S, Rapa I, Danenberg KD, Danenberg PV, Cambieri A, Selvaggi G, Saviozzi S, Calogero R, Papotti M, Scagliotti GV. ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann Oncol. 2006;17:1818–1825. doi: 10.1093/annonc/mdl300. [DOI] [PubMed] [Google Scholar]
- 20.Ohtaka K, Kohya N, Sato K, Kitajima Y, Ide T, Mitsuno M, Miyazaki K. Ribonucleotide reductase subunit M1 is a possible chemoresistance marker to gemcitabine in biliary tract carcinoma. Oncol Rep. 2008;20:279–286. [PubMed] [Google Scholar]
- 21.Schirmer M, Stegmann AP, Geisen F, Konwalinka G. Lack of cross-resistance with gemcitabine and cytarabine in cladribine-resistant HL60 cells with elevated 5′-nucleotidase activity. Exp Hematol. 1998;26:1223–1228. [PubMed] [Google Scholar]
- 22.Bergman AM, Adema AD, Balzarini J, Bruheim S, Fichtner I, Noordhuis P, Fodstad O, Myhren F, Sandvold ML, Hendriks HR, Peters GJ. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs. 2011;29:456–466. doi: 10.1007/s10637-009-9377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen P, Chien PY, Khan AR, Sheikh S, Ali SM, Ahmad MU, Ahmad I. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anticancer Drugs. 2006;17:53–61. doi: 10.1097/01.cad.0000185182.80227.48. [DOI] [PubMed] [Google Scholar]
- 24.Alexander RL, Greene BT, Torti SV, Kucera GL. A novel phospholipid gemcitabine conjugate is able to bypass three drug-resistance mechanisms. Cancer Chemother Pharmacol. 2005;56:15–21. doi: 10.1007/s00280-004-0949-0. [DOI] [PubMed] [Google Scholar]
- 25.Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10:836–841. doi: 10.2174/138920009790274540. [DOI] [PubMed] [Google Scholar]
- 26.Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J Control Release. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Sloat BR, Sandoval MA, Li D, Chung WG, Lansakara PD, Proteau PJ, Kiguchi K, DiGiovanni J, Cui Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409:278–288. doi: 10.1016/j.ijpharm.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz van Haperen VW, Veerman G, Eriksson S, Boven E, Stegmann AP, Hermsen M, Vermorken JB, Pinedo HM, Peters GJ. Development and molecular characterization of a 2′,2′-difluorodeoxycytidine-resistant variant of the human ovarian carcinoma cell line A2780. Cancer Res. 1994;54:4138–4143. [PubMed] [Google Scholar]
- 29.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Chung WG, Miranda CL, Maier CS. Detection of carbonyl-modified proteins in interfibrillar rat mitochondria using N′-aminooxymethylcarbonylhydrazino-D-biotin as an aldehyde/keto-reactive probe in combination with Western blot analysis and tandem mass spectrometry. Electrophoresis. 2008;29:1317–1324. doi: 10.1002/elps.200700606. [DOI] [PubMed] [Google Scholar]
- 31.Marley K, Mooney DT, Clark-Scannell G, Tong TT, Watson J, Hagen TM, Stevens JF, Maier CS. Mass tagging approach for mitochondrial thiol proteins. J Proteome Res. 2005;4:1403–1412. doi: 10.1021/pr050078k. [DOI] [PubMed] [Google Scholar]
- 32.Chavez J, Wu J, Han B, Chung WG, Maier CS. New role for an old probe: affinity labeling of oxylipid protein conjugates by N′-aminooxymethylcarbonylhydrazino d-biotin. Anal Chem. 2006;78:6847–6854. doi: 10.1021/ac0607257. [DOI] [PubMed] [Google Scholar]
- 33.Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22:7524–7536. doi: 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- 34.Clarke ML, Mackey JR, Baldwin SA, Young JD, Cass CE. The role of membrane transporters in cellular resistance to anticancer nucleoside drugs. Cancer Treat Res. 2002;112:27–47. doi: 10.1007/978-1-4615-1173-1_2. [DOI] [PubMed] [Google Scholar]
- 35.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3:415–424. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 36.Leisewitz AV, Zimmerman EI, Jones SZ, Yang J, Graves LM. Imatinib-resistant CML cells have low ENT activity but maintain sensitivity to gemcitabine. Nucleosides Nucleotides Nucleic Acids. 2008;27:779–786. doi: 10.1080/15257770802145892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 38.Wisse E, Braet F, Luo D, De Zanger R, Jans D, Crabbe E, Vermoesen A. Structure and function of sinusoidal lining cells in the liver. Toxicol Pathol. 1996;24:100–111. doi: 10.1177/019262339602400114. [DOI] [PubMed] [Google Scholar]
- 39.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 40.Cecconi D, Donadelli M, Scarpa A, Milli A, Palmieri M, Hamdan M, Areces LB, Rappsilber J, Righetti PG. Proteomic analysis of pancreatic ductal carcinoma cells after combined treatment with gemcitabine and trichostatin A. J Proteome Res. 2005;4:1909–1916. doi: 10.1021/pr050154j. [DOI] [PubMed] [Google Scholar]
- 41.Wu W, Tang X, Hu W, Lotan R, Hong WK, Mao L. Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin Exp Metastasis. 2002;19:319–326. doi: 10.1023/a:1015515119300. [DOI] [PubMed] [Google Scholar]
- 42.Moldave K. Eukaryotic protein synthesis. Annu Rev Biochem. 1985;54:1109–1149. doi: 10.1146/annurev.bi.54.070185.005333. [DOI] [PubMed] [Google Scholar]
- 43.Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, Ouelaa R, Bracke M, De Wever O, Gespach C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat. 2008;11:123–151. doi: 10.1016/j.drup.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 44.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 45.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629–3637. doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 46.Wright JA, Chan AK, Choy BK, Hurta RA, McClarty GA, Tagger AY. Regulation and drug resistance mechanisms of mammalian ribonucleotide reductase, and the significance to DNA synthesis. Biochem Cell Biol. 1990;68:1364–1371. doi: 10.1139/o90-199. [DOI] [PubMed] [Google Scholar]
- 47.Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, Yamasaki M, Marubashi S, Takemasa I, Miyamoto A, Takeda Y, Nagano H, Dono K, Umeshita K, Sakon M, Monden M. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer. 2007;120:1355–1363. doi: 10.1002/ijc.22390. [DOI] [PubMed] [Google Scholar]
- 48.Rosell R, Sanchez JM, Taron M, O’Brate A, Gutierrez JL, Monzo M, Felip E, Sanchez JJ, Alberola V. Novel approaches in the treatment of non-small-cell lung cancer. Oncology (Williston Park) 2001;15:52–60. [PubMed] [Google Scholar]
- 49.Reid JM, Qu W, Safgren SL, Ames MM, Krailo MD, Seibel NL, Kuttesch J, Holcenberg J. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J Clin Oncol. 2004;22:2445–2451. doi: 10.1200/JCO.2004.10.142. [DOI] [PubMed] [Google Scholar]
- 50.Bender DM, Bao J, Dantzig AH, Diseroad WD, Law KL, Magnus NA, Peterson JA, Perkins EJ, Pu YJ, Reutzel-Edens SM, Remick DM, Starling JJ, Stephenson GA, Vaid RK, Zhang D, McCarthy JR. Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J Med Chem. 2009;52:6958–6961. doi: 10.1021/jm901181h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.