

Abstract
The CLC-1 chloride channel, a member of the CLC-channel/transporter family, plays important roles for the physiological functions of skeletal muscles. The opening of this chloride channel is voltage dependent and is also regulated by protons and chloride ions. Mutations of the gene encoding CLC-1 result in a genetic disease, myotonia congenita, which can be inherited as an autosmal dominant (Thomsen type) or an autosomal recessive (Becker type) pattern. These mutations are scattered throughout the entire protein sequence, and no clear relationship exists between the inheritance pattern of the mutation and the location of the mutation in the channel protein. The inheritance pattern of some but not all myotonia mutants can be explained by a working hypothesis that these mutations may exert a “dominant negative” effect on the gating function of the channel. However, other mutations may be due to different pathophysiological mechanisms, such as the defect of protein trafficking to membranes. Thus, the underlying mechanisms of myotonia are likely to be quite diverse, and elucidating the pathophysiology of myotonia mutations will require the understanding of multiple molecular/cellular mechanisms of CLC-1 channels in skeletal muscles, including molecular operation, protein synthesis, and membrane trafficking mechanisms.
1. Introduction
Chloride (Cl−) channels are membrane proteins that consist of Cl−-permeation pores. Different types of human cells express Cl− channels in the cell membrane for various physiological purposes. In the last several decades, ion channels that conduct cations, such as sodium (Na+), potassium (K+) or calcium (Ca2+) channels, have been studied more extensively than anion channels. Nonetheless, Cl− channels are as abundant as cation channels, and they also participate in many important physiological tasks, including the maintainence of normal cellular excitability, the control of neurotransmitter release, and the transport of ions across epithelial cells, to name a few. The aim of this paper is to provide an up-to-date overview of the mechanism and the consequence of the disruption of Cl− channel function. We will focus on the physiology and pathophysiology of a Cl− channel critical for the function of skeletal muscles, the CLC-1 Cl− channel.
Cl− is the most abundant anion in most organisms. In adult mammalian cells, the extracellular concentration of Cl− is significantly higher than its intracellular counterpart, resulting in a negative Cl− equilibrium potential (ECl) that is exquisitely determined by an intracellular Cl− concentration. Two secondary active transport systems contribute the most to the regulation of the cytoplasmic Cl− level. The Na+-K+-Cl− cotransporter (NKCC) normally accumulates Cl− in the cell [1], whereas the K+-Cl− co-transporter (KCC) usually transports Cl− out of the cell [2]. Most epithelial cells express predominantly NKCC and display an ECl positive to the resting potential [3, 4]. In contrast, the majority of mature neurons have an enhanced expression of KCC, and therefore manifest a quite negative ECl—sometimes even more negative than the resting potential [3, 5]. In skeletal muscle, despite the presence of both NKCC and KCC, the contribution of secondary active transporters to ECl is rather small, mainly due to the presence of an extraordinarily high Cl− permeability that is virtually impossible to be counteracted by active transporters [6–9]. The ECl in skeletal muscle is thus mainly set by passive electrochemical equilibrium of Cl− according to the resting membrane potential which is primarily determined by K+ equilibrium potential (EK). Recent evidence, however, supports the idea that the secondary active transporter NKCC may modulate the membrane potential of skeletal muscle via its Cl− import function [10–12]. Indeed, the muscle resting potential is never the same as the value of EK while the ECl has been shown to be slightly more positive than the resting membrane potential [13]. Furthermore, the value of the intracellular Cl− activity has been shown to be slightly higher than would be expected for a passive Cl− distribution [13]. These observations underscore the contribution of the chloride conductance in determining the resting membrane potential of skeletal muscles.
2. Physiological Roles of CLC-1 Channels in Skeletal Muscles
Because of a large membrane Cl− conductance, up to 80% of the resting sarcolemmal conductance [14–16], a relatively negative ECl explains the physiological role of Cl− channels in the cell membrane of skeletal muscles (sarcolemma). For example, activation of Cl− channels is essential for ensuring the electrical stability of skeletal muscle by resetting its membrane excitability to the resting state after firing an action potential. Furthermore, a significant Cl− conductance is also located in the transverse-tubule system [15, 17–19], indicating that the presence of an effective Cl− homeostasis system is crucial for the generation and propagation of action potential in both the sarcolemmal and the transverse-tubule system. Finally, emerging evidence suggests that disruptions of the balance of ion channel functions in sarcolemma may contribute to skeletal muscle fatigue [19–21]. During intensive firing of muscle action potentials, K+ ions tend to accumulate in the extracellular space up to 10 mM as the extracellular volume of skeletal muscles is limited. The increase of extracellular K+ concentration results in depolarization of membrane potential, and, consequently, a partial inactivation of voltage-gated Na+ channels. If the Na+ channels that remain active fail to generate a sufficient Na+ inward current to overcome the shunting currents mediated by the resting sarcolemmal Cl− conductance, the firing of action potential is not possible, thereby leading to muscle fatigue.
Although various types of Cl− channels are expressed in skeletal muscles, the most abundant Cl− channel in the sarcolemma is CLC-1 [4, 22], which is a member of the CLC-channel/transporter family. The mammalian CLC family consists of nine members: CLC-1, CLC-2, CLC-3, CLC-4, CLC-5, CLC-6, CLC-7, CLC-Ka, and CLC-Kb [22–24]. Among these members, CLC-1, CLC-2, CLC-Ka, and CLC-Kb are Cl− channels, predominantly residing in the plasma membrane. The rest of the CLC members (CLC-3 to CLC-7) are thought to be transporters, mostly located in intracellular organelles. Like bacterial CLC proteins [25], these mammalian CLC transporters are thought to mediate the counter transport of H+ and Cl−; that is, they are Cl−/H+ antiporters [26, 27].
Among the four plasma membrane CLC-channels, CLC-Ka and CLC-Kb channels are involved in transepithelial transport in the kidney and the inner ear [4, 28]. CLC-2 channels can be activated by hyperpolarization, cell swelling, and extracellular acidification [29, 30]. Northern analysis indicates that brain, kidney, and intestine express relatively high levels of CLC-2 channels, although these channel are broadly expressed in various tissues as well [31]. In contrast, CLC-1 channels are most abundantly expressed in the skeletal muscle [32]. A very low level of CLC-1 expression, however, has been reported in kidney, heart, smooth muscle, and, more recently, glial cells [32, 33]. Since the CLC-1 channel is the major sarcolemmal Cl− conductance, mutations of the gene encoding this Cl− channel lead to a significant muscle hyperexcitability in humans, mice, and other animals [34–38], a situation known as “myotonia” [32, 39]. Myotonia is a muscle disease due to hyper-excitability of skeletal muscles. Therefore, this disease can be caused either by the gain of function of Na+ channels or a loss of function of Cl− channels in the sarcolemma of skeletal muscles. In the following sections we will focus on the defect of CLC-1, starting with the CLC-1's molecular properties.
3. Molecular Biophysics of CLC-1 Channels
CLC-1 is a voltage-gated channel, and the open probability of CLC-1 channels increases with membrane depolarization. The functional study of CLC-1 at the single-channel level is challenging due to the small single-channel conductance of this channel. Therefore, many functional properties of CLC-1 are inferred from those found in its fish homologue, the Torpedo CLC-0 Cl− channel [23]. One unique functional feature in CLC-channels is that the channel opens to two current levels separated by equidistance in the single-channel recording trace (Figure 1). This feature has been identified as the consequence of a “double-barreled” channel opening, first found in CLC-0 in early 1980s [40, 41]. Later single-channel recordings confirmed that the opening of CLC-1 channels also fluctuates between three different conductance levels (Figure 1(a)), corresponding to the three functional states: two pores closed; one open and one closed; and, finally, both pores open [42]. These functional recordings of CLC-0 and CLC-1 channels foretold the recent structural findings from bacterial CLC proteins in which two identical Cl−-transport pathways were found in one CLC functional unit [43, 44].
Figure 1.
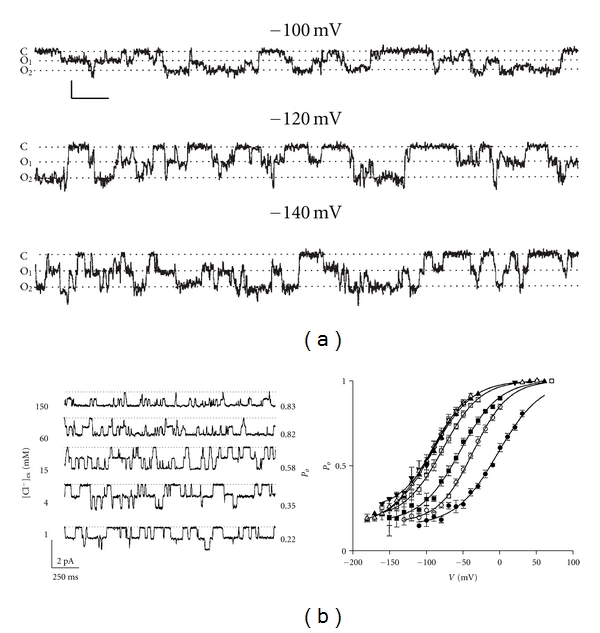
Molecular functions of CLC-channels. (a) Single-channel recordings of CLC-1 showing the “double-barreled” behavior. Dotted lines depict the three current levels: C: closed state, O1: one protopore open, and O2: both protopores open. Horizontal and vertical scale bars represent 200 ms and 0.2 pA, respectively. Notice that the three current levels are separated in equi-distance. Figure, taken from Saviane et al. [42] (© Rockefeller University Press, 1999). (b) Effects of extracellular Cl− on the fast-gate open probability of the Torpedo CLC-0 Cl− channel. Left panel shows single-channel recordings of CLC-0 at different extracellular Cl− concentrations indicated on the left. The calculated open probabilities of the fast gate in each Cl− concentration are shown on the right. Membrane potentials in all recordings are −60 mV. Right panel shows a summarized result for the Cl− effect on the fast-gate Po-V curve. The extracellular Cl− concentrations are 300 mM and those indicated in the left panel. As the extracellular Cl− concentration is reduced (from 300 mM to 1 mM), the fast-gate Po-V curve is shifted to the more depolarized membrane potential. A similar Cl− effect on the fast-gate Po-V curve has been observed in CLC-1. Figures, taken from Chen and Miller [45] (© Rockefeller University Press, 1996).
The opening and closure of the two pores in CLC-0 and CLC-1 channels are controlled by two distinct gating mechanisms [23]. One of these gating mechanisms controls the opening and closure of two pores simultaneously, and is therefore called “common gate”. In addition, each pore is also controlled by a “fast gate” that operates independently from the partner fast-gate. Thus, the activation of the Cl− conducting pathway of CLC-1 channels requires the opening of both the common gate and the fast gate. The open-close transition of the fast gate operates at a time scale of milliseconds at negative membrane potentials. At the peak of the action potential, the opening kinetics of CLC-1 can be in the submillisecond range. Thus, the opening of CLC-1's fast-gate can counteract the depolarization generated by the opening of Na+ channels during an action potential. This gating mechanism is thus important for CLC-1 channels to control the action potential in skeletal muscles. Mutations that reverse the voltage dependence of CLC-1 channels result in certain forms of myotonia (see below) because these mutant channels are unable to open after membrane depolarization. In addition to the control by membrane potentials, the fast-gating is also regulated by Cl− and H+ [46, 47]. The regulation of CLC-1 and CLC-0 channels by Cl− and H+ is thought to bear an evolutional relationship to the Cl−/H+ counter transport function of their CLC transporter counterparts [48], although the exact link between the channel-gating mechanism and the Cl−/H+-antiporter mechanism is not known. Interestingly, a recent crystallographic study of a prokaryotic CLC protein provided a potential mechanism for the exchange stoichiometry of 2 Cl− for 1 H+ [49].
The voltage dependence of the fast gating is similar to that found in voltage-gated cation channels; namely, the open probability (Po) is higher at more depolarized membrane potentials [40, 41, 45, 50, 51]. However, unlike voltage-gated cation channels with the “S4” transmembrane segment serving as the “voltage sensor” [52], there is no such structure in CLC-0 and CLC-1 channels. The voltage-dependent activation of the fast gate of CLC-0 and CLC-1 is likely to arise from the coupling of Cl− transport with the gating process [45, 46, 53]. This gating-permeation coupling mechanism was first proposed by Pusch and his colleague [53], who demonstrated that the Po-V curve of the fast gating of CLC-0 channels was shifted toward a more depolarized membrane potential by reducing the extracellular Cl− concentration. At a constant voltage, therefore, reducing the extracellular Cl− concentration inhibits the opening of the fast gate. More detailed experiments at the single-channel level (Figure 1(b)) further showed that extracellular Cl− increases the open probability of the fast-gate by increasing the opening rate of the fast gate [45]. Later experiments also revealed the same dependent fast gating mechanism for CLC-1 channels [46]. To explain this gating effect, investigators have proposed a model in which the binding of Cl− to the channel pore opens the fast gate and the movement of Cl− across membrane electric field constitutes the fundamental mechanism for the observed voltage dependence [23]. This hypothesis was formulated before the crystal structure of CLC molecules became available. Later structural information from crystallographic studies of bacterial CLC proteins revealed that the transport pathway of CLC molecules appear to be obstructed by the negatively charged side chain of a glutamate residue, and Cl− in the pore may compete with this glutamate side chain [43, 44].
4. Structural/Functional Relationship of CLC-1 Channels
The gene of the human CLC-1 channel encodes a transmembrane protein consisting of 991 amino acids (AA). The protein can be roughly divided into two parts, the amino (N)-terminal transmembrane portion (up to ~590 AA.) and the carboxyl (C)-terminal cytoplasmic portion (Figure 2). Although the molecular structure of CLC-1 has not been solved, recent breakthroughs in obtaining the crystal structure of bacteria CLC proteins [43, 44] and the crystal structures of the C-terminal cytoplasmic domain of several vertebrate CLC molecules, such as CLC-0 [54], CLC-5 [55] and CLC-K [56], have provided insightful structural information for other homologous CLC molecules. The CLC protein from E. coli (CLC-ec1) consists of only ∼460 AA, which form a structure corresponding to the N-terminal transmembrane portion of CLC-1 [44] (Figure 2(a), upper panel). This part of the channel protein is composed of eighteen α-helices (helices 1 to 18, or helices A to R), seventeen of which being membrane associated (helix A is not inserted into the membrane). Most of these helices are not perpendicular to the membrane, but severely tilted. Moreover, many of these helices do not span the entire width of the lipid membrane (Figure 2(a), upper panel). The most interesting feature of the transmembrane portion of CLC molecules is that a glutamate residue located at the beginning of helix N (helix 14) protrudes its negatively charged side chain into the Cl−-permeation pathway (Figure 2(a), red residues in the upper panel). With the glutamate side chain blocking the Cl−-permeation pore, Cl− permeation is not possible [44]. Mutation of this glutamate residue to a neutral amino acid in CLC-channels results in channels that appear to have a fully open pore. The side chain of this glutamate residue is therefore thought to be the gate that controls each individual protopore. It is also thought that the competition of Cl− with this glutamate side-chain may underlie the aforementioned gating permeation mechanism thoroughly characterized for CLC-0 and CLC-1 channels [23, 57–59].
Figure 2.
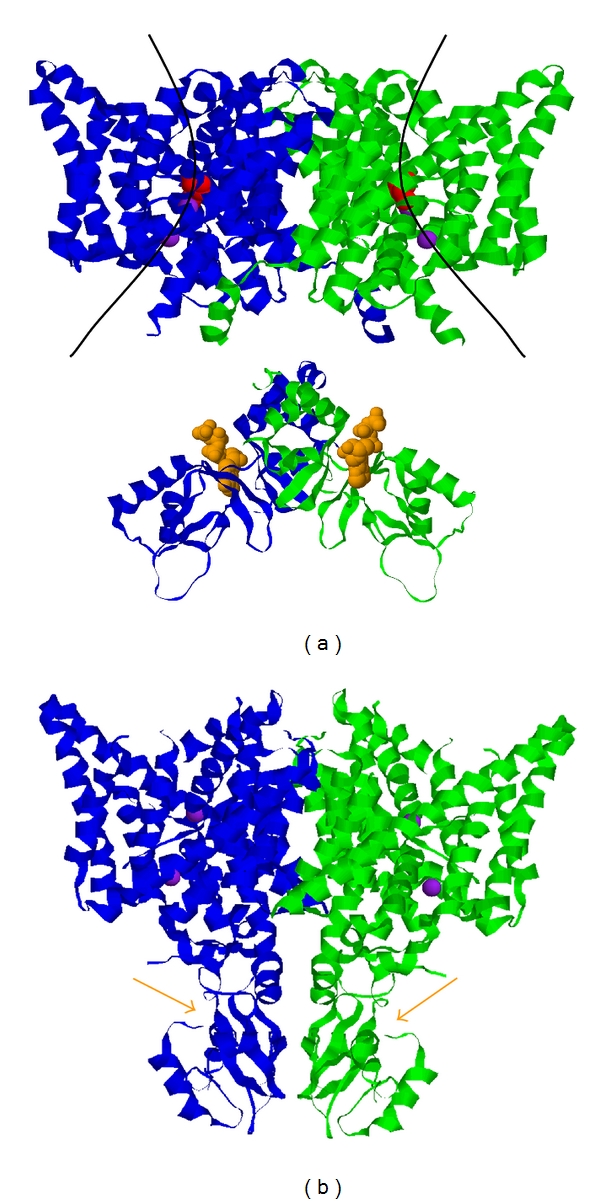
Molecular architecture of mammalian CLC molecules. (a) The composite structure of a generic CLC molecule consists of two parts: the membrane region, represented by the crystal structure of E. coli CLC molecule (CLC-ec1) (top), and the cytoplasmic domain represented by the crystal structure of the cytoplasmic domain of CLC-5. The two subunits are colored in green and blue, respectively. The two curve lines in the membrane portion roughly depict the transport pathways of Cl− ions (purple spheres). Red residues are Glu 148 of CLC-ec1, which correspond to Glu 232 of CLC-1. The negatively charged side chain of this residue obstructs the ion-transport pathway, and therefore is hypothesized to be the fast gate of CLC-channels. The two space-filled molecules in orange color in the cytoplasmic domains (one in each subunit) are ATP molecules seen in the crystal structure of the CLC-5's cytoplasmic domain. Binding of ATP to CLC-1 inhibits the common gating of CLC-1. (b) X-ray crystal structure of CmCLC, a CLC protein from a thermophilic red alga Cyanidioschyzon merolae. Orange arrows point to the ATP-binding sites.
The C-terminal half of CLC-1 (from ∼AA 591 to the C-terminus) is believed to be entirely located in the cytoplasmic side of the membrane (Figure 2(a), lower panel). This structure of the C-terminal cytoplasmic domain was initially solved in several mammalian CLC molecules independent of the transmembrane domain. A most recent crystallographic study revealed the structure of CmCLC, a CLC protein from thermophilic alga that consists of a transmembrane region and a C-terminal cytoplasmic domain [49]. The transmembrane and cytoplasmic domains of the CmCLC structure are similar to those solved previously in other CLC proteins, including the extensive helical architecture in the transmembrane region and the characteristic cystathionine β-synthase (CBS) domains in the cytoplasmic region (Figure 2(b)). Thus, it is very likely that the C-terminal cytoplasmic domain of CLC-1, like those in other mammalian CLC molecules, also contains two tandem CBS domains that are folded into a potential ATP-binding site. It has been shown that cytoplasmic ATP can inhibit the current of CLC-1 channels in acidic pH conditions [60–62], and the crystal structure of the C-terminal region of the CLC-5 protein revealed an ATP molecule bound to the predicted ATP-binding site formed by the two tandem CBS domains [55] (Figure 2(a), lower panel). The inhibition of CLC-1 channels by ATP is therefore thought to be due to a direct ATP binding to the C-terminus of protonated CLC-1 channels. Experimental evidence shows that the effect of intracellular ATP is to make the opening of the common gate more difficult [61]. The mechanism of common gating of CLC-channels is not clear, but it has been proposed that this gating mechanism may involve the relative motion of the two channel subunits, including the movement of the C-terminal cytoplasmic domain [63]. The inhibition of the common gating by cytoplasmic ATP is consistent with the structural feature that the ATP-binding site is located in the C-terminal cytoplasmic region of CLC-1 channels.
During vigorous muscle activities, ATP level in fast-twitch muscle fibers is significantly lowered [64], which in turn reduces ATP inhibition of CLC-1 channels. This enhanced activation of CLC-1 channels is expected to decrease muscle excitability, a potential cell protection mechanism during metabolic stress that may contribute to the development of muscle fatigue [65]. As discussed above, muscle fatigue may also be caused by partial Na+ channel inactivation as a result of the accumulation of extracellular k+ ions after multiple action potentials. Moreover, intensive exercise leads to muscle acidosis [66, 67], which in the presence of ATP, may result in CLC-1 channel inhibition [60]. This down regulation of membrane Cl− conductance will notably reduce the input conductance of sarcolemma and consequently increase the likelihood of spike induction for the Na+ channels that remain active, and may therefore serve as a physiological response to prevent the development of muscle fatigue.
5. Pathophysiology of Myotonia Congenita
In humans, mutations in the skeletal muscle CLC-1 gene (CLCN1) on chromosome 7 have been linked to a hereditary muscle disease, myotonia congenita [68]. Myotonia can be defined as a hyperexcitability of the plasma membrane of skeletal muscle fibers. Myotonia is due to an electrical instability of the muscle membrane itself, leading to repetitive action potentials with a single stimulus (“myotonic runs”). Myotonia congenita was one of the first human diseases proven to be caused by an ion-channel defect (channelopathy). This discovery was based on studies in goats with hereditary myotonia that closely resembled myotonia congenita in humans [69–71]. Subsequent studies also demonstrated that there was indeed a reduced Cl− conductance in goat and human myotonic muscle fibers and that normal muscle fibers exhibited myotonic features when Cl− was replaced with an impermeant anion [14, 72]. This myotonia-like phenomenon induced by low concentrations of Cl− can be accounted for by the aforementioned Cl−-dependent gating mechanism for CLC-1 channels.
Human myotonia congenita can be inherited in an autosomal recessive (Becker type) or autosomal dominant (Thomsen type) manner [73]. By now, more than 100 different mutations in the CLCN1 gene have been identified in patients with myotonia congenita [74–76]. Myotonia-causing mutations are scattered over the entire sequence of the channel protein, both in the transmembrane region and in the cytosolic N-terminal and C-terminal parts. They include nonsense, splice-site, and frameshift mutations that truncate the channel protein. Truncation mutations are always associated with recessive myotonia, except when they are very close to the C-terminus. Missense mutations can be associated with either recessive or dominant inheritance. Mutations with dominant inheritance are less frequent. With the exception of truncations very close to the C-terminus of CLC-1 channels, all dominant mutations are missense mutations. Recessive mutations are more diverse; they can be associated with truncations, insertions, splice defects, missense, or nonsense/stop errors. Therefore, it is not possible to predict on the basis of the mutation location or the mutation type whether the inheritance will be dominant or recessive.
For the autosomal dominant form of myotonia, patients are expected to carry the heterozygous CLCN1 genotype: one copy each of the wild-type and the mutant allele. Regarding the molecular mechanism of myotonia congenita, a loss-of-function phenotype of the mutant CLCN1 gene certainly supports haploinsufficiency as a reasonable mechanism causing the malfunction of muscles. For many other cases of disease-related mutations, however, a total loss of functional CLC-1 channels on only one allele does not lead to myotonia [32, 36]. It has been suggested that dominant myotonia is due to dominant-negative effects of the mutant subunit on the wild-type subunit coexpressed in the muscles of heterozygous patients. On the other hand, lots of CLCN1 gene mutations result in recessive myotonia, and the mutant proteins do not have dominant negative effects. A likely reason for a lack of dominant negative effects for these recessive mutants is the inability of truncated proteins to associate with the wild-type subunit [74].
Therefore, a current working hypothesis on the molecular basis for the inheritance of myotonia congenita is that the inheritance pattern of a mutation is predominantly determined by the functional consequence of the mutation on the gating of CLC-1 channels: those mutations that affect the common gate lead to an autosomal-dominant inheritance pattern, whereas those affecting individual protopores only result in a recessive pattern [42, 77]. As described above, a functional CLC-1 channel is a homo-dimer. With the exception of truncations very close to the C-terminus of CLC-1 channels, all dominant mutations are missense mutations. Almost all these mutations shift the voltage-dependence of gating of the channel towards positive voltages so that the activity of the mutant channels is insufficient to cause membrane repolarization [53]. The dominant-negative effect of these mutations on the hetero-dimeric channel is due to the fact that the common-gate controlling both protopores is affected by the mutation in the mutant subunit [42]. Indeed, many, but not all, mutations in dominant myotonia are due to mutations of residues close to the subunit interface [74, 75, 78]. Consistent with this observation, site-directed mutagenesis of residues lining subunit interface affects the common gate of CLC-1 [79, 80]. On the other hand, as the ion-conducting pore of CLC-1 is entirely contained within each subunit of the dimer [43, 81], mutations affecting the function of one protopore are unlikely to affect the conductance of the second subunit in wild-type/mutant hetero dimeric channels and therefore will generally lack dominantnegative effects. For example, the equivalent residue of CLC-1's M485 in bacterial CLC proteins is located in the ion-transport pathway. In heterologous expression systems, the mutation M485 V in CLC-1 drastically changed the single-channel conductance and the voltage-dependent gating of homodimeric mutant CLC-1 channels [82]. This mutation, however, displays a recessive inheritance pattern. As both alleles are mutated in patients with recessive myotonia, a total loss of CLC-1 channel function may ensue. In contrast, by assuming equal association affinity for both wild-type and mutant subunits in the dimeric channel architecture, at least 25% of wild-type currents will still remain in heterozygous patients carrying dominant-negative mutations. Accordingly, recessive myotonia is clinically more severe than the dominant Thomsen form.
In addition to faulty channel gating, other mechanisms may also contribute to the pathophysiology of myotonia congenita [83]. For example, several recessive CLCN1 mutations (e.g., Y150C, V165G, F167L, V236L, Y261C, V327I, and F413C) have been shown to yield functional CLC-1 channels with biophysical properties either only slightly different or virtually indistinguishable from those of wild-type channels [75]. Similarly, some dominant CLCN1 mutations (e.g., R338Q, F428S, and T550M) have been shown to display no detectable gating defects upon forming heterodimers with their wild-type counterparts [84, 85]. By no means can the foregoing dominant negative mechanism explain the inheritance patterns of these mutations. These examples clearly demonstrate that the effect of myotonia-related mutations cannot be simply attributed to the disruption of the gating of CLC-1 channels. Several novel mutations in the CLCN1 gene have recently been identified in Taiwanese patients suffering from myotonia congenita [86, 87]. Interestingly, one of the detected mutations, fs793X, was found in a Taiwanese family with dominant inheritance pattern [87]. However, the same mutation was previously found in an recessive Italian pedigree [88]. This is only one of the several examples showing that the same mutations are associated with recessive myotonia in some families, but with dominant myotonia in others [77, 89, 90]. This dual inheritance pattern again demonstrates the inadequacy of the gating hypothesis and further highlights the importance of other pathophysiological mechanisms of myotonia congenita. It is likely that some myotonia congenita-related mutations may (i) result in aberrant biogenesis and subunit assembly and/or (ii) lead to a defective membrane targeting subcellular localization patterns of CLC-1 channels. Indeed, three myotonia-related mutations in the distal C-terminus of CLC-1 channels have been shown to have a reduction of protein expression in the surface membrane [83]. Thus, the underlying mechanisms of myotonia due to CLC-1 channelopathy are likely to be quite diverse.
6. Clinical Correlation
Myotonia is characterized by the impaired relaxation of skeletal muscle following sudden voluntary contraction. As discussed above, CLC-1 conductance contributes up to 80% of the resting membrane conductance in normal skeletal muscle. Myotonia-causing mutations in the CLCN1 gene therefore lead to a significant reduction in resting membrane conductance, thereby increasing the input resistance of skeletal muscle [72, 91]. Consequently, a smaller membrane depolarization (threshold potential) will be sufficient to trigger an action potential, that is, muscle excitability will be enhanced. This scenario explains why a single nerve stimulus elicited a train of action potentials in muscle fibers from myotonic goats; in contrast, the same stimulus only induced a single action potential in normal muscle fibers [70].
Another important role of CLC-1 conductance in muscle is to counteract the depolarizing effect of tubular K+ accumulation during intensive firing of action potentials [70]. As the extracellular volume in the transverse-tubule system of skeletal muscles is limited, K+ ions tend to accumulate in the extracellular space during intensive firing of muscle action potentials. This increase in extracellular K+ normally has little effect on the membrane potential due to the presence of high CLC-1 conductance. In myotonic muscle, however, a small accumulation of tubular K+ will result in a significant membrane depolarization. In the presence of rapid successions of action potentials, summation of these K+ accumulation-induced membrane depolarization may trigger spontaneous muscle action potentials, thereby manifesting myotonia symptoms such as muscle stiffness after voluntary contraction. Hence, the medication of choice for myotonic patients usually involves drugs that suppress muscle excitability via inhibition of voltage-gated Na+ channels [92].
The muscle stiffness of myotonia can gradually be relieved by exercise (the so-called “warm-up” phenomenon) [93]. One plausible mechanism of the warm-up is the enhanced activity of the muscle Na+/K+-ATPase induced by exercise, which facilitates the clearance of extracellular K+ from transverse-tubules. A recent study in myotonic patients, however, failed to support this hypothesis [94], indicating that the precise mechanism of warm-up is still unclear.
Also elusive are the rationales for several other clinical manifestations of myotonia congenita. For example, the recessive myotonia is usually more common in men than in women, and for female patients of dominant myotonia, the symptoms become worse during pregnancy [75]. It has been suggested that the observed gender difference may arise from the modulation of CLC-1 channel function by sex hormones [95]. In addition, recessive but not dominant myotonia is often associated with transient muscle weakness on the initiation of movement [75, 96], a defect that is not predicted by enhanced muscle excitability. Obviously, a combination of biophysical and cell biological studies in both in vitro and in vivo models will be required for better understanding of the clinical symptoms of myotonia congenita.
7. Concluding Remarks
ClC-1 channels play a crucial role in setting membrane excitability in skeletal muscle. Despite of the numerous documentations of the association between CLCN1 mutations and myotonia congenita, elucidation of the mechanistic link between genetic defects and pathogenesis is still at the primitive stage. One major limitation to our better understanding of this issue lies in the fact that protein biosynthetic pathways as well as subcellular localization patterns of CLC-1 channels in situ remain obscure. For example, even though a significant Cl− conductance has been identified in the transverse-tubule system, it remains inconclusive whether CLC-1 channels are actually expressed in the transverse-tubule system. In the ADR (arrested development of righting response) mouse that has been used as a model for recessive myotonia [34, 69], immuonhistochemistry of muscle cryosection located CLC-1 channels primarily in the outer, sarcolemmal membrane, but not in the transverse-tubule of skeletal muscle [97]. A similar conclusion on sarcolemmal localization of CLC-1 channels was recently reported in flexor digitorum brevis muscle fibers of wild-type mice as well [98]. Biophysical and pharmacological studies in a skinned rat skeletal muscle, however, demonstrated that the transverse-tubule Cl− channel conductance was blocked by 9-AC, low intracellular pH, and protein kinase C activators [18, 19, 96], all of which are known to affect the properties of CLC-1 channels observed in the heterologous expression system [32, 60, 61, 99]. As has been previously proposed [19, 100], this apparent discrepancy may arise from the possibility that the transverse-tubule system expresses certain splice variants of CLC-1 channels that lack the epitope for the antibody used in the immunofluorescence study, or that the surrounding microenvironment in the transverse-tubule system prevents the antibody from properly recognizing the epitope in CLC-1 channels in situ. The field thus requires more extensive studies on not only the gating mechanisms but also the biosynthetic process and subcellular localization of CLC-1 channels.
Myotonia congenita, therefore, is still in lack of a standard, effective treatment. The field thus begs for further research efforts in multiple directions. At the molecular level, the mechanistic principles underlying the operation of CLC-1 need to be further examined. At the cellular level, the protein biosynthesis mechanisms of CLC-1 (protein biogenesis, membrane trafficking, as well as subcellular localization patterns in situ), although drawing much research attention recently, remain obscure and need more in-depth investigations. At the clinical level, many myotonia-associated symptoms require better understanding of their pathophysiological mechanisms. Through elucidations of the physiological roles of CLC-1 and the pathophysiological mechanisms of the CLC-1 channelopathy, the therapeutic strategies for myotonia congenita will eventually be illuminated.
Acknowledgments
The authors thank Dr. Tzyh-Chang Hwang for critical readings of the paper. The research in Dr. Chih-Yung Tang's laboratory is supported by a Grant (NSC 96-2320-B-002-069-MY3) from National Science Council of Taiwan, while the work of Dr. Tsung-Yu Chen's laboratory is supported by a Grant (R01GM065447) from National Institutes of Health of USA.
References
- 1.Russell JM. Sodium-potassium-chloride cotransport. Physiological Reviews. 2000;80(1):211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 2.Adragna NC, di Fulvio M, Lauf PK. Regulation of K-Cl cotransport: from function to genes. Journal of Membrane Biology. 2004;201(3):109–137. doi: 10.1007/s00232-004-0695-6. [DOI] [PubMed] [Google Scholar]
- 3.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiological Reviews. 2005;85(2):423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 4.Jentsch TJ, Maritzen T, Zdebik AA. Chloride channel diseases resulting from impaired transepithelial transport or vesicular function. Journal of Clinical Investigation. 2005;115(8):2039–2046. doi: 10.1172/JCI25470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends in Neurosciences. 2003;26(4):199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 6.Hodgkin AL, Horowicz P. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. Journal of Physiology. 1959;148:127–160. doi: 10.1113/jphysiol.1959.sp006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adrian RH. Potassium chloride movement and the membrane potential of frog muscle. Journal of Physiology. 1960;151:154–185. [PMC free article] [PubMed] [Google Scholar]
- 8.Adrian RH. Internal chloride concentration and chloride efflux of frog muscle. Journal of Physiology. 1961;156:623–632. doi: 10.1113/jphysiol.1961.sp006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dulhunty AF. The dependence of membrane potential on extracellular chloride concentration in mammalian skeletal muscle fibres. Journal of Physiology. 1978;276:67–82. doi: 10.1113/jphysiol.1978.sp012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foppen RJG, van Mil HGJ, van Heukelom JS. Effects of chloride transport on bistable behaviour of the membrane potential in mouse skeletal muscle. Journal of Physiology. 2002;542(1):181–191. doi: 10.1113/jphysiol.2001.013298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foppen RJG. In skeletal muscle the relaxation of the resting membrane potential induced by K(+) permeability changes depends on Cl(-) transport. Pflugers Archiv. 2004;447(4):416–425. doi: 10.1007/s00424-003-1165-1. [DOI] [PubMed] [Google Scholar]
- 12.Gallaher J, Bier M, van Heukelom JS. The role of chloride transport in the control of the membrane potential in skeletal muscle—theory and experiment. Biophysical Chemistry. 2009;143(1-2):18–25. doi: 10.1016/j.bpc.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Aickin CC, Betz WJ, Harris GL. Intracellular chloride and the mechanism for its accumulation in rat lumbrical muscle. Journal of Physiology. 1989;411:437–455. doi: 10.1113/jphysiol.1989.sp017582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryant SH, Morales-Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. Journal of Physiology. 1971;219(2):367–383. doi: 10.1113/jphysiol.1971.sp009667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dulhunty AF. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. Journal of Membrane Biology. 1979;45(3-4):293–310. doi: 10.1007/BF01869290. [DOI] [PubMed] [Google Scholar]
- 16.Bretag AH. Muscle chloride channels. Physiological Reviews. 1987;67(2):618–724. doi: 10.1152/physrev.1987.67.2.618. [DOI] [PubMed] [Google Scholar]
- 17.Palade PT, Barchi RL. Characteristics of the chloride conductance in muscle fibers of the rat diaphragm. Journal of General Physiology. 1977;69(3):325–342. doi: 10.1085/jgp.69.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coonan JR, Lamb GD. Effect of transverse-tubular chloride conductance on excitability in skinned skeletal muscle fibres of rat and toad. Journal of Physiology. 1998;509, part 2:551–564. doi: 10.1111/j.1469-7793.1998.551bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dutka TL, Murphy RM, Stephenson DG, Lamb GD. Chloride conductance in the transverse tubular system of rat skeletal muscle fibres: importance in excitation-contraction coupling and fatigue. Journal of Physiology. 2008;586(3):875–887. doi: 10.1113/jphysiol.2007.144667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedersen TH, Nielsen OB, Lamb GD, Stephenson DG. Intracellular acidosis enhances the excitability of working muscle. Science. 2004;305(5687):1144–1147. doi: 10.1126/science.1101141. [DOI] [PubMed] [Google Scholar]
- 21.Cairns SP, Lindinger MI. Do multiple ionic interactions contribute to skeletal muscle fatigue? Journal of Physiology. 2008;586(17):4039–4054. doi: 10.1113/jphysiol.2008.155424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiological Reviews. 2002;82(2):503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 23.Chen TY. Structure and function of CLC channels. Annual Review of Physiology. 2005;67:809–839. doi: 10.1146/annurev.physiol.67.032003.153012. [DOI] [PubMed] [Google Scholar]
- 24.Jentsch TJ, Poët M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annual Review of Physiology. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- 25.Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature. 2004;427(6977):803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 26.Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436(7049):420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 27.Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436(7049):424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 28.Devuyst O, Guggino WB. Chloride channels in the kidney: lessons learned from knockout animals. American Journal of Physiology—Renal Physiology. 2002;283(6):F1176–F1191. doi: 10.1152/ajprenal.00184.2002. [DOI] [PubMed] [Google Scholar]
- 29.Cuppoletti J, Tewari KP, Sherry AM, Malinowska DH. Activation of human ClC-2 Cl- channels: implications for cystic fibrosis. Clinical and Experimental Pharmacology and Physiology. 2000;27(11):896–900. doi: 10.1046/j.1440-1681.2000.03357.x. [DOI] [PubMed] [Google Scholar]
- 30.Strange K. Of mice and worms: novel insights into CIC-2 anion channel physiology. News in Physiological Sciences. 2002;17(1):11–16. doi: 10.1152/physiologyonline.2002.17.1.11. [DOI] [PubMed] [Google Scholar]
- 31.Thiemann A, Grunder S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356(6364):57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- 32.Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991;354(6351):301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XD, Morishima S, Ando-Akatsuka Y, et al. Expression of novel isoforms of the ClC-1 chloride channel, in astrocytic glial cells in vitro. GLIA. 2004;47(1):46–57. doi: 10.1002/glia.20024. [DOI] [PubMed] [Google Scholar]
- 34.Steinmeyer K, Klocke R, Ortland C, et al. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 1991;354(6351):304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- 35.Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257(5071):797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- 36.Gronemeier M, Condie A, Prosser J, Steinmeyer K, Jentsch TJ, Jockusch H. Nonsense and missense mutations in the muscular chloride channel gene Clc- 1 of myotonic mice. Journal of Biological Chemistry. 1994;269(8):5963–5967. [PubMed] [Google Scholar]
- 37.Beck CL, Fahlke C, George AL., Jr. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):11248–11252. doi: 10.1073/pnas.93.20.11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhodes TH, Vite CH, Giger U, Patterson DF, Fahlke C, George AL., Jr. A missense mutation in canine ClC-1 causes recessive myotonia congenita in the dog. FEBS Letters. 1999;456(1):54–58. doi: 10.1016/s0014-5793(99)00926-6. [DOI] [PubMed] [Google Scholar]
- 39.Fahlke C, Rüdel R, Mitrovic N, Zhou M, George AL., Jr. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 1995;15(2):463–472. doi: 10.1016/0896-6273(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 40.Miller C. Open-state substructure of single chloride channels from Torpedo electroplax. Philosophical Transactions of the Royal Society of London B. 1982;299(1097):401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- 41.Miller C, White MM. Dimeric structure of single chloride channels from Torpedo electroplax. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(9):2772–2775. doi: 10.1073/pnas.81.9.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saviane C, Conti F, Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. Journal of General Physiology. 1999;113(3):457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a CIC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415(6869):287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 44.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300(5616):108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 45.Chen TY, Miller C. Nonequilibrium gating and voltage dependence of the ClC-0 Cl- channel. Journal of General Physiology. 1996;108(4):237–250. doi: 10.1085/jgp.108.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rychkov GY, Pusch M, Astill DSJ, Roberts ML, Jentsch TJ, Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel C1C-1. Journal of Physiology. 1996;497, part 2:423–435. doi: 10.1113/jphysiol.1996.sp021778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen MF, Chen TY. Different fast-gate regulation by external Cl(-) and H(+) of the muscle-type ClC chloride channels. Journal of General Physiology. 2001;118(1):23–32. doi: 10.1085/jgp.118.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440(7083):484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- 49.Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330(6004):635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanke W, Miller C. Single chloride channels from Torpedo electroplax. Activation by protons. Journal of General Physiology. 1983;82(1):25–45. doi: 10.1085/jgp.82.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin YW, Lin CW, Chen TY. Elimination of the slow gating of C1C-0 chloride channel by a point mutation. Journal of General Physiology. 1999;114(1):1–12. doi: 10.1085/jgp.114.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hille B. Ion Channels of Excitable Membranes. Sunderland, Mass, USA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- 53.Pusch M, Steinmeyer K, Koch MC, Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron. 1995;15(6):1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 54.Meyer S, Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14(2):299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 55.Meyer S, Savaresi S, Forster IC, Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nature Structural and Molecular Biology. 2007;14(1):60–67. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 56.Markovic S, Dutzler R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure. 2007;15(6):715–725. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 57.Chen TY. Coupling gating with ion permeation in ClC channels. Science’s STKE. 2003;2003(188):p. e23. doi: 10.1126/stke.2003.188.pe23. [DOI] [PubMed] [Google Scholar]
- 58.Dutzler R. The structural basis of ClC chloride channel function. Trends in Neurosciences. 2004;27(6):315–320. doi: 10.1016/j.tins.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 59.Dutzler R. Structural basis for ion conduction and gating in ClC chloride channels. FEBS Letters. 2004;564(3):229–233. doi: 10.1016/S0014-5793(04)00210-8. [DOI] [PubMed] [Google Scholar]
- 60.Bennetts B, Parker MW, Cromer BA. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. Journal of Biological Chemistry. 2007;282(45):32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- 61.Tseng PY, Bennetts B, Chen TY. Cytoplasmic ATP inhibition of CLC-1 is enhanced by low pH. Journal of General Physiology. 2007;130(2):217–221. doi: 10.1085/jgp.200709817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang XD, Tseng PY, Chen TY. ATP inhibition of CLC-1 is controlled by oxidation and reduction. Journal of General Physiology. 2008;132(4):421–428. doi: 10.1085/jgp.200810023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bykova EA, Zhang XD, Chen TY, Zheng J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nature Structural and Molecular Biology. 2006;13(12):1115–1119. doi: 10.1038/nsmb1176. [DOI] [PubMed] [Google Scholar]
- 64.Karatzaferi C, de Haan A, Ferguson RA, van Mechelen W, Sargeant AJ. Phosphocreatine and ATP content in human single muscle fibres before and after maximum dynamic exercise. Pflugers Archiv. 2001;442(3):467–474. doi: 10.1007/s004240100552. [DOI] [PubMed] [Google Scholar]
- 65.Bennetts B, Rychkov GY, Ng HL, et al. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. Journal of Biological Chemistry. 2005;280(37):32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- 66.Roos A, Boron WF. Intracellular pH transients in rat diaphragm muscle measured with DMO. American Journal of Physiology. 1978;235(1):C49–C54. doi: 10.1152/ajpcell.1978.235.1.C49. [DOI] [PubMed] [Google Scholar]
- 67.Wilson JR, McCully KK, Mancini DM, Boden B, Chance B. Relationship of muscular fatigue to pH and diprotonated P(i) in humans: a 31P-NMR study. Journal of Applied Physiology. 1988;64(6):2333–2339. doi: 10.1152/jappl.1988.64.6.2333. [DOI] [PubMed] [Google Scholar]
- 68.Jurkat-Rott K, Lerche H, Lehmann-Horn F. Skeletal muscle channelopathies. Journal of Neurology. 2002;249(11):1493–1502. doi: 10.1007/s00415-002-0871-5. [DOI] [PubMed] [Google Scholar]
- 69.Lipicky RJ, Bryant SH. Sodium, potassium, and chloride fluxes in intercostal muscle from normal goats and goats with hereditary myotonia. Journal of General Physiology. 1966;50(1):89–111. doi: 10.1085/jgp.50.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adrian RH, Bryant SH. On the repetitive discharge in myotonic muscle fibres. Journal of Physiology. 1974;240(2):505–515. doi: 10.1113/jphysiol.1974.sp010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adrian RH, Marshall MW. Action potentials reconstructed in normal and myotonic muscle fibres. Journal of Physiology. 1976;258(1):125–143. doi: 10.1113/jphysiol.1976.sp011410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipicky RJ, Bryant SH, Salmon JH. Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. Journal of Clinical Investigation. 1971;50(10):2091–2103. doi: 10.1172/JCI106703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ptacek LJ, Johnson KJ, Griggs RC. Genetics and physiology of the myotonic muscle disorders. The New England Journal of Medicine. 1993;328(7):482–489. doi: 10.1056/NEJM199302183280707. [DOI] [PubMed] [Google Scholar]
- 74.Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Human Mutation. 2002;19(4):423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- 75.Colding-Jørgensen E. Phenotypic variability in myotonia congenita. Muscle and Nerve. 2005;32(1):19–34. doi: 10.1002/mus.20295. [DOI] [PubMed] [Google Scholar]
- 76.Lossin C, George AL., Jr. Myotonia congenita. Advances in Genetics. 2008;63:25–55. doi: 10.1016/S0065-2660(08)01002-X. [DOI] [PubMed] [Google Scholar]
- 77.Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH, Jentsch TJ. CIC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Human Molecular Genetics. 1998;7(11):1753–1760. doi: 10.1093/hmg/7.11.1753. [DOI] [PubMed] [Google Scholar]
- 78.Fialho D, Schorge S, Pucovska U, et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007;130(12):3265–3274. doi: 10.1093/brain/awm248. [DOI] [PubMed] [Google Scholar]
- 79.Accardi A, Ferrera L, Pusch M. Drastic reduction of the slow gate of human muscle chloride channel (CIC-1) by mutation C277S. Journal of Physiology. 2001;534(3):745–752. doi: 10.1111/j.1469-7793.2001.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duffield M, Rychkov G, Bretag A, Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. Journal of General Physiology. 2003;121(2):149–161. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weinreich F, Jentsch TJ. Pores formed by single subunits in mixed dimers of different CLC chloride channels. Journal of Biological Chemistry. 2001;276(4):2347–2353. doi: 10.1074/jbc.M005733200. [DOI] [PubMed] [Google Scholar]
- 82.Wollnik B, Kubisch C, Steinmeyer K, Pusch M. Identification of functionally important regions of the muscular chloride channel CIC-1 by analysis of recessive and dominant myotonic mutations. Human Molecular Genetics. 1997;6(5):805–811. doi: 10.1093/hmg/6.5.805. [DOI] [PubMed] [Google Scholar]
- 83.Macías MJ, Teijido O, Zifarelli G, et al. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochemical Journal. 2007;403(1):79–87. doi: 10.1042/BJ20061230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu FF, Ryan A, Devaney J, et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002;125(11):2392–2407. doi: 10.1093/brain/awf246. [DOI] [PubMed] [Google Scholar]
- 85.Zhang J, Bendahhou S, Sanguinetti MC, Ptáček LJ. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology. 2000;54(4):937–942. doi: 10.1212/wnl.54.4.937. [DOI] [PubMed] [Google Scholar]
- 86.Jou SB, Chang LI, Pan H, Chen PR, Hsiao KM. Novel CLCN1 mutations in Taiwanese patients with myotonia congenita. Journal of Neurology. 2004;251(6):666–670. doi: 10.1007/s00415-004-0383-6. [DOI] [PubMed] [Google Scholar]
- 87.Kuo HC, Hsiao KM, Chang LI, You TH, Yeh TH, Huang CC. Novel mutations at carboxyl terminus of CIC-1 channel in myotonia congenita. Acta Neurologica Scandinavica. 2006;113(5):342–346. doi: 10.1111/j.1600-0404.2006.00589.x. [DOI] [PubMed] [Google Scholar]
- 88.Sangiuolo F, Botta A, Mesoraca A, et al. Identification of five new mutations and three novel polymorphisms in the muscle chloride channel gene (CLCN1) in 20 Italian patients with dominant and recessive myotonia congenita. Mutations in brief no. 118. Online. Human Mutation. 1998;11(4):p. 331. doi: 10.1002/(SICI)1098-1004(1998)11:4<331::AID-HUMU12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 89.George AL, Jr., Sloan-Brown K, Fenichel GM, Mitchell GA, Spiegel R, Pascuzzi RM. Nonsense and missense mutations of the muscle chloride channel gene in patients with myotonia congenita. Human Molecular Genetics. 1994;3(11):2071–2072. [PubMed] [Google Scholar]
- 90.Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ, Koch MC. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. American Journal of Human Genetics. 1995;57(6):1325–1334. [PMC free article] [PubMed] [Google Scholar]
- 91.Bryant SH. Cable properties of external intercostal muscle fibres from myotonic and nonmyotonic goats. Journal of Physiology. 1969;204(3):539–550. doi: 10.1113/jphysiol.1969.sp008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meola G, Sansone V. Therapy in myotonic disorders and in muscle channelopathies. Neurological Sciences. 2000;21(5):S953–S961. doi: 10.1007/s100720070009. [DOI] [PubMed] [Google Scholar]
- 93.Rudel R, Lehmann-Horn F. Membrane changes in cells from myotonia patients. Physiological Reviews. 1985;65(2):310–356. doi: 10.1152/physrev.1985.65.2.310. [DOI] [PubMed] [Google Scholar]
- 94.van Beekvelt MCP, Drost G, Rongen G, Stegeman DF, van Engelen BGM, Zwarts MJ. Na+-K+-ATPase is not involved in the warming-up phenomenon in generalized myotonia. Muscle and Nerve. 2006;33(4):514–523. doi: 10.1002/mus.20483. [DOI] [PubMed] [Google Scholar]
- 95.Fialho D, Kullmann DM, Hanna MG, Schorge S. Non-genomic effects of sex hormones on CLC-1 may contribute to gender differences in myotonia congenita. Neuromuscular Disorders. 2008;18(11):869–872. doi: 10.1016/j.nmd.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 96.Rayan DLR, Hanna MG. Skeletal muscle channelopathies: nondystrophic myotonias and periodic paralysis. Current Opinion in Neurology. 2010;23(5):466–476. doi: 10.1097/WCO.0b013e32833cc97e. [DOI] [PubMed] [Google Scholar]
- 97.Gurnett CA, Kahl SD, Anderson RD, Campbell KP. Absence of the skeletal muscle sarcolemma chloride channel ClC-1 in myotonic mice. Journal of Biological Chemistry. 1995;270(16):9035–9038. doi: 10.1074/jbc.270.16.9035. [DOI] [PubMed] [Google Scholar]
- 98.Lueck JD, Rossi AE, Thornton CA, Campbell KP, Dirksen RT. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. Journal of General Physiology. 2010;136(6):597–613. doi: 10.1085/jgp.201010526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosenbohm A, Rüdel R, Fahlke C. Regulation of the human skeletal muscle chloride channel hClC-1 by protein kinase C. Journal of Physiology. 1999;514, part 3:677–685. doi: 10.1111/j.1469-7793.1999.677ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aromataris EC, Rychkov GY. ClC-1 chloride channel: matching its properties to a role in skeletal muscle. Clinical and Experimental Pharmacology and Physiology. 2006;33(11):1118–1123. doi: 10.1111/j.1440-1681.2006.04502.x. [DOI] [PubMed] [Google Scholar]