

Abstract
Familial hypertrophic cardiomyopathy (FHC) is a disease of cardiac sarcomeres. To identify molecular mechanisms underlying FHC pathology, functional and structural differences in three FHC-related mutations in recombinant α-Tm (V95A, D175N, and E180G) were characterized using both conventional and modified in vitro motility assays and circular dichroism spectroscopy. Mutant Tm's exhibited reduced α-helical structure and increased unordered structure. When thin filaments were fully occupied by regulatory proteins, little or no motion was detected at pCa 9, and maximum speed (pCa 5) was similar for all tropomyosins. Ca2+-responsiveness of filament sliding speed was increased either by increased pCa50 (V95A), reduced cooperativity n (D175N), or both (E180G). When temperature was increased, thin filaments with E180G exhibited dysregulation at temperatures ~10°C lower, and much closer to body temperature, than WT. When HMM density was reduced, thin filaments with D175N required fewer motors to initiate sliding or achieve maximum sliding speed.
1. Introduction
Familial hypertrophic cardiomyopathy (FHC) is a disease of cardiac sarcomeres, with causal mutations identified in a variety of myofilament proteins including thin filament Ca2+-regulatory proteins [1–5]. Many mutations in cardiac troponin I (cTnI, the inhibitory subunit of the troponin complex, Tn) and cardiac troponin T (cTnT, Tn's tropomyosin-binding subunit) enhance contractile function at subsaturating, and in some instances at saturating [Ca2+] [6–13]. Cardiac α-tropomyosin (α-Tm) is a third component of the thin filament associated with FHC [14, 15] that could have a central role in functional enhancement by FHC mutations, at least at subsaturating levels of [Ca2+], although wild-type Tm by itself (i.e., in the absence of Tn) does not enhance function [7, 16, 17].
The Tm molecule is an α-helical coiled-coil dimer that binds head to tail with adjacent Tm's to form a continuous strand, with two Tm strands associated with an F-actin [18]. Each Tm is associated with seven actin monomers and one ternary complex of Tn to form a structural regulatory unit that blocks strong myosin cross-bridge formation in the absence of Ca2+ (blocked state) [19]. Ca2+ binding to troponin C (TnC, Tn's Ca2+-binding subunit) permits Tm movement on the thin filament to partially expose strong cross-bridge binding sites on actin (closed state), with further movement to the open state upon binding of myosin [19]. Some FHC mutations in α-Tm have been shown to reduce Tm's affinity for actin in vitro in the absence of Tn [20, 21] and may additionally destabilize Tn binding [22]; they further reduce the thermal stability of α-helix structure content in isolated Tm [21, 23, 24] or in actin-Tm [24]. It is not known, however, to what extent these structural changes and changes in thermal stability of Tm influence the function of regulated thin filaments.
Several FHC mutations in Tm may enhance some aspects of actomyosin function, but only at subsaturating and not saturating [Ca2+]. V95A increased Ca2+-sensitivity but also caused small decreases in the maximum solution ATPase activity, maximum speed of filament sliding in motility assays, and maximum Ca2+-activatable tension in thin filament-reconstituted cardiac preparations [15, 22, 25]. E180G increased Ca2+-sensitivity of tension in thin filament-reconstituted cardiac preparations [25], and both D175N and E180G increased Ca2+-sensitivity of the fraction of motile filaments in motility assays [26]. Maximum filament sliding speed was unchanged by D175N or E180G when the ternary complex of cardiac Tn (cTn) was used [26] but increased with skeletal Tn (sTn) [20]. Vastus lateralis fibers from patients with the D175N mutation exhibited increased Ca2+-sensitivity of isometric force compared with control fibers, with no change in maximum force or shortening velocity [27]. Maximum Ca2+-activatable tension in thin filament-reconstituted cardiac preparations was decreased for D175N but not E180G, while total maximum tension was unchanged for D175N but increased for E180G; this was attributable to increases in Ca2+-independent tension for both D175N and E180G, and V95A, too [25]. In a transgenic (Tg) mouse model, the E180G mutation leads to cardiac hypertrophy and increased mortality [28, 29]. These results from disparate assays indicate that FHC-related mutations require more thorough functional analysis at the level of single thin filaments to understand molecular mechanisms of the disease, and to allow for testing of potential therapeutic agents.
We therefore examined the influence of three FHC mutants in α-Tm on structure and Ca2+ activation of cardiac thin filaments under unloaded conditions. Thin filaments were reconstituted from recombinant, human α-GS-Tm WT, mutants E180G, D175N, or V95A, and cTn, and in vitro motility assays were used to examine function at the level of individual filaments. The three mutants increased Ca2+-responsiveness of filament sliding speed at 30°C with no change in the maximum or minimum speeds at pCa 5 or pCa 9, respectively, when thin filaments were saturated with regulatory proteins. Increased Ca2+-responsiveness by FHC mutations was associated with greater structural disorder by circular dichroism spectroscopy (CD) at 25°C; E180G exhibited reduced functional stability at ~10°C lower than WT when temperature was varied in motility assays at pCa 5. Cross-bridge duty ratio was lower by ~30% with regulated thin filaments at pCa 5 and 30°C than with unregulated F-actin; duty ratio was similarly lower for thin filaments reconstituted with cTn and mutants D175N and E180G. In addition, enhancement of sliding speed for thin filaments reconstituted with D175N occurred at lower HMM densities than for WT or E180G. These results support the hypothesis that structural destabilization of α-Tm may play a role in functional enhancement by FHC-related mutations when there are few cross-bridges and could in some cases lead to loss of regulation (diastolic dysfunction) under normally minor perturbations of cellular conditions such as slight elevation of body core temperature during exercise.
2. Materials and Methods
2.1. Tropomyosin
Full-length human α-Tm (accession no. M19713) and, for comparison in structural assays, β-Tm (accession no. X06825) cDNAs were PCR amplified from a Marathon-Ready cDNA Library (CLONTECH Laboratories Inc., Palo Alto, Calif, USA) made from human cardiac tissue. Both PCR products were confirmed by sequencing. The α- and β-Tm constructs were engineered with a BamHI site just before the start codon and a SalI site just after the stop codon by PCR using Pfu Turbo DNA polymerase (Stratagene Inc., Cedar Creek, Tex, USA). Inserts were ligated to maltose-binding protein (MBP) fusion expression vector pMAL-c2 (New England BioLabs Inc., Beverly, Mass, USA) in which a thrombin cleavage site had been engineered. DH5α cells were transformed with the ligation mixtures, and the positive clones were identified by PCR with the same primers used for cloning. The recombinant plasmids were extracted for expression and mutagenesis (α-Tm only).
Three α-Tm mutants, α-TmV95A, D175N, and E180G, were generated by PCR using QuikChange XL Site-Directed Mutagenesis system (Stratagene). Reactions were carried out per manufacturer's instructions with the template of α-Tm/pMAL-c2 plasmid and primer pairs:
pTmV95Af: 5′-CGCATCCAGCTGGCTGAGGAAGAGTTGG-3′
pTmV95Ar: 5′-CCAACTCTTCCTCAGCCAGCTGGATGCG-3′
pTmD175Nf: 5′-GGTCATCATTGAGAGCAACCTGGAACGTGC-3′
pTmD175Nr: 5′-GCACGTTCCAGGTTGCTCTCAATGATGACC-3′
pTmE180Gf: 5′-CGACCTGGAACGTGCAGGAGAGCGGGCTGAGCTCTC-3′
pTmE180Gr: 5′-GAGAGCTCAGCCCGCTCTCCTGCACGTTCCAGGTCG-3′
Plasmids extracted from three transformed mutants were sequenced with primers targeted at the flanks of MCS on pMAL-c2 vector to confirm sequence changes in the mutants.
For expression, competent E. coli BL21 (DE3) cells (Novagen Inc., Madison, Wis, USA) were transformed with cDNA/pMAL-c2 plasmids for α-Tm, β-Tm, or the α-Tm mutants. Single colonies were grown at 37°C to an OD600 of 0.5, followed by IPTG induction. The cells were harvested, resuspended in column buffer (20 mM Tris.Cl, pH 7.4, 10 mM β-mercaptoethanol, 200 mM NaCl, and 1 mM EDTA), lysed with mild sonication, centrifuged at 12,000 g, passed over an amylose affinity column (New England BioLabs Inc.), and eluted with column buffer containing 10 mM maltose. Fusion proteins were cleaved with thrombin (Sigma, St. Louis, Mo, USA) at 1 unit of thrombin per mg of protein in column buffer for 4 hrs at room temperature. The liberated recombinant Tm homodimers were purified on a Mono Q column (Pharmacia Biotech, Uppsala, Sweden) by high performance liquid chromatography (HPLC) with a gradient elution (buffer A: 20 mM Tris·Cl, pH 8.0, 25 mM NaCl; buffer B: buffer A containing 500 mM NaCl). The BamHI site on pMAL-c2 vector is integral to the thrombin cleavage region so all recombinant Tm's have two extra-amino acids (GS-) at the N-terminus; GS-represents a conservative alternative to the AS-dipeptide in bacterially expressed Tm that substitutes functionally for N-terminal acetylation [21, 30]. The purified recombinant proteins (Figure 1) were concentrated, aliquoted, and stored at −80°C in HPLC elution buffer until use [22, 25, 31].
Figure 1.
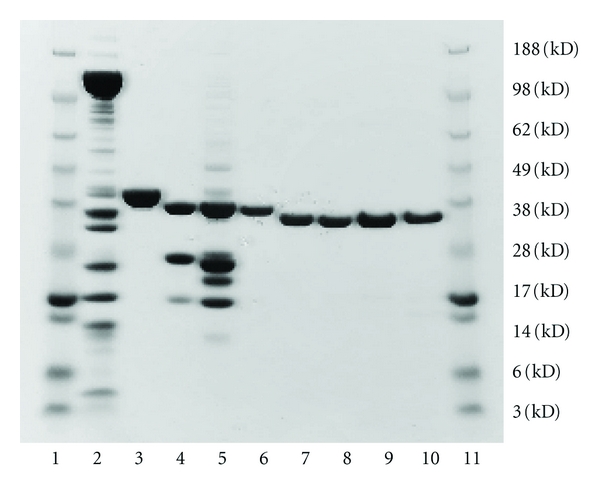
SDS-PAGE analysis of representative protein preparations. Gel was 4–12% polyacrylamide gradient (Invitrogen) and was stained with Coomassie brilliant blue. Lanes 1 and 11: MW markers (Invitrogen See Blue Plus 2; MWs given at right). Lane 2: rabbit skeletal muscle HMM. Lane 3: rabbit skeletal muscle actin. Lane 4: recombinant rat cardiac troponin (for reference only). Lane 5: human cTn (HcTn; Research Diagnostics). Lane 6: recombinant β-GS-Tm WT. Lane 7: recombinant α-GS-Tm WT. Lane 8: recombinant α-GS-Tm E180G. Lane 9: recombinant α-GS-Tm D175N. Lane 10: recombinant α-GS-Tm V95A.
2.2. Myosin, Heavy Meromyosin, Actin, and Troponin
Myosin and muscle acetone powder were made from rabbit back and leg muscles as previously described [8, 31–34]. Animal handling was in accordance with the current US National Institutes of Health/National Research Council Guide for the Care and Use of Laboratory Animals. All procedures and protocols were approved by Florida State University's Institutional Animal Care and Use Committee. Adult male New Zealand white rabbits were anesthetized with 10 mg/kg Xylazine + 50 mg/kg of Ketamine + 3 mg Acepromazine in physiological saline (IM). Following verification of appropriate surgical depth of anesthesia, the animal was exsanguinated, skinned, eviscerated, and chilled on ice. Back and leg muscles were removed for the isolation of the desired proteins. F-actin was prepared from muscle acetone powder; aliquots of F-actin were labeled with rhodamine-phalloidin (RhPh) for fluorescence microscopy [8, 31, 32, 35]. Heavy meromyosin (HMM) was prepared by chymotryptic digestion of myosin [8, 31, 32, 35]. Native human cardiac troponin (HcTn) was obtained from Research Diagnostics (Flanders, NJ, USA). SDS-PAGE analysis of representative batches of proteins is shown in Figure 1.
2.3. SDS-PAGE and Western Blotting
Proteins were analyzed by SDS-PAGE and Western blotting. SDS-PAGE was conducted with 4–12% SDS-PAGE gels (Invitrogen Inc., Carlsbad, Calif, USA). Gels were either stained with Coomassie brilliant blue (Sigma) to visualize proteins (Figure 1) and analyzed with an EDAS-290 digital imaging system (Kodak, Rochester, NY, USA), or were used to transfer proteins to nitrocellulose (NC) membranes (BIO-RAD, Hercules, Calif, USA) at 30 mA, 4°C for overnight. NC membranes were rinsed for 2 min in TBS-T buffer (20 mM Tris·Cl, 150 mM NaCl, 0.05% Tween-20, pH 7.5), blocked for 1 hr at room temperature in TBS-T containing 2% BSA (Sigma), then incubated with rabbit anti-Tm antibody (Sigma, T3651) in TBS-T (1 : 3000) for 1 hr at room temperature. Bound antibody was detected using horseradish peroxidase-conjugated anti-rabbit IgG, followed by enhanced chemiluminescence reaction with ECL Plus Kit (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Finally, the blots were exposed to Kodak X-ray film (Kodak, Rochester, NY, USA).
2.4. Circular Dichroism Spectroscopy
α-Tm, β-Tm, and three α-Tm mutants were dialyzed against CD buffer (20 mM sodium phosphate, 20 mM NaCl, pH 7.4) and concentrated using Centricon concentrators of 30,000 MW cut off (Millipore Corporation, Bedford, Mass, USA). Protein concentration was determined in CD buffer (in absence or presence [36] of 6 M urea) from A280 using an extinction coefficient of 8700 M−1 cm−1 (http://us.expasy.org/). Data were collected at Tm concentrations of ~3–9 μM on an AVIV model 202 spectrometer (Aviv Biomedical Inc, Lakewood, NJ, USA) using a 1 mm cuvette and wavelength range of 190–260 nm at 25°C. Each sample was equilibrated for 10 min prior to data collection and scanned three times with 0.4 or 0.5 nm wavelength steps and averaging time of 1 s. Molar ellipticity at 222 nm ([θ]222) was used to calculate α-helix content [37], and the elements of the secondary structure were analyzed with CDPro [38] using basis set 4. CD data represent the average of 2-3 independent experiments. The instrument was calibrated with ammonium (+)-camphor-10-sulfonate (CSA) using the two-point method [39].
2.5. In Vitro Motility Assays
In vitro motility assays with regulated thin filaments were carried out essentially as described [8, 31, 32, 35, 40]. HMM was deposited in flow cells constructed with NC-coated coverslips. Motion of RhPh-labeled filaments was visualized by fluorescence microscopy and a SIT camera and recorded on VHS videocassettes. Motility assays were conducted at 30°C unless otherwise indicated. Control assays with unregulated F-actin were conducted in actin buffer (AB: 25 mM KCl, 25 mM imidazole, 4 mM MgCl2, 1 mM EGTA, 1 mM DTT, and pH 7.4) [35] with 2 mM ATP and 0.3% methylcellulose (MC), or at pCa 5 without cTn or Tm. Solution composition for assays with regulated thin filaments was calculated as described [8, 31, 40]. Solutions contained 2 mM MgATP, 1 mM Mg2+, 10 mM EGTA, sufficient Ca(CH3COO)2 to achieve the desired pCa (pCa 9–4) (pCa = −log[Ca2+], where [Ca2+] is in molar), 50 mM K+, 15 mM Na+, 20 mM MOPS, pH 7.00 at 30°C, 0.5% MC, and cTn and Tm (see below). Γ/2 was adjusted to 0.085 M with TrisOH and acetic acid. To minimize fluorophore photobleaching and photo-oxidative damage to the proteins, 3 mg/mL glucose, 100 μg/mL glucose oxidase, 18 μg/mL catalase, and 40 mM DTT were added to motility buffers.
Regulated thin filaments were reconstituted in the flow cell [8, 31, 40]. Regulatory protein concentrations were 125 nM in the reconstitution buffer and motility buffer. At 125 nM HcTn plus 125 nM Tm, filaments were “well regulated” as demonstrated by the absence of movement at pCa 9 and essentially all filaments moving rapidly at pCa 5.
2.6. Temperature Dependence of Filament Sliding Speed
Motility data were collected during continuously varying temperature transients as described [41, 42]. Flow cells were modified by photolithographic fabrication of a heater (thin Au film, 80–100 nm thick) and a gold resistive thermometer (10 mm wide and 80 nm thick stripes) onto opposite sides of the glass coverslip. The heater and thermometer were independently biased by two DC source meters (Keithley, MODEL 2400) that also measured the voltage drop over the thermometer stripe in a four-probe configuration. Experimental control and data acquisition were accomplished with a custom LabVIEW program. Thermometers were electrically insulated with GE varnish and calibrated over the temperature range 10–70°C. The thermoelectric heater covered most of the flow cell except for a small central window for observation of filament sliding, with the thermometer stripe located ~500 μm from the window's edge. Flow cell heating was achieved using 0.8-0.9 A current which was turned off during cooling. The low starting temperature (14–18°C) and the cooling process were realized by chilled water flowing through a copper coil wrapped around the microscope objective.
2.7. Motion Analysis
Two approaches were used to quantify the speed of filament sliding. The first semiautomated approach was used to determine Ca2+-dependence of sliding speed. Digital movies were recorded directly from the video signal or from videotape using a DC2 interface w/B&W adapter hardware (Miglia Tech., Ltd., Chesham, UK) connected to a Macintosh G4 with iMovie software. Movies were stored in QuickTime format. Custom software for filament tracking (motion analysis) was developed in the Linux environment and consists of image analysis software written in C++ with scripting and interface modules written in Python.
The image analysis software reads as input a series of images and outputs a list of filament paths. Initially, each frame of the digitized sequence was converted into a binary image using the Canny edge detection algorithm. For all detected edges, a centroid was computed and compared to the previous frame. Paths were constructed for objects whose current centroid was within a user-defined measure of tolerance of its predicted value. If a centroid was outside of these bounds, a new path was created and a current path may be ended. Each output path thus lists every member edge's centroid, length, head, and tail for all constituent frames. User-defined parameters control variables such as edge thresholding and minimum path length. Individual filament paths were retained when the centroid could be unambiguously tracked for ≥2 s. Speed statistics were calculated for each retained path. The ratio of SD/mean speed was calculated for each path as an indicator of uniformity of motion [32, 43], and filaments were accepted as moving uniformly when this ratio was <0.3. For each flow cell at a given condition, the fraction of uniformly moving filaments and their unweighted mean speed (±SD) was computed from all retained paths; the typical number of paths analyzed per flow cell was in the hundreds.
The second approach for motion analysis was used to determine the sliding speed of filaments during short time intervals (e.g., during continual variation of temperature) and to correlate sliding speed with the length of individual filaments (e.g., duty ratio experiments). This procedure utilized MetaMorph (Universal Imaging) and was carried out as described [41, 42]. Stacks of frames were created from digitized movies, and filament paths were visualized by superimposing all frames of one stack to form a projection and subtracting the image of the first frame. For each stack, 4–8 filaments were randomly selected, and the distance traveled (d) was determined by measuring the residual contour line for each filament path. Speed was determined as d/(Δt(#frames − 1)), where Δt (Δt = 0.0333 s) is determined by the 30 frames-per-second video recording. Contour length of each filament was also recorded.
2.8. Cross-Bridge Duty Ratio during Unloaded Filament Sliding
Cross-bridge duty ratio (f) during unloaded filament sliding was estimated at pCa 5 using the method of Uyeda et al. [44]. Motility data were obtained as described above for regulated thin filaments and unregulated F-actin as a function of the density, ρ, of competent HMM on the flow cell surface, where ρ was varied by applying different concentrations of HMM in the initial sequence of solutions added to each flow cell. In separate experiments, ρ was determined from K-EDTA ATPase assays [16, 44]. Speeds of individual filaments and their contour lengths were measured as described above. The filament length dependence of speed was fitted to an equation that describes the interaction between myosin and actin in a stochastic manner, based on the idea that f is equal to the probability that a myosin head is in the strong actin-binding state of its cross-bridge cycle, and that filaments will move at their maximum speed only if there is at least one cross-bridge at any given time [44]:
| (1) |
where s is the experimentally measured speed, sm is the intrinsic maximum sliding speed, and η(ρ) is a number between 0 and 1 that represents the efficiency by which force generated by a single myosin head is translated into sliding speed at a given ρ; the regression parameters are f and η(ρ) × sm. The number of myosin motor domains (N) available for interaction with a filament of a given length l was calculated according to either the band model [44]:
| (2) |
where the constant 30 has units of nm and represents twice the reach of a myosin motor domain, or the nearest neighbor model [44, 45]:
| (3) |
2.9. Statistical Analyses
Ca2+-dependence of filament sliding speed was assessed by fitting data to a modified form of the Hill equation (4) by nonlinear least squares regression (SigmaPlot ver. 8.0; SPSS Inc., Richmond, Calif, USA):
| (4) |
Equation (4) has four regression parameters: smax is the maximum Ca2+-activated speed obtained at high [Ca2+] (low pCa); smin is the speed at low [Ca2+] (high pCa); pCa50 is an indicator of Ca2+-sensitivity and is the pCa needed for the value su − smin to reach 50% of smax; n is an estimate of the slope around pCa50 and is an indicator of cooperativity. Regression parameter estimates are given ±standard error of the regression. Statistical differences between regression parameter estimates (e.g., mutant versus WT) were evaluated for (4) by determining whether the difference deviated significantly from zero (SigmaPlot); parameters were considered to be significantly different when P < 0.05.
3. Results and Discussion
3.1. Maximum and Minimum Speeds of Filament Sliding
All motility assays with regulated thin filaments reconstituted with HcTn and α-GS-Tm WT, β-GS-Tm WT, or α-GS-Tm mutants were considered to be well regulated [31, 32, 40], that is, there was little or no motion at pCa 9, and fast uniform sliding at pCa 5 (Figure 2), indicating that both WT and mutants support complete Ca2+-regulation when thin filaments are saturated with regulatory proteins. These unloaded motility data contrast with isometric force measurements in thin filament-reconstituted cardiac preparations which exhibited active force generation in the absence of Ca2+ when V95A, D175N, or E180G replaced WT α-Tm [25] although this difference may at least partially reflect altered affinity of mutant Tm for F-actin and/or Tn [22] and experimental conditions where thin filaments may not have been fully saturated with regulatory proteins; it remains to be seen which condition more closely reflects diastole in FHC patients with these mutations. Unregulated F-actin exhibited similar speeds at both high and low [Ca2+] (Figure 2) as reported previously [32, 43]. At pCa 5, smax values for regulated thin filaments were 53–72% faster than for unregulated F-actin Figure 2; this result with HcTn and α-Tm agrees with our previous report [31] and compares with the enhancement of sliding speed by regulatory proteins from rabbit skeletal muscle [32, 46]. There was no significant difference between the speed at pCa 5 for thin filaments reconstituted with α-GS-Tm WT compared with any of the three mutants or β-GS-Tm WT (Figure 2). Tm alone (i.e., nonmutant Tm in the absence of troponin) does not enhance smax [7, 16, 17], and, thus, this effect may be more intimately linked to troponin than Tm [7, 47], as further implicated by FHC mutant data (Figure 2) [8]. In support of this idea, mutations D175N and E180G in human α-Tm were previously reported to increase smax relative to WT, but only when coupled with rabbit sTn [20, 26] and not with human cTn (Figure 2) [26].
Figure 2.
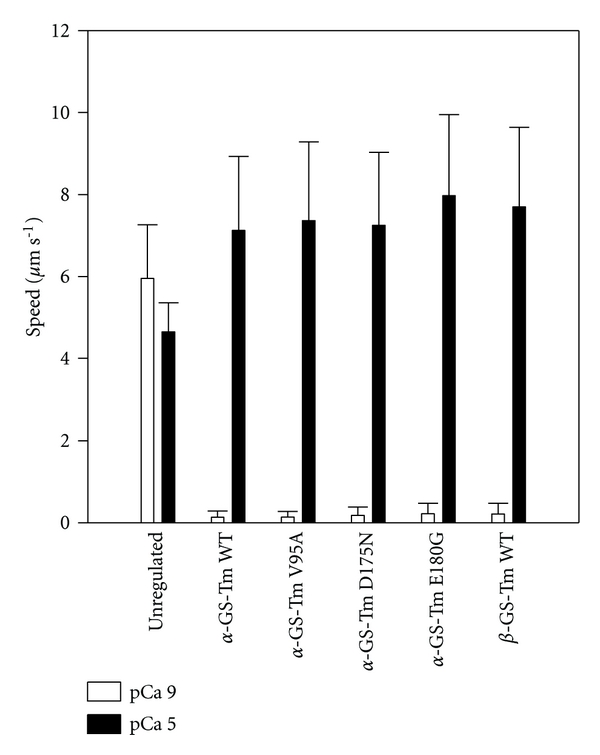
In vitro motility speeds at pCa 5 (solid bars) or pCa 9 (open bars) for regulated thin filaments (α-GS-Tm WT, α-GS-Tm V95A, α-GS-Tm D175N, α-GS-Tm E180G, or β-GS-Tm WT and HcTn), or at pCa 5 (solid bars) or in actin buffer (AB)-based motility buffer (pCa ~9; see Section 2.5) for unregulated F-actin. Values are the weighted mean + S.D. for 4–10 flow cells (weighted by the number of filament paths tracked in each flow cell).
The data in Figure 2 are generally compatible with reports in which disparate experimental systems were used, with the exception of thin filament-reconstituted cardiac preparations at low Ca2+, that these FHC-related mutations in α-Tm have generally small effects on function at very low or at saturating Ca2+ levels [15, 22, 25–27, 48, 49]. Decreased maximum ATPase activity and smax were found with V95A [15, 22] although these changes were small (<10%) and are not incompatible with our data (Figure 2). Increased maximum isometric force was previously reported in preparations from α-Tm E180G Tg mice [50] and thin filament-reconstituted cardiac preparations at saturating Ca2+ [25] although FHC-related changes in unloaded smax in motility assays or Vmax in fibers are not necessarily associated with corresponding changes in maximum isometric force [8, 11]. Thus, in conditions where thin filaments were saturated with Tn and Tm, the three mutations did not significantly affect the regulatory proteins' ability to inhibit actomyosin cycling at low [Ca2+] and also had little or no effect on maximum unloaded filament sliding speed.
3.2. Ca2+-Dependence of Filament Sliding
Sliding speed increased monotonically as [Ca2+] increased (pCa decreased) for filaments reconstituted with HcTn and α-GS-Tm WT (Figure 3(a)). The data were well fit by the Hill relation, (4) (R2 = 0.928; Figure 3(a)) and regression parameter estimates for pCa50 and n are given in Table 1. Ca2+-dependence of sliding speed for regulated thin filaments containing mutant Tm was shifted leftward (increased Ca2+-sensitivity) relative to α-GS-Tm WT (Figures 3(b)–3(d)). Regression estimates of pCa50 (4) increased by 0.11–0.64 pCa units relative to α-GS-Tm WT and were significant (P < 0.05) for the V95A and E180G mutations, although not for the D175N mutation (Table 1). The cooperativity parameter n (4) was significantly reduced for both the D175N and E180G mutations compared with WT (Table 1); this result translated into an increase in Ca2+-responsiveness for the D175N mutant, too, even though pCa50 was not significantly affected (Figure 3; Table 1).
Figure 3.
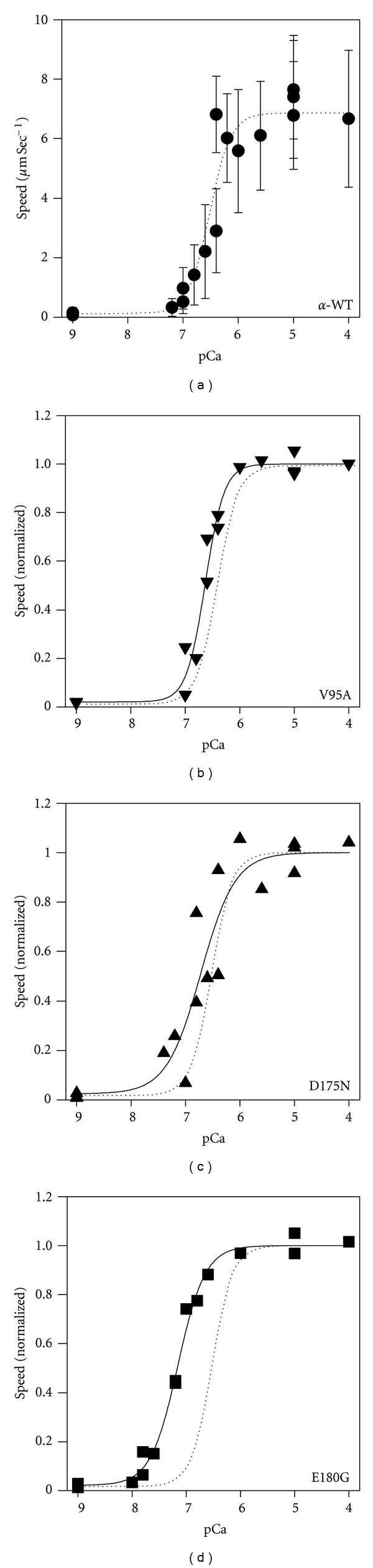
Ca2+-dependence of in vitro motility speed for regulated thin filaments reconstituted with HcTn and either, (a) α-GS-Tm WT (•), (b) α-GS-Tm V95A (▾), (c) α-GS-Tm D175N (▴), or (d) α-GS-Tm E180G (■). In (b–d), data and regressions for each Tm were normalized to smax + smin from regression (4) for the same Tm. Each point represents the mean for one flow cell; error bars in (b–d) are comparable to those in (a). Solid lines are nonlinear least squares regression fits of the mutant Tm data to (4), and parameter estimates are given in Table 1. For comparison, the regression line for α-GS-Tm WT (a) was normalized as described above and plotted in panels (b–d) as a dotted line for reference.
Table 1.
Motility speed-pCa regression parameter estimates for regulated thin filaments containing HcTn and WT or mutant tropomyosin.
| α-GS-Tm | α-GS-Tm | α-GS-Tm | α-GS-Tm | |
|---|---|---|---|---|
| WT | V95A | D175N | E180G | |
| pCa50 | 6.52 ± 0.07 | 6.63 ± 0.03* | 6.72 ± 0.12 | 7.16 ± 0.03* |
| N | 2.3 ± 0.7 | 2.5 ± 0.4 | 1.4 ± 0.5* | 1.7 ± 0.2* |
Our results with regulated thin filaments containing human α-Tm and cTn (Figure 3; Table 1) are in general agreement with previous studies when direct comparisons can be made (see Section 1), but there are also specific differences in reported effects of these Tm mutations on contractile performance. Rat cardiac myocytes transfected with human α-Tm D175N exhibited no effect on the isometric force-pCa relation or maximum force [48] although Tg rats expressing human D175N exhibited a reduction in Ca2+-sensitivity of force compared with WT [51]. Thin filament-reconstituted cardiac preparations exhibited increased Ca2+-sensitivity of isometric force with V95A and E180G [25], as observed in motility assays (Figure 3; Table 1), and cooperativity was reduced for all three mutants [25]. Where differences were observed, they may depend on whether the assays test isometric or isotonic function which have different rate-limiting steps although another source of variability may derive from combining protein isoforms from different species. Differences could also stem from the use of homo-dimers of Tm (both with respect to isoforms and mutations) in most in vitro experiments, while varying proportions of homo- and heterodimers are expected in many experiments with Tg animal tissues or transfection of living cells. Where this has been examined however, the functional effects were linearly related to the amount of mutant present [49].
The E180G mutation increased pCa50 of motility by 0.64 pCa units (Figure 3(d); Table 1), which is a much greater extent than most other mutants, and similar to that observed for the same mutant's effect on Ca2+-sensitivity of isometric force in thin filament-reconstituted cardiac preparations [25]. In some other reports, the E180G mutant also increased Ca2+-sensitivity of various functional assays [26, 48, 50] but by lesser extents (~0.1–0.14 pCa units). An exception was ventricular strips from Tg rats expressing human α-Tm E180G mutation (~5% of Tm mRNA) that exhibited no difference in Ca2+-sensitivity of force relative to WT and also exhibited no cardiac hypertrophy [51]. Ca2+-sensitivity of force increased more (~0.3 pCa units) in cardiac myocytes from Tg mouse hearts expressing ~65% α-Tm E180G, with a linear correlation between ΔpCa50 and fraction of α-Tm E180G that extrapolated to a large, ~0.4 pCa unit shift at 100% α-Tm E180G [49].
These differences in Ca2+-sensitivity due to FHC-related mutations are in the same direction as observed with three C-terminal FHC-related mutations in cTnI although the cTnI mutants also exhibited substantial increases in smax [8]. α-Tm and cTnI mutants could affect smax differently because of differences in the molecular mechanisms by which cTnI and α-Tm mutants alter thin filament activation, or because of different sources for the regulatory proteins.
3.3. CD Spectroscopy
Ca2+-sensitizing effects of the FHC mutations (Figure 3; Table 1) may be related to changes in Tm structure and are thus downstream in the signaling pathway from Ca2+ binding to TnC. Figure 4 shows CD spectra obtained from the five Tm samples. Each spectrum in Figure 4 is the average of 2-3 independent determinations from different batches of protein and each determination is the average of 3 scans. Spectra were quantified from [θ]222 and also by more extensive analysis (see Section 2) with CDPro software (Table 2). Both methods of analysis yielded similar, high estimates of α-helix content for both WT α- and β-Tm (78–85%) as expected for a protein that is largely α-helical coiled coil. CDPro and [θ]222 analyses indicated that α-helix content was lower in the three mutant proteins (61–73%; Table 2). Lower α-helix content of the mutant Tm's was correlated with higher fractions of unordered structure in CDPro analysis (Table 2). The V95A mutant had the lowest α-helix content, and reduced α-helix content of the E180G mutant is consistent with prediction from the COILS algorithm [52] of local destabilization around the mutation.
Figure 4.
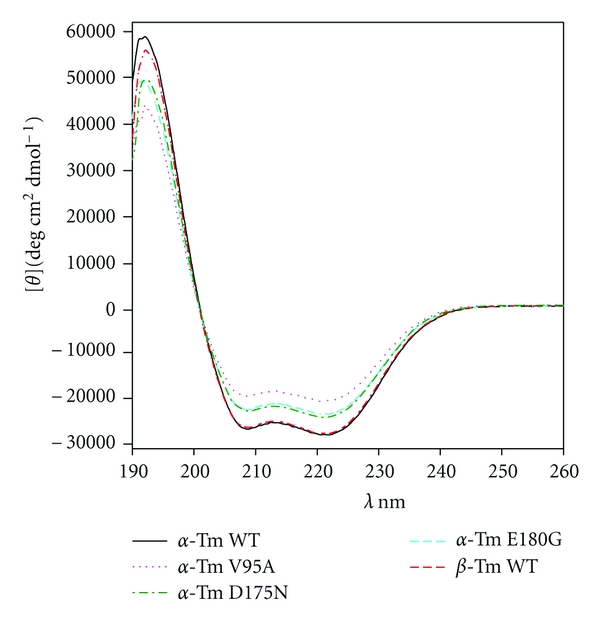
Circular dichroism spectra of WT and mutant recombinant tropomyosins in solution. Each spectrum is the average of 2-3 independent determinations. Estimates of structural content are given in Table 2. Note that curves for α-GS-Tm WT and β-GS-Tm WT overlap over most of the wavelength range.
Table 2.
Estimates of structural content of WT and mutant tropomyosins.
| α-helix (%) | β-sheet (%) | β-turn (%) | Unordered (%) | ||
|---|---|---|---|---|---|
| [θ]222 | CDPro | CDPro | CDPro | CDPro | |
| α-GS-Tm WT | 84.5 ± 4.6 | 79.1 ± 10.8 | 4.3 ± 3.4 | 7.0 ± 6.7 | 10.4 ± 5.8 |
| α-GS-Tm V95A | 60.6 ± 10.4 | 62.7 ± 6.4 | 8.2 ± 3.2 | 12.2 ± 3.5 | 17.0 ± 5.1 |
| α-GS-Tm D175N | 72.1 ± 0.7 | 72.9 ± 7.0 | 5.6 ± 3.7 | 11.3 ± 5.5 | 13.3 ± 6.0 |
| α-GS-Tm E180G | 69.7 ± 3.3 | 67.9 ± 5.6 | 5.9 ± 2.7 | 9.5 ± 4.7 | 17.3 ± 3.7 |
| β-GS-Tm WT | 83.6 ± 6.4 | 77.8 ± 13.1 | 5.2 ± 4.4 | 7.8 ± 6.5 | 10.1 ± 8.8 |
Estimates of structural content for each of the five recombinant Tm's were obtained from analyses of the individual CD spectra that were averaged for Figure 4. α-helical content was estimated from [θ]222 (Methods). Content of α-helix, β-sheet, and unordered structure was obtained using CDPro software (Methods). Values are mean ± S.D. (N = 2-3 spectra); note that, for CDPro estimates, S.D. is the mean of S.D. values from CDPro.
3.4. α-Tropomyosin E180G Mutation Alters Temperature Sensitivity of Filament Sliding at pCa 5
In addition to reduced α-helix content at 25°C (Figure 4), the E180G mutant exhibited decreased stability to thermal unfolding in the presence or absence of actin [21, 24]. To test whether these structural changes are associated with functional effects in addition to the marked increase in Ca2+-sensitivity (Figure 3(d)), we used a novel method for continuously varying temperature in motility assays [41, 42].
Speed of thin filaments reconstituted with α-Tm WT at pCa 5 increased with increasing temperature up to at least 50°C (Figure 5, open circles; note that these control data are a subset of the WT-regulated thin filament data replotted from Brunet et al. [42]). Above ~54°C, thin filaments reconstituted with WT Tm/Tn exhibit an anomalous decline in sliding speed [42] that is likely due to dissociation of regulatory proteins from actin [24] and/or denaturation of Tm or Tn at elevated temperatures. Filament speed declines to that of unregulated F-actin [42] because actin and HMM denature at higher temperatures than Tm/Tn during these brief excursions to high temperatures.
Figure 5.
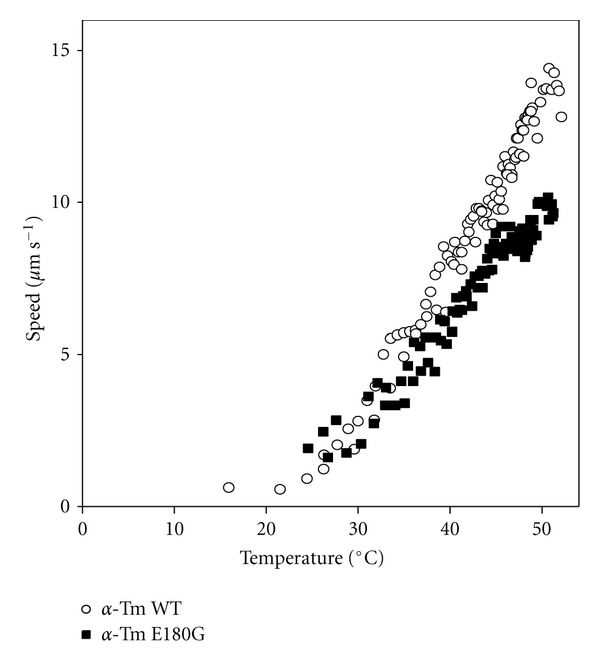
Temperature dependence of sliding speed at pCa 5 for regulated thin filaments reconstituted with HcTn and either α-GS-Tm E180G (■) or α-GS-Tm WT (○) [note that the control WT data are a subset of regulated thin filament data replotted from [42]]. Data were obtained during the heating phase of two cycles of heating and cooling in flow cells configured with a microfabricated thermo-electric heater for continuous variation of temperature (see Section 2). Points represent mean of the sliding speed for 3–8 filaments during one second. Note the deviation of mutant Tm data from WT as temperature increases.
When the assay was repeated with thin filaments reconstituted with α-Tm E180G (Figure 5, solid squares), the temperature dependence of speed deviated from WT at temperatures slightly above body temperature. At these higher temperatures, sliding speed was slower with mutant Tm than with WT. Above ~44°C, there is a deflection in the speed-temperature relation for thin filaments reconstituted with α-Tm E180G (Figure 5). This deflection is likely indicative of structural destabilization, denaturation, and/or dissociation of mutant regulatory proteins at a substantially lower temperature (~10°C lower) than for WT and is also indicative of a strong functional correlate of differential scanning calorimetry (DSC) measurements in solution [24].
The substantial increase in Ca2+-sensitivity associated with the E180G mutation (Figure 3; Table 1) is thus correlated with reduced affinity for actin in vitro in the absence of Tn [20–22], destabilization of Tn binding to F-actin-Tm [22], reduced α-helix content (Figure 4; Table 2), and reduced thermal stability of structure [21, 24] and function (Figure 5). Structural destabilization of α-Tm by FHC mutations could reduce flexural rigidity, as suggested by Heller et al. [23] when considering other FHC-related mutations in α-Tm; modeling suggests that changes in protein compliance within sarcomeres could markedly affect muscle function [53–56]. The details of this mechanism are therefore distinct from the likely molecular basis for Ca2+-sensitization by FHC-related mutations in the COOH-terminus of cTnI [8, 57]. Flexibility of Tm is clearly important for its structure and function [58] and changes related to FHC mutations could lead to a decrease in the already low-energy barrier for movement of Tm on actin [59]; detailed structural studies are needed to clarify the influence of FHC mutations on Tm structure and flexibility. In addition, coupling between adjacent regulatory units could be affected, as suggested by reduced cooperativity with the E180G mutation (Figure 3(d); Table 1) [25]. Significantly, the reduced thermal stability of thin filaments containing the E180G mutant α-Tm illustrated in Figure 5 suggests that patients with the FHC mutation could be more susceptible to diastolic dysfunction, and possibly sudden cardiac death, due to thin filament dysregulation during heavy exercise [24].
3.5. Dependence of Regulated Thin Filament Sliding at pCa 5 on Cross-Bridge Density and Number
Mutations in α-Tm could modulate the dependence of thin filament activation on cross-bridges and cross-bridge number [60]. To examine this possibility for the D175N and E180G mutations—the two mutations associated with the largest effects on Ca2+-responsiveness (Figure 3)—we measured sliding speed of regulated thin filaments at pCa 5 by varying the density of functional HMM (ρ) on the flow cell surface [44]. For each condition (Tm and ρ), speed (s) and the length (l) of individual filaments were used to determine η(ρ) × sm and actomyosin duty ratio (f; (1)) as described (see Section 2) [44]. Figure 6 shows that f was lower for WT-reconstituted thin filaments than for unregulated F-actin, and that f was similar for reconstituted thin filaments containing WT Tm or either of the two Tm mutants examined. Estimates of f in Figure 6 were obtained using the band model (2) and were averaged for all ρ. These results show that the structural and functional changes associated with mutants D175N and E180G of α-Tm do not lead to substantial changes in actomyosin duty ratio relative to WT-regulated thin filaments, consistent with regulated thin filament sliding speed being similar at saturating HMM density (Figure 2), and the distribution of cross-bridge states from sinusoidal analysis of isometric force in thin filament-reconstituted cardiac preparations [25].
Figure 6.
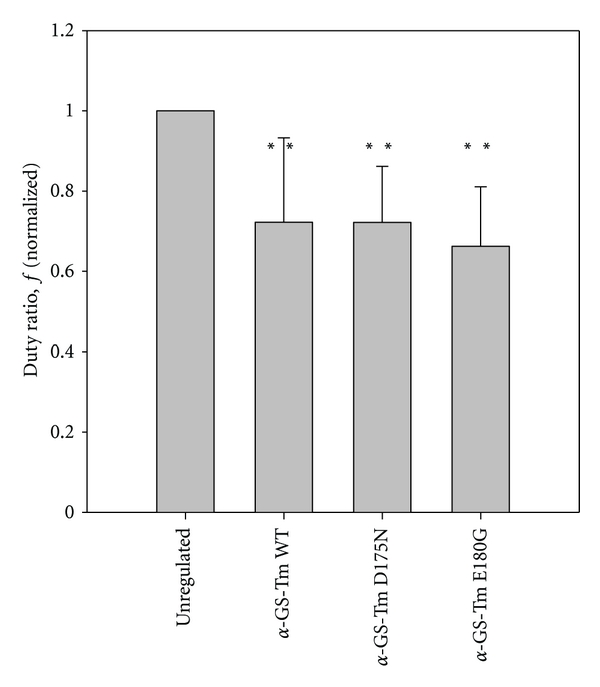
Cross-bridge duty ratio (f) during unloaded sliding of regulated thin filaments reconstituted with HcTn and either α-GS-Tm WT, α-GS-Tm D175N, or α-GS-Tm E180G is lower than for unregulated F-actin. Duty ratio was estimated using the method of Uyeda et al. [61] by varying HMM density (ρ) as described in Materials and Methods (see Section 2.8). Estimates of f were obtained by applying the band model and were normalized to the value obtained for WT at the same ρ. Bars indicate average normalized f for all ρ (n = 5 − 8); error bars indicate S.D. Duty ratios for all three types of regulated thin filaments were significantly lower than for unregulated F-actin (**P < 0.01) but were not statistically different from each other (P > 0.05).
For regulated thin filaments at pCa 5, sm is a hyperbolic function of ρ (Figure 7). The ρ-dependence of η(ρ) × sm was indistinguishable for α-Tm E180G compared with α-Tm WT (Figure 7). Thin filaments reconstituted with α-Tm D175N, on the other hand, exhibited motility and achieved maximum sliding speed at lower ρ's than either E180G or WT (Figure 7). These results indicate that the D175N and E180G mutations increase Ca2+-responsiveness of filament sliding (Figure 3; Table 1) by very different mechanisms even though the two mutations reside in close proximity and have similar effects on overall α-helical content as detected by CD spectroscopy (Figure 4). The data in Figures 3 and 7 suggest that effects of the D175N would be most evident at low levels of thin filament activation where the number of cross-bridges is small, and additional cross-bridges have the greatest impact.
Figure 7.
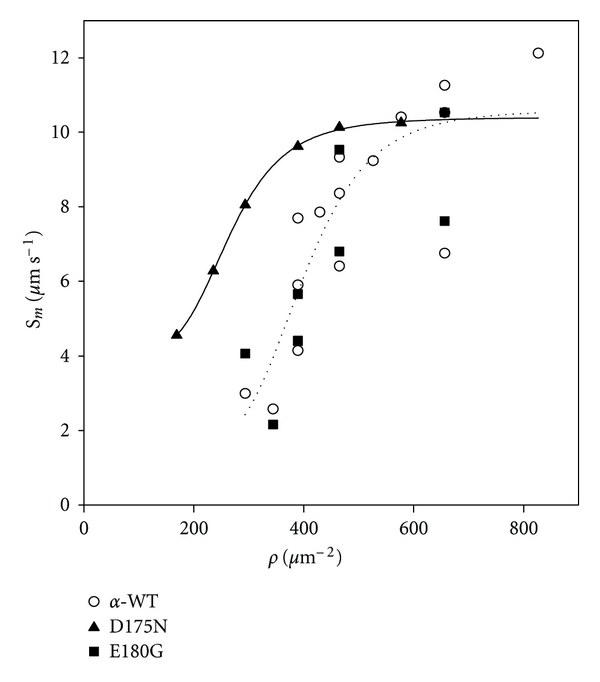
HMM density (ρ) dependence of maximum unloaded filament sliding speed (sm) of regulated thin filaments reconstituted with HcTn and either α-GS-Tm WT (○), α-GS-Tm D175N (▲), or α-GS-Tm E180G (■). sm was obtained at each ρ according to the method of Uyeda et al. [61] as described in Section 2.8; each point is from one flow cell. Note that sm(ρ) for thin filaments containing α-GS-Tm D175N is shifted leftward of that for thin filaments containing either α-GS-Tm WT or α-GS-Tm E180G.
3.6. Conclusions
Functional and structural differences in three FHC-related mutations in α-Tm (V95A, D175N, and E180G) were found using both conventional and modified in vitro motility assays and CD spectroscopy. Mutant Tm's exhibited lower α-helical content and more unordered structure than α-GS-Tm WT; these structural changes could result in alterations in flexural rigidity that, in turn, could be responsible for increased Ca2+-responsiveness of regulated filament sliding that was observed with the mutants. α-GS-Tm E180G exhibited the greatest enhancement of Ca2+-responsiveness of the mutants studied due to both a large increase in pCa50 and a reduction in n, and dysregulation at ~10°C lower temperature than WT. α-GS-Tm V95A exhibited a smaller but significant increase in pCa50. α-GS-Tm D175N exhibited lower n and a marked reduction in the minimum density of HMM necessary for initiating filament sliding and for achieving maximum sliding speed at saturating Ca2+. These structural and functional changes provide insights into molecular mechanisms of pathological cardiac hypertrophy.
Increased Ca2+-responsiveness of cardiac myofibrillar function appears to be a common feature of FHC-related mutations in α-Tm, cTnI, and cTnT (Figure 3) [8, 13, 15, 23, 25–27, 29, 48, 50, 62]. The extent of enhancement of Ca2+-sensitivity is generally associated with clinical prognosis in FHC patients [57, 63] although it is not generally known the extent to which function in vivo with variable ratios of WT and mutant homo-dimers, and also presumptive hetero-dimers, might differ from that found using in vitro assays with pure homo-dimers such as those described here. Functional consequences of increased Ca2+-sensitivity in the living heart, in the absence of adaptive responses, are expected to be slowed relaxation and increased duration of systole because the intracellular Ca2+ transient does not normally achieve sufficiently high Ca2+ concentrations to saturate TnC [56]. Observations on intact hearts from Tg mice with α-Tm mutation E180G are consistent with these expectations [28, 29, 49]. Thus, there are clear possibilities for direct links between thin filament mutations and hypertrophic signaling through altered mechanical and energetic load, and/or altered Ca2+-cycling in myocytes [56].
Acknowledgments
The authors gratefully acknowledge Shanedah Williams Brown, Lori McFadden and Alyson Barnes for preparation of myosin and actin, Margaret Seavy for HPLC analyses, Kim Riddle and Tom Fellers from FSU's Biological Science Imaging Resource for assistance with MetaMorph, Drs. Peng Xiong and Stephan von Molnár in Florida State University's Department of Physics and Ingetrative NanoScience Institute for expertise necessary to accomplish the experiments involving microfabrication and for critical comments, Campion Loong and Dr. Robin Maytum for helpful discussions; and Janina Bhuvasorakul for assistance with preparation of figures and tables. They also thank the Physical Biochemistry Facility at the FSU Institute of Molecular Biophysics for use of the circular dichroism spectrometer. This work was supported by NIH/NHLBI Grant HL63974 (PBC), American Heart Association FL/PR Affiliate Predoctoral Fellowships 0315097B (NMB) and 0415104B (LAC), and NSF NIRT Grant ECS-0210332 (PX).
References
- 1.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415(6868):227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 2.Redwood CS, Moolman-Smook JC, Watkins H. Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovascular Research. 1999;44(1):20–36. doi: 10.1016/s0008-6363(99)00213-8. [DOI] [PubMed] [Google Scholar]
- 3.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiological Reviews. 2002;82(4):945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 4.Parmacek MS, Solaro RJ. Biology of the troponin complex in cardiac myocytes. Progress in Cardiovascular Diseases. 2004;47(3):159–176. doi: 10.1016/j.pcad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annual Review of Genomics and Human Genetics. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 6.Elliott K, Watkins H, Redwood CS. Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2000;275(29):22069–22074. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 7.Homsher E, Lee DM, Morris C, Pavlov D, Tobacman LS. Regulation of force and unloaded sliding speed in single thin filaments: effects of regulatory proteins and calcium. Journal of Physiology. 2000;524(1):233–243. doi: 10.1111/j.1469-7793.2000.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Köhler J, Chen Y, Brenner B, et al. Familial hypertrophic cardiomyopathy mutations in troponin I (K183Δ, G203S, K206Q) enhance filament sliding. Physiological Genomics. 2003;14:117–128. doi: 10.1152/physiolgenomics.00101.2002. [DOI] [PubMed] [Google Scholar]
- 9.Lang R, Gomes AV, Zhao J, Housmans PR, Miller T, Potter JD. Functional analysis of a troponin I (R145G) mutation associated with familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2002;277(14):11670–11678. doi: 10.1074/jbc.M108912200. [DOI] [PubMed] [Google Scholar]
- 10.Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. Journal of Clinical Investigation. 1996;97(12):2842–2848. doi: 10.1172/JCI118740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney HL, Feng HS, Yang Z, Watkins H. Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: insights into disease pathogenesis and troponin function. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(24):14406–14410. doi: 10.1073/pnas.95.24.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2000;275(1):624–630. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi-Yanaga F, Morimoto S, Harada K, et al. Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2001;33(12):2095–2107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 14.Thierfelder L, Watkins H, MacRae C, et al. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77(5):701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 15.Karibe A, Tobacman LS, Strand J, et al. Hypertrophic cardiomyopathy caused by a novel α-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation. 2001;103(1):65–71. doi: 10.1161/01.cir.103.1.65. [DOI] [PubMed] [Google Scholar]
- 16.Gordon AM, Chen Y, Liang B, LaMadrid M, Luo Z, Chase PB. Skeletal muscle regulatory proteins enhance F-actin in vitro motility. Advances in Experimental Medicine and Biology. 1998;453:187–197. doi: 10.1007/978-1-4684-6039-1_22. [DOI] [PubMed] [Google Scholar]
- 17.VanBuren P, Palmiter KA, Warshaw DM. Tropomyosin directly modulates actomyosin mechanical performance at the level of a single actin filament. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(22):12488–12493. doi: 10.1073/pnas.96.22.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry SV. Vertebrate tropomyosin: distribution, properties and function. Journal of Muscle Research and Cell Motility. 2001;22(1):5–49. doi: 10.1023/a:1010303732441. [DOI] [PubMed] [Google Scholar]
- 19.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological Reviews. 2000;80(2):853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 20.Bing W, Redwood CS, Purcell IF, Esposito G, Watkins H, Marston SB. Effects of two hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on Ca2+ regulation of thin filament motility. Biochemical and Biophysical Research Communications. 1997;236(3):760–764. doi: 10.1006/bbrc.1997.7045. [DOI] [PubMed] [Google Scholar]
- 21.Golitsina N, An Y, Greenfield NJ, et al. Effects of two familial hypertrophic cardiomyopathy-causing mutations on α-tropomyosin structure and function. Biochemistry. 1999;38(12):p. 3850. doi: 10.1021/bi9950701. [DOI] [PubMed] [Google Scholar]
- 22.Mathur MC, Chase PB, Chalovich JM. Several cardiomyopathy causing mutations on tropomyosin either destabilize the active state of actomyosin or alter the binding properties of tropomyosin. Biochemical and Biophysical Research Communications. 2011;406(1):74–78. doi: 10.1016/j.bbrc.2011.01.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heller MJ, Nili M, Homsher E, Tobacman LS. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. Journal of Biological Chemistry. 2003;278(43):41742–41748. doi: 10.1074/jbc.M303408200. [DOI] [PubMed] [Google Scholar]
- 24.Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI. Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glut180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophysical Journal. 2004;87(6):3922–3933. doi: 10.1529/biophysj.104.048793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophysical Journal. 2011;100(4):1014–1023. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bing W, Knott A, Redwood C, et al. Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle α-tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. Journal of Molecular and Cellular Cardiology. 2000;32(8):1489–1498. doi: 10.1006/jmcc.2000.1182. [DOI] [PubMed] [Google Scholar]
- 27.Bottinelli R, Coviello DA, Redwood CS, et al. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circulation Research. 1998;82(1):106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- 28.Prabhakar R, Petrashevskaya N, Schwartz A, et al. A mouse model of familial hypertrophic cardiomyopathy caused by a α-tropomyosin mutation. Molecular and Cellular Biochemistry. 2003;251(1-2):33–42. [PubMed] [Google Scholar]
- 29.Prabhakar R, Boivin GP, Grupp IL, et al. A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. Journal of Molecular and Cellular Cardiology. 2001;33(10):1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 30.Monteiro PB, Lataro RC, Ferro JA, Reinach FDC. Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. Journal of Biological Chemistry. 1994;269(14):10461–10466. [PubMed] [Google Scholar]
- 31.Schoffstall B, Brunet NM, Williams S, et al. Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform. Journal of Physiology. 2006;577(3):935–944. doi: 10.1113/jphysiol.2006.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB. Calcium regulation of skeletal muscle thin filament motility in vitro. Biophysical Journal. 1997;72(3):1295–1307. doi: 10.1016/S0006-3495(97)78776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods in Enzymology. 1982;85:55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 34.Pardee JD, Aspudich J. Purification of muscle actin. Methods in Enzymology. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 35.Kron SJ, Toyoshima YY, Uyeda TQP, Spudich JA. Assays for actin sliding movement over myosin-coated surfaces. Methods in Enzymology. 1991;196:399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 36.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6(7):1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 37.Chen YH, Yang JT, Martinez HM. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 1972;11(22):4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 38.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Analytical Biochemistry. 2000;287(2):252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 39.Chen GC, Yang JT. Two-point calibration of circular dichrometer with d-10-camphorsulfonic acid. Analytical Letters. 1977;10:1195–1207. [Google Scholar]
- 40.Liang B, Chen Y, Wang CK, et al. Ca2+ regulation of rabbit skeletal muscle thin filament sliding: role of cross-bridge number. Biophysical Journal. 2003;85(3):1775–1786. doi: 10.1016/S0006-3495(03)74607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mihajlović G, Brunet NM, Trbović J, Xiong P, Von Molnár S, Chase PB. All-electrical switching and control mechanism for actomyosin-powered nanoactuators. Applied Physics Letters. 2004;85(6):1060–1062. [Google Scholar]
- 42.Brunet NM, Mihajlovi ć G, Aledealat K, et al. Micro-mechanical thermal assays of Ca2+-regulated thin filament function and modulation by hypertrophic cardiomyopathy mutants of human cardiac troponin. doi: 10.1155/2012/657523. Journal of Biomedicine and Biotechnology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Homsher E, Kim B, Bobkova A, Tobacman LS. Calcium regulation of thin filament movement in an in vitro motility assay. Biophysical Journal. 1996;70(4):1881–1892. doi: 10.1016/S0006-3495(96)79753-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uyeda TQP, Kron SJ, Spudich JA. Myosin step size estimation from slow sliding movement of actin over low densities of heavy meromyosin. Journal of Molecular Biology. 1990;214(3):699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 45.Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T. Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay. Journal of Molecular Biology. 1990;216(1):49–68. doi: 10.1016/S0022-2836(05)80060-9. [DOI] [PubMed] [Google Scholar]
- 46.Fraser IDC, Marston SB. In vitro motility analysis of actin-tropomyosin regulation by troponin and calcium. The thin filament is switched as a single cooperative unit. Journal of Biological Chemistry. 1995;270(14):7836–7841. doi: 10.1074/jbc.270.14.7836. [DOI] [PubMed] [Google Scholar]
- 47.Schoffstall B, LaBarbera VA, Brunet NM, et al. Interaction between troponin and myosin enhances contractile activity of myosin in cardiac muscle. doi: 10.1089/dna.2010.1163. DNA and Cell Biology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michele DE, Albayya FP, Metzger JM. Direct, convergent hypersensitivity of calcium-activated force generation produced by hypertrophic cardiomyopathy mutant α-tropomyosins in adult cardiac myocytes. Nature Medicine. 1999;5(12):1413–1417. doi: 10.1038/70990. [DOI] [PubMed] [Google Scholar]
- 49.Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by β-blockade. Circulation Research. 2002;91(3):255–262. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 50.Burkart EM, Arteaga GM, Sumandea MP, Prabhakar R, Wieczorek DF, Solaro RJ. Altered signaling surrounding the C-lobe of cardiac troponin C in myofilaments containing an α-tropomyosin mutation linked to familial hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2003;35(10):1285–1293. doi: 10.1016/s0022-2828(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 51.Wernicke D, Thiel C, Duja-Isac CM, et al. α-tropomyosin mutations Asp175 Asn and Glu180 Gly affect cardiac function in transgenic rats in different ways. American Journal of Physiology. 2004;287(3):R685–R695. doi: 10.1152/ajpregu.00620.2003. [DOI] [PubMed] [Google Scholar]
- 52.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252(5010):1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 53.Daniel TL, Trimble AC, Chase PB. Compliant realignment of binding sites in muscle: transient behavior and mechanical tuning. Biophysical Journal. 1998;74(4):1611–1621. doi: 10.1016/s0006-3495(98)77875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martyn DA, Chase PB, Regnier M, Gordon AM. A simple model with myofilament compliance predicts activation-dependent crossbridge kinetics in skinned skeletal fibers. Biophysical Journal. 2002;83(6):3425–3434. doi: 10.1016/S0006-3495(02)75342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chase PB, Macpherson JM, Daniel TL. A spatially explicit nanomechanical model of the half-sarcomere: myofilament compliance affects Ca2+-activation. Annals of Biomedical Engineering. 2004;32(11):1559–1568. doi: 10.1114/b:abme.0000049039.89173.08. [DOI] [PubMed] [Google Scholar]
- 56.Kataoka A, Hemmer C, Chase PB. Computational simulation of hypertrophic cardiomyopathy mutations in Troponin I: influence of increased myofilament calcium sensitivity on isometric force, ATPase and [Ca2+]i. Journal of Biomechanics. 2007;40(9):2044–2052. doi: 10.1016/j.jbiomech.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 57.Gomes AV, Potter JD. Molecular and cellular aspects of troponin cardiomyopathies. Annals of the New York Academy of Sciences. 2004;1015:214–224. doi: 10.1196/annals.1302.018. [DOI] [PubMed] [Google Scholar]
- 58.Brown JH, Kim KH, Jun G, et al. Deciphering the design of the tropomyosin molecule. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8496–8501. doi: 10.1073/pnas.131219198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehman W, Hatch V, Korman V, et al. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. Journal of Molecular Biology. 2000;302(3):593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 60.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. Journal of Molecular Biology. 1997;266(1):8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 61.Uyeda TQP, Warrick HM, Kron SJ, Spudich JA. Quantized velocities at low myosin densities in an in vitro motility assay. Nature. 1991;352(6333):307–311. doi: 10.1038/352307a0. [DOI] [PubMed] [Google Scholar]
- 62.Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, Chalovich JM. The Δ14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry. 2004;43(48):15276–15285. doi: 10.1021/bi048646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomes AV, Potter JD. Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Molecular and Cellular Biochemistry. 2004;263(1):99–114. doi: 10.1023/B:MCBI.0000041852.42291.aa. [DOI] [PubMed] [Google Scholar]