

Abstract
In parasitology, particularly in helminthes studies, several methods have been used to look for the expression of specific molecules, such as RT-PCR, western blot, 2D-electrophoresis, and microscopy, among others. However, these methods require homogenization of the whole helminth parasite, preventing evaluation of individual cells or specific cell types in a given parasite tissue or organ. Also, the extremely high interaction between helminthes and host cells (particularly immune cells) is an important point to be considered. It is really hard to obtain fresh parasites without host cell contamination. Then, it becomes crucial to determine that the analyzed proteins are exclusively from parasitic origin, and not a consequence of host cell contamination. Flow cytometry is a fluorescence-based technique used to evaluate the expression of extra-and intracellular proteins in different type cells, including protozoan parasites. It also allows the isolation and recovery of single-cell populations. Here, we describe a method to isolate and obtain purified helminthes cells.
1. Introduction
Flow cytometry is a technique that uses the principles of light scattering, light excitation, and fluorescence, to analyze size, complexity, molecular or cellular characteristics of single cells or particles by suspending them in a stream of fluid [1]. This technique is widely used to analyze the expression of both extracellular and intracellular proteins, secreted molecules and DNA-content, among others [2]. It is also used in basic and clinical research (immunology, hematology, molecular biology) and recently in other research areas such as microbiology, algae, and plant biology. Among the main advantages of flow cytometry are (1) the multi-parametric analysis of mixed populations by identifying specific markers on these cells and (2) the separation of particular subpopulations by electrical or mechanical means to divert cells with measured characteristics within the range specified by the user [1].
Helminthes present a formidable challenge to the mammalian defense mechanisms. They are large, meaning that their size exceeds that one of the host defense cells by several orders of magnitude. Furthermore, the parasite and the host interphase is outside the cells, partially because most helminthes have a relatively impermeable cuticle composed of proteins that can be structurally arranged so they become “hard”. It is unclear how the mammalian defense mechanisms could incapacitate these macroscopic animals. Furthermore, it is worth noting that tissue dwelling nematodes poses a completely different problem than the better studied gastrointestinal nematodes [3]. In the latter instances, live parasites can be successfully eliminated from within the mammal by such effectors as mucin of increased viscosity and enhanced peristaltic activity. Such mechanisms are not available in the case of tissue dwelling nematodes, which have to be killed and perhaps broken down and digested before they can be successfully eliminated. Thus, many host cells are literally covering all helminth body, as well as inside the parasite. This characteristic of helminthes makes it really difficult to isolate and characaterize antigens, or proteins that belong exclusively to the parasite, without host interference or contamination. Thus, finding a method that is capable to render cells of only parasite origin is a challenge for researchers.
For the past years, in our laboratory, we have been trying out to describe the role of sex steroids on different helminthes parasites. Also, the specific receptors of the parasite able to respond to these molecules have been partially characterized [4, 5]. Previously, have been detected mRNA and protein, respectively, for steroid hormone receptors by using real-time reverse-transcriptase polymerase chain reaction (RT-PCR) and fluorescent microscopy methods in several parasites. However, as previously mentioned, due to the extremely high interaction between helminthes and immune or other host cell types, which may eventually lead to host cell invasion into several parasitic tissues, it is critical to determine that the detected and analyzed protein(s) are exclusively found in the helminth parasitic cells, and not as a consequence of host immune (or any other type) of cell contamination. Thus, it was crucial to develop a method that could sort out very specifically, and without any possible error, the cross-contamination between host and parasite cells. To date, the available methods to dissect single parasite cells are quite expensive and complicated (microscopic laser dissection, for instance). Here we describe a method to dissect different helminthes cells, and by using flow cytometry, we were able to determine that there was no host cell contamination and provide information that demonstrate that, these cells are from only helminthes cell population based on both proportions and size of cells, as well as microscopic characterization.
2. Materials and Methods
2.1. Ethics Statement
Animal care and experimentation practices at the Instituto de Investigaciones Biomédicas are frequently evaluated by the Institute's Animal Care and Use Committee, according to the official Mexican regulations (NOM-062-ZOO-1999). Mexican regulations are in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH and The Weatherall Report) of the USA, to ensure compliance with established international regulations and guidelines. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Instituto de Investigaciones Biomédicas (Permit Number: 2011 –16). Mouse, rats, and pigs sacrifice to obtain parasites was performed under anesthesia, and all efforts were made to minimize animal suffering.
2.2. Harvesting and Preparing T. crassiceps and T. solium cysticerci, and Trichinella spiralis Larvae for Experimentation
Taenia crassiceps cysticerci for each experimental session were obtained from intraperitoneally infected female mice and placed in tubes containing sterile PBS (1x) supplemented with 100 U/mL of antibiotics-fungizone (Gibco, Grand Island). The tubes were centrifuged for 10 min at 1,500 rpm at 4°C and the supernatants were discarded. The packed cysticerci were incubated in DMEM serum-free medium (Sigma, St. Louis, Missouri). They were then centrifuged 3 times for 10 min at 1500 rpm for washing. After the final wash, the numbers of viable (complete, translucent and motile cystic structures) cysticerci were counted under a binocular microscope. Ten viable nonbudding cysticerci of approximately 2 mm in diameter were selected and dispensed in 24-well culture plates (Falcon, BD Labware, Franklin Lakes, New Jersey) in 1 mL DMEM serum-free medium (Gibco BRL) and maintained at 37°C under 5% CO2 until used. Taenia solium cysticerci were dissected from the muscle of infected pigs in independent experiments. The fibrous capsule surrounding each parasite was carefully separated under a dissection microscope. Once dissected, cysticerci were placed in tubes containing sterile PBS (1x) supplemented with 100 U/mL of antibiotics-fungizone (Gibco, Grand Island, NY). The tubes were centrifuged for 10 min., at 800 g at 4°C, and the supernatant was discarded. The packed cysticerci were incubated in DMEM serum-free medium (Gibco, BRL, Rockville, MD). They were then centrifuged 3 times for 10 min at 800 g for washing. After the final wash, the numbers of viable (complete and translucent cystic structures) cysticerci were counted using a binocular microscope and placed in 24-well culture plates (Falcon, BD Labware, Franklin Lakes, New Jersey) in 1 mL DMEM serum-free medium (Gibco BRL) and maintained at 37°C under 5% CO2 until used. Trichinella spiralis (ISS 406) was maintained in the laboratory by serial passage infections in BALB/c mice and/or Sprague-Dawley rats. The infective-stage muscular larvae (ML) were recovered from experimentally infected mice at 30 days p.i. by a standard pepsin-hydrochloric acid digestion method. Larvae were washed several times with PBS, and 4,000 ML were used to infect Sprague-Dawley rats. Adult parasites were recovered from 1 to 6 days p.i. from the small intestine. The NBL (24 –72 h old) were obtained by incubating 4-, 5-, and 6-day-old adult parasites in RPMI medium (Gibco BRL) supplemented with 10% FBS (Hyclone) and antibiotics (Gentamicin 50 mg/L and Amphotericin B, Sigma, 2 .5 mg/L).
2.3. T. crassiceps, T. solium, and T. spiralis Cell Isolation
T. crassiceps, T. solium and T. spiralis cells were extracted by tissue disruption according to the following protocols. T. crassiceps parasites were macerated using a nylon mesh “sandwich” (150 mm. Small Parts) and a syringe plunger in 1 mL of RPMI media. Meshes were washed with media, and cell suspension was centrifuged at 300 ×g for 5 min, and cells in pellet were recovered in PBS. T. solium parasites were disaggregated initially by passing them through a 3 mL syringe to recover internal cells. Disrupted cysticerci were macerated as described for T. crassiceps cells. Cells recovered in RPMI media, centrifuged at 300 ×g for 5 min and pellets were recovered in PBS. T. spiralis larvae were disaggregated using a micropestle (Eppendorf, USA) until no more clumps were visible. Cells were centrifuged at 300 ×g for 5 min and cells in pellet recovered in PBS. Parasite cell viability was >80% for all three parasites.
2.4. Analysis of Host-Derived Cells in T. crassiceps and T. solium cysticerci, and T. spiralis Larvae Cells by Flow Cytometry
T. crassiceps and T. spiralis larvae cells were stained with the following antibodies for 10 min at 4°C: anti-mouse CD3-FITC, anti-mouse CD4 FITC, anti-mouse CD8-PE-Cy5, anti-mouse CD19-PE, anti-mouse Mac-1, and anti-mouse Mac-3 (all from BD Biosciences), and washed with 500 μL of staining buffer (PBS pH.7.4, 2% Fetal Bovine Serum, 0 .02% NaN2). Cells were fixed in 2% paraformaldehyde solution and stored until analysis protected from light. T. solium cysticerci-derived cells were stained with anti-human MHC-I-biotin antibody, washed once, and stained with APC-coupled Streptavidin (BD Biosciences). All samples were analyzed by flow cytometry using an FACS Calibur (BD, Biosciences) and data analyzed using the FlowJo© software.
2.5. Specific Determination of T. crassiceps and T. solium cysticerci, and T. spiralis Larvae Cells by Flow Cytometry
Helmith-derived cells were fixed in 2% paraformaldehyde solution for 10 min at 37°C and centrifuged at 300 ×g for 5 min. Afterwards, they were incubated in absolute methanol for 30 min at 4°C and centrifuged at 300 ×g for 5 minutes and then washed twice with 500 μL of staining buffer (PBS pH.7.4, 2% Fetal Bovine Serum, 0 .02% NaN2) and resuspended in same buffer (100 μL/test). T. crassiceps and T. solium cells were incubated in presence of mouse antiparamyosin (1 μg/test) (kindly provided by Dr. Pedro Ostoa, Instituto de Investigaciones Biomédicas, UNAM). T. spiralis cells were incubated in the presence of mouse anticaveolin (1 μg/test) (kindly provided by Dr. Guadalupe Ortega-Pierres, CINVESTAV, IPN.) at room temperature for 20 min, and subsequently washed with 1 mL of staining buffer. Immediately after, cells were centrifuged at 300 ×g for 5 min. Cell pellets were resuspended separately in presence of the secondary antibody FITC or Alexa488-conjugated goat anti-mouse (Zymed) and incubated at 4°C for 30 min in the dark. After this second incubation, cells were washed twice in staining buffer and centrifuged at 300 ×g for 5 min. Cell pellets were resuspended in 500 μL of staining buffer in absence of light and analyzed by flow cytometry using an FACS Calibur (BD, Biosciences). Data was analyzed with FlowJo software.
2.6. Nuclei Staining with Diamidino-Phenylindole (DAPI) of Isolated Cells
For DAPI fluorescent staining, in vitro cultivated primary cells were collected, fixed in paraformaldehyde (Sigma) for 10 min, and permeabilized with 0 .2% Triton X-100 (Merck) for 5 min. DAPI at a concentration of 0 .5 μg/mL was added to the fixed cells on the slide, followed by an incubation for 20 min in the dark. Slides were then rinsed in PBS, Fluoprep (bioMerieux) was added, and a cover slip applied. The stained biological substrates were visualized using an optical microscope. For light microscopy applications, we used a Nikon Eclipse E600 microscope equipped with a Nikon DXM1200 F CCD (Nikon Corp.; Tokyo, Japan). Immunofluorescence staining experiments were carried out using a Nikon Eclipse 80 i microscope and a Nikon DXM1200 C CCD (Nikon Corp.). For DAPI visualization, a 350 LP filter was used (Nikon, Corp.). Image processing and analysis was carried out using Adobe Photoshop CS3 (Adobe Systems Inc., San Jose, CA, USA) and Image Pro Plus 6 .2 (Media Cybernetics, Bethesda, MD, USA).
3. Results
3.1. Taenia solium, Taenia crassiceps, and Trichinella spiralis Single Cells
In Figure 1, it is clear that we were able to obtain isolated single cells of the cestodes (a) Taenia solium, (b) Taenia crassiceps, and the nematode (c) Trichinella spiralis. The composition shown in the first photograph of every row shows the complete parasites, before of the isolation process. In the second picture of every row, the first step, by disrupting the parasites and, showing clumps of parasites, and, debris of pieces of the same. Finally, in the third picture of every row, the isolated single cells of the three parasites are depicted. It is interesting to note that, Taenia crassiceps (b) cells are multinucleated, and, due to the size (10 μm) it cannot be a complete parasite. The same can be said also for Taenia solium (a) and Trichinella spiralis (c). Cell viability was >80% for all three parasites after disaggregation (data not shown).
Figure 1.

Imaging of the process of getting single cells from T. solium, T. crassiceps, and T. spiralis after mechanical disaggregation. (a) T. solium cysticerci (first picture), pieces of T. solium after disgregation (middle picture), and T. solium isolated cells (third picture of panel (a)). In (b) T. crassiceps (first picture), pieces of T. crassiceps after disaggregation (middle picture), and T. crassiceps isolated cells (third picture of panel (b)) and (c) Trichinella spiralis (first picture), pieces of T. spiralis after disaggregation (middle picture) and T. spiralis isolated cells (third picture of panel (c)). Pictures were taken using an inverted microscope (Olympus, MO21, Tokyo) at 10x and 100x magnification.
3.2. DAPI Stained Normal Isolated Parasite Cells
Often, one may wish to monitor the presence or expression of several different molecules in cell culture. One component that is almost always monitored is the cell nucleus, which is stained with DAPI, a molecular probe characterized by λex = 358 nm and λem = 461 nm. DAPI binds to the inner groove of DNA present in cell nuclei and results in a blue emission that can be seen in the fluorescent microscope. Staining of DAPI in these parasite-isolated cells not only allows us to visualize the cell nuclei, but also allows an easy quantification of the number of cells in a given field of view. A representative example of the images processing steps and subsequent, phase contrast, counting of cell nuclei stained with DAPI, and the overlay using both techniques are shown in Figure 2, where (a) is the composition to show T. solium cells, (b) T. crassiceps cells, and (c) T. spiralis cells.
Figure 2.
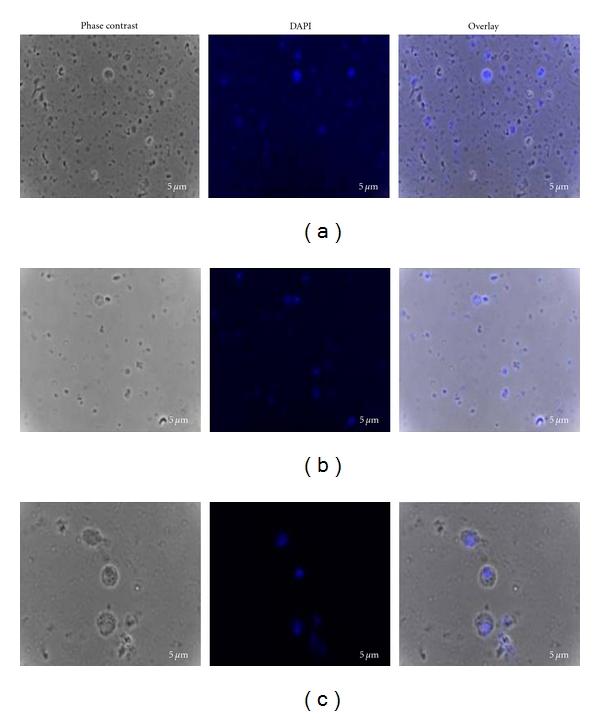
Disruption of parasite tissue and staining of primary cells. After isolation, primary cells were fixed and nuclei were stained with DAPI. Composed figure depicts stained cells with DAPI (produces blue color) of Taenia solium, Taenia crassiceps, and Trichinella spiralis cells.
3.3. Size and Complexity of T. crassiceps, T. solium, and T. spiralis Are Different from the Host Cells
Flow cytometry analysis firstly showed that (b) T. solium, (c) T. crassiceps and (d) T. spiralis cells were different in size and complexity from mouse spleen cells (a). In fact, parasite cells were approximately 3-fold smaller and exhibited less complexity (Figures 3(b), 3(c), and 3(d)) than the mouse spleen cells (Figure 3(a)).
Figure 3.
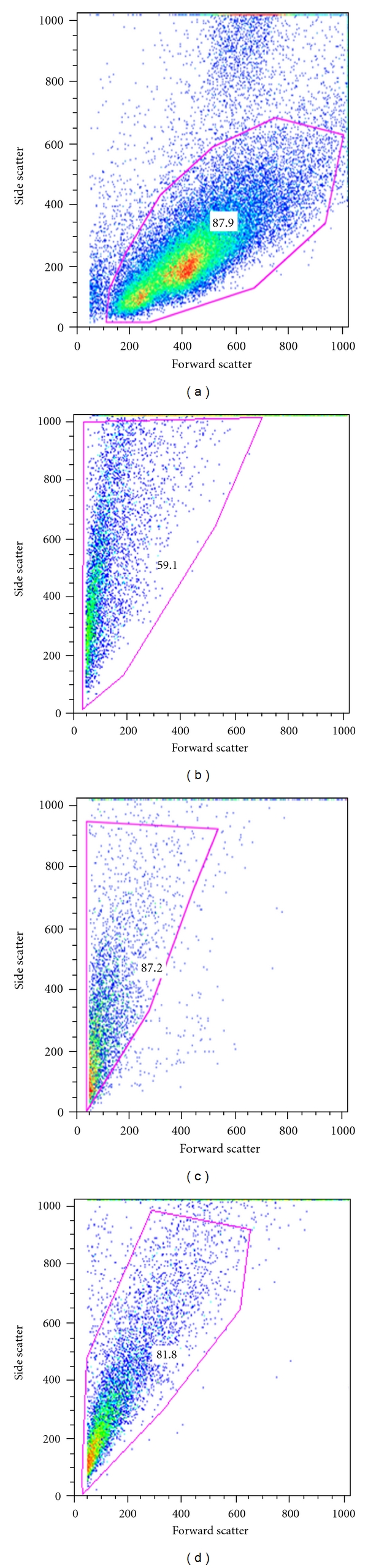
Forward/Side scatter of splenocytes and parasites isolated cells. (a) Spleen cells, (b) T. solium cells, (c) T. crassiceps cells and (d) T. spiralis cells were disaggregated by tissue disruption, washed twice with FC buffer, and fixed using Lyse/Fix buffer (BD Biosciences).
In addition, parasite cells showed no expression of the membrane markers CD3, CD4, CD8 which are typically present in some types of mammalian leukocytes or MHC I, marker of all types of mammalian cells (Figure 4).
Figure 4.
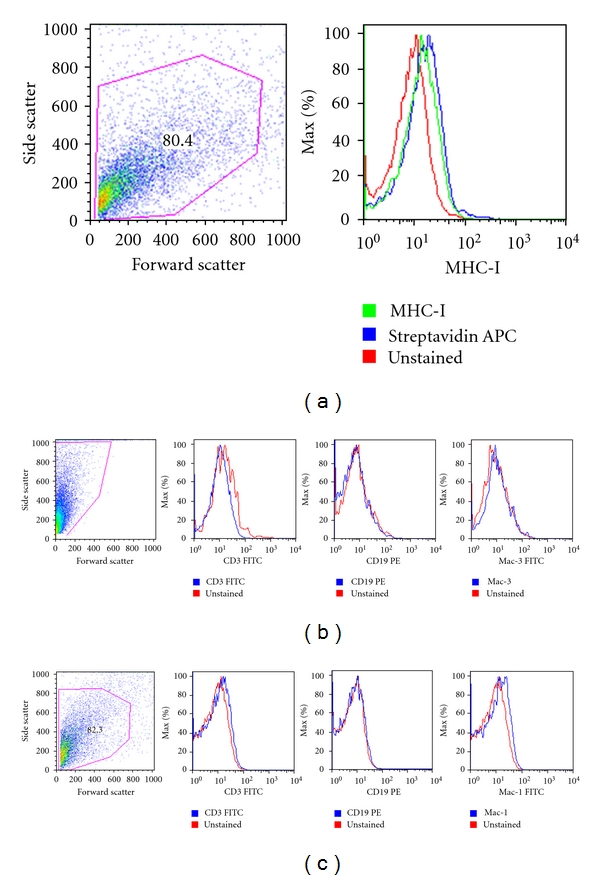
Non-host-contaminated parasite isolated cells. Cells from (a) T. solium cells, (b) T. crassiceps cells, and (c) T. spiralis cells were disaggregated by tissue disruption and stained with anti-hMHC-I, anti-mCD3, anti-mCD19, anti-mCD11 b, and anti-mMac-3 antibodies.
3.4. Expression of Paramyosin and Caveolin-1 Is Only on Parasites and Not Host Cells
On the other hand, in Figure 5, the FACS analysis showed that T. solium isolated cells expressed calreticulin (a) and T. crassiceps isolated cells expressed paramyosin (Ag-B) (b), an exclusive component of the cytoskeleton of cestodes, nematodes, and insects, while the host cells did not showed expression of this molecule (not shown). In the case of T. spiralis isolated single cells, there was a clear expression of caveolin-1 (cloned, sequenced, and expressed exclusively of this parasite), while the host cells analyzed to look for the expression of this protein have a negative dying (not shown).
Figure 5.
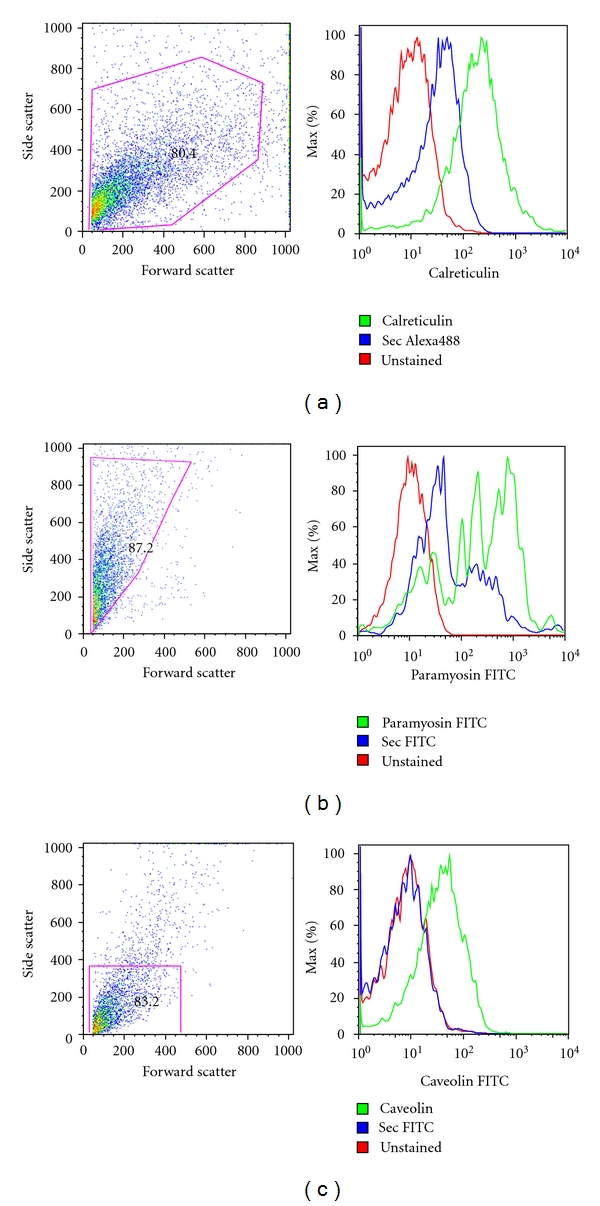
Expression of parasite-specific proteins on (a) T. solium, (b) T. crassiceps, and (c) T. spiralis. Anticalreticulin and antiparamyosin antibodies were obtained by mice immunization with cloned and expressed proteins from T. solium and T. crassiceps. Anticaveolin antibodies were obtained by mice immunization with a cloned and expressed protein from T. spiralis.
4. Discussion
Several methods are currently employed to evaluate expression of specific molecules in helminthes, including real-time reverse-transcriptase polymerase chain reaction (real-time RT-PCR), western blot, double-dimension gels, and several microscopic assays. However, all these methods require homogenization of the whole helminth parasite cells, thereby preventing evaluation of individual cells or specific cell types in a given parasite tissue or area and cannot differentiate from parasite cells and host cell contamination cells. The problem with analyzing helminths proteins is that helminth tissues are usually highly contaminated by host immune cells, thus preventing researchers to determine that the molecule they are trying to characterize truly belong to their parasites. Our method of isolation of helminth cells is highly specific and is able to determine and sort specifically the parasite cells from the host cells. In addition, parasite cells can be sorted out and recovered alive to perform experiments in these isolated cells, like, in vitro culture, transfection, and possibly regeneration of complete parasites derived from single cells.
The disaggregation, and separation for further flow cytometry analysis shown in here, is an important tool for determining protein expression of cell surface molecules and has also been shown to be useful for identifying expression of intracellular proteins in permeabilized cells [6]. Therefore, our method can be a potentially useful tool in the field of new protein helminth parasite discovery biology by providing a technique for analyzing all type of proteins from helminth parasite origin within intact parasite cells, with no host cell contamination [7]. This type of analysis has not been previously reported and may pose increased difficulties due to nonspecific binding of antibodies to intracellular proteins.
In addition to providing information on cells expressing specific proteins, once that we have the disaggregated parasites, the cells specifically detected as parasites cells, by size and granularity and by using a single specific protein marker (as paramyosin and caveolin-1 in our case), are gated to use to determine the problem protein. This gives an indication of the relative amount of the searched protein expressed by an individual cell, thus providing a means to quantify that protein on each cell. Another advantage of this technique is that expression of proteins of interest can be correlated with the degree of activation, maturation, or differentiation of given parasite cell types. Finally, if complex mixtures of parasite helminthes cells are present (as is the case), flow cytometry can be used to sort subpopulations of cells and, therefore, identify proteins expressed by specific parasite cell types. This method is, however, limited in its inability to specify the location within the cell (nucleus, cytoplasm, mitochondria) of intracellular proteins that are identified.
Specifically in the field of parasite hormone receptors, flow cytometry can be used to identify intracellular expression of steroid hormone receptors. We have been able to demonstrate and characterize expression of estrogen hormone receptors, from T. crassiceps [4] Using real-time reverse-transcriptase polymerase chain reaction (RT-PCR) and fluorescent microscopy methods, we were able to determine that it was present, respectively, for this receptor in the whole parasite [8]. This method could, therefore, be useful to answer a variety of scientific questions related to hormone receptors biology in the complex-host parasite interaction, including determining the responsiveness of specific cell types of parasites to steroid hormone treatment, for instance. This method would, therefore, be a very useful tool for rapid, high throughput measurement of not only hormone receptors, or receptors in general, but at the protein level in single, intact helminthes isolated cells.
On the other hand, it was critical to determine that the obtained cell populations were exclusively found in the T. crassiceps and T. solium cysticercus, and T. spiralis larvae, and not a consequence of host immune, or other type of cell contamination, because, again, as shown elsewhere, there is extremely high interaction between parasites and host cells, which may eventually lead to host cell invasion into several parasitic tissues [9]. For this reason, an alternative use of flow cytometry was employed to differentiate proteins from T. crassiceps, T. solium, and T. spiralis and their murine host by identifying exclusive molecules of the parasite, which are neither synthesized nor expressed by the host. This is the case of paramyosin, a muscle protein found only in invertebrates, such as Drosophila melanogaster, Caenorhabditis elegans [10, 11], Taenia solium [12, 13] and T. saginata [14], and caveolin-1 a protein that is implicated in T. spiralis differentiation [15]. The flow cytometry studies showed that presence of the analyzed cells protein belonged specifically to the parasites, because paramyosin was only detected in T. crassiceps and T. solium cells, and caveolin-1 was only expressed in T. spiralis. In contrast, the αnti-paramyosin or αnti-caveolin-1 antibody did not recognize cells extracted from mouse, but they were positive for CD3, CD4, CD8, CD19, and macrophage antibodies, contrary to parasite cells. These results demonstrate that the analyzed parasite cells are in fact from the three parasite's origin and not from other sources, and simultaneously accentuate the potential use of flow cytometry for differential identification of molecules from organisms with extremely close contact, such as helminthes parasites and their hosts.
Acknowledgments
Financial support was provided by Grant no. IN 214011 -3 from Programa de Apoyo a Proyectos de Innovación Tecnológica, Dirección General de Asuntos del Personal Académico, (PAPIIT, DGAPA), Universidad Nacional Autónoma de México to J. Morales-Montor. R. Hernández-Bello has a postdoctoral fellowship from Red Farmed.
References
- 1.Shaphiro H. Practical Flow Cytometry. New York, NY, USA: Wiley-Liss; 2003. [Google Scholar]
- 2.Cunningham RE. Overview of flow cytometry and fluorescent probes for flow cytometry. Methods in Molecular Biology. 2010;588:319–326. doi: 10.1007/978-1-59745-324-0_31. [DOI] [PubMed] [Google Scholar]
- 3.Rajan TV, Ganley L, Paciorkowski N, Spencer L, Klei TR, Shultz LD. Brugian infections in the peritoneal cavities of laboratory mice: kinetics of infection and cellular responses. Experimental Parasitology. 2002;100(4):235–247. doi: 10.1016/s0014-4894(02)00015-2. [DOI] [PubMed] [Google Scholar]
- 4.Ibarra-Coronado EG, Escobedo G, Nava-Castro K, et al. A helminth cestode parasite express an estrogen-binding protein resembling a classic nuclear estrogen receptor. Steroids. 2011;76(10-11):1149–1159. doi: 10.1016/j.steroids.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Hernández-Bello R, Ramirez-Nieto R, Sánchez-Acosta AG, et al. Sex steroids effects on the molting process of the helminth human parasite Trichinella spiralis. doi: 10.1155/2011/625380. Journal of Biomedicine and Biotechnology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krutzik PO, Irish JM, Nolan GP, Perez OD. Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clinical Immunology. 2004;110(3):206–221. doi: 10.1016/j.clim.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Thomas P, Pang Y, Dong J, et al. Steroid and G protein binding characteristics of the seatrout and human progestin membrane receptor α subtypes and their evolutionary origins. Endocrinology. 2007;148(2):705–718. doi: 10.1210/en.2006-0974. [DOI] [PubMed] [Google Scholar]
- 8.Butts CL, Shukair SA, Duncan KM, et al. Progesterone inhibits mature rat dendritic cells in a receptor-mediated fashion. International Immunology. 2007;19(3):287–296. doi: 10.1093/intimm/dxl145. [DOI] [PubMed] [Google Scholar]
- 9.Huerta L, Lopez-Balderas N, Larralde C, Lamoyi E. Discriminating in vitro cell fusion from cell aggregation by flow cytometry combined with fluorescence resonance energy transfer. Journal of Virological Methods. 2006;138(1-2):17–23. doi: 10.1016/j.jviromet.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Deitiker PR, Epstein HF. Thick filament substructures in Caenorhabditis elegans: evidence for two populations of paramyosin. Journal of Cell Biology. 1993;123(2):303–311. doi: 10.1083/jcb.123.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein HF, Casey DL, Ortiz I. Myosin and paramyosin of Caenorhabditis elegans embryos assemble into nascent structures distinct from thick filaments and multi-filament assemblages. Journal of Cell Biology. 1993;122(4):845–858. doi: 10.1083/jcb.122.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solis CF, Ostoa-Saloma P, Lugo-Martinez VH, Johnston SA, Laclette JP. Genetic vaccination against murine cysticercosis by using a plasmid vector carrying Taenia solium paramyosin. Infection and Immunity. 2005;73(3):1895–1897. doi: 10.1128/IAI.73.3.1895-1897.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vargas-Parada L, Laclette JP. Gene structure of Taenia solium paramyosin. Parasitology Research. 2003;89(5):375–378. doi: 10.1007/s00436-002-0761-x. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer E, Moyano E, Benitez L, et al. Cloning and characterization of Taenia saginata paramyosin cDNA. Parasitology Research. 2003;91(1):60–67. doi: 10.1007/s00436-003-0895-5. [DOI] [PubMed] [Google Scholar]
- 15.Hernández-Bello R, Bermúdez-Cruz RM, Fonseca-Liñán R, et al. Identification, molecular characterisation and differential expression of caveolin-1 in Trichinella spiralis maturing oocytes and embryos. International Journal for Parasitology. 2008;38(2):191–202. doi: 10.1016/j.ijpara.2007.07.009. [DOI] [PubMed] [Google Scholar]