

Abstract
Selected reaction monitoring (SRM)-MS is an emerging technology for high throughput targeted protein quantification and verification in biomarker discovery studies; however, the cost associated with the application of stable isotope-labeled synthetic peptides as internal standards can be prohibitive for screening a large number of candidate proteins as often required in the preverification phase of discovery studies. Herein we present a proof of concept study using an 18O-labeled proteome reference as global internal standards (GIS) for SRM-based relative quantification. The 18O-labeled proteome reference (or GIS) can be readily prepared and contains a heavy isotope (18O)-labeled internal standard for every possible tryptic peptide. Our results showed that the percentage of heavy isotope (18O) incorporation applying an improved protocol was >99.5% for most peptides investigated. The accuracy, reproducibility, and linear dynamic range of quantification were further assessed based on known ratios of standard proteins spiked into the labeled mouse plasma reference. Reliable quantification was observed with high reproducibility (i.e. coefficient of variance <10%) for analyte concentrations that were set at 100-fold higher or lower than those of the GIS based on the light (16O)/heavy (18O) peak area ratios. The utility of 18O-labeled GIS was further illustrated by accurate relative quantification of 45 major human plasma proteins. Moreover, quantification of the concentrations of C-reactive protein and prostate-specific antigen was illustrated by coupling the GIS with standard additions of purified protein standards. Collectively, our results demonstrated that the use of 18O-labeled proteome reference as GIS provides a convenient, low cost, and effective strategy for relative quantification of a large number of candidate proteins in biological or clinical samples using SRM.
Selected reaction monitoring (SRM),1 also known as multiple reaction monitoring, is a promising technology for reliable quantification of targeted analytes in complex biological and clinical applications (1–10). SRM measurements provide increased sensitivity and selectivity compared with conventional LC-MS-based shotgun proteomics mainly caused by the two-stage (Q1 and Q3) mass filtering of a triple quadrupole instrument for precursor and product ions (in Q1 and Q3, respectively) (1, 3, 9). The precision and reproducibility of SRM-based protein quantification in human plasma applying labeled synthetic peptides as internal standards have been recently demonstrated through a multi-site assessment (11). However, the technology is presently limited in the number of candidate proteins that can be screened often because of the costs associated with stable isotope-labeled synthetic peptides (1, 3, 7, 12, 13) or artificial protein concatemers (1, 14, 15) that are typically utilized as internal standards. Recently the use of 18O labeling for individual synthetic peptides as internal standards for SRM has also been recently reported (16). In this work, we investigated the concept of using an 18O-labeled proteome reference to generate global internal standards (GIS) as an alternative to stable isotope-labeled synthetic peptides for broad SRM-based relative quantification.
The initial concept of employing internal standards derived from a stable isotope-labeled whole proteome has been demonstrated previously for global quantitative proteomics (17–19). In one case, a “universal” reference was created by pooling aliquots of tryptically digested individual samples, and then the peptides in the pooled sample were labeled with 18O. 18O labeling is ideal for generating a labeled reference sample because of its simplicity, versatility in labeling any kind of biological sample, and low cost. When spiked into individual samples, the 18O-labeled proteome reference served as GIS for all tryptic peptides that enabled relative abundance comparisons across any number of samples based on light/heavy (16O/18O) ratios (18).
In the present study, known ratios of standard proteins spiked into mouse plasma were used to evaluate the accuracy, reproducibility, and dynamic range of SRM-based quantification using the 18O-labeled GIS. The benefits of the labeled proteome reference were further illustrated by accurate relative quantification of 45 major human plasma proteins without the use of synthetic peptides (1, 6). Quantification of endogenous C-reactive protein (CRP) and spiked prostate-specific antigen (PSA) concentrations in nondepleted human female plasma was achieved by using a standard addition method with purified protein standards and the 18O-labeled reference. Our results demonstrate that the 18O-labeled proteome reference affords sufficient quantification accuracy for SRM analysis of many protein targets in biological samples.
EXPERIMENTAL PROCEDURES
Plasma Samples and Standard Proteins
Mouse plasma (∼40 mg/ml determined by BCA protein assay (Pierce)) obtained from Equitech-Bio, Inc. (Kerrville, TX), human female plasma (∼50 mg/ml) obtained from BioChemed Services (Winchester, VA), six nonhuman standard proteins obtained from Sigma-Aldrich, i.e. bovine carbonic anhydrase II, bovine β-lactoglobulin, Escherichia coli β-galactosidase, equine skeletal muscle myoglobin, chicken ovalbumin, and bovine cytochrome c, and two human standard proteins PSA obtained from Sigma, and CRP obtained from EMD Chemicals (Gibbstown, NJ) were used in this study. In the experiment for quantifying “endogenous” protein concentrations, three human female plasma samples were prepared with PSA and CRP at three different concentrations (2, 4, and 10 μg/ml), respectively; another human female plasma sample spiked with 0.50 μg/ml PSA only was used as a “hypothetical” endogenous sample.
Protein Digestion
All of the protein samples including the nonhuman standard proteins, mouse plasma, and human plasma were digested individually. The protein samples were initially denatured and reduced with 8 m urea and 10 mm dithiothreitol in 50 mm NH4HCO3 buffer (pH 8.2) for 1 h at 37 °C and followed by alkylation of cysteine residues with 40 mm iodoacetamide for 1 h at 37 °C in the dark. Following a 10-fold dilution with 50 mm NH4HCO3, each resulting sample was digested separately using sequencing grade modified porcine trypsin (Promega, Madison, WI) at a trypsin-to-protein ratio of 1:50 (w/w) for 5 h at 37 °C. The digested samples were loaded individually onto a 1-ml solid phase extraction C18 column (Supelco, Bellefonte, PA) and washed with 4 ml of 0.1% trifluoroacetic acid, 5% acetonitrile. Peptides were eluted from the solid phase extraction column with 1 ml of 0.1% trifluoroacetic acid, 80% acetonitrile and then lyophilized. After reconstituting the resulting peptide samples in 25 mm NH4HCO3, the residual trypsin activity was quenched by boiling for 10 min, and then the samples were immediately placed on ice for 30 min. The final peptide concentration for each sample was measured with the BCA protein assay.
Preparation of 18O-Labeled Plasma Reference Samples
Two 18O-labeled mouse plasma reference samples were generated by spiking six standard protein digests into mouse plasma digests at 0.1% and 0.01% (w/w) of the total mouse plasma protein mass. The corresponding standard protein concentrations in original plasma were ∼40 and ∼4 μg/ml, respectively. Trypsin-catalyzed 18O labeling at the peptide level was performed using a recently improved protocol (20). Briefly, the peptide sample was lyophilized to dryness and reconstituted in 100 μl of 50 mm NH4HCO3 in H218O (97%; ISOTEC, Miamisburg, OH), pH 7.8. One μl of 1 m CaCl2 and solution phase trypsin dissolved in H218O at a 1:50 trypsin/peptide ratio (w/w) were added to the samples. The tubes were wrapped in parafilm and mixed continuously for 5 h at 37 °C. The reaction was stopped by boiling the sample in a water bath for 10 min. After snap-freezing the sample in liquid nitrogen, the samples were acidified by adding 5 μl of formic acid, and final peptide concentrations were measured using a BCA assay. Similarly, a human female plasma sample spiked with PSA (2 μg/ml) and CRP (2 μg/ml) was digested and labeled as the 18O-reference. In the experiments for quantifying 45 human plasma proteins, a labeled human plasma digest was prepared with 99% 18O-enriched water (Cambridge Isotope Laboratories, Andover, MA) rather than the initial stock of 97% 18O-enriched water.
Preparation of Calibration Mixtures
Individually digested standard proteins were spiked into the two labeled mouse plasma references to generate 11 calibration mixtures from 400 ng/ml to 400 μg/ml for the unlabeled spiked proteins in the labeled plasma references with the unlabeled versus labeled protein concentration ratios ranging from 0.01 to 100 (see Table I). For quantifying 45 major human plasma proteins, a labeled human plasma reference and unlabeled plasma digest were prepared separately. The unlabeled human plasma digest and the labeled reference were then mixed 1:10, 1:3, 1:1, 3:1, and 10:1 peptide mass ratios of the unlabeled versus the labeled reference to make five mixtures. All of the samples were analyzed by LC-SRM-MS in three technical replicates (triplicate injections).
Table I. A series of calibration mixtures made of mouse plasma and six standard proteins. The concentration values denote the concentrations in original plasma.
| Calibration mixture | Concentration of each standard protein in 18O-reference (% w/w) | Concentration of each unlabeled standard protein | Standard protein concentration ratio (unlabeled: 18O-reference) |
|---|---|---|---|
| M0 | 40 μg/ml (0.10%) | Blank | Background |
| M1 | 40 μg/ml (0.10%) | 400 ng/ml | 1:100 |
| M2 | 40 μg/ml (0.10%) | 800 ng/ml | 1:50 |
| M3 | 40 μg/ml (0.10%) | 1.6 μg/ml | 1:25 |
| M4 | 40 μg/ml (0.10%) | 4 μg/ml | 1:10 |
| M5 | 40 μg/ml (0.10%) | 8 μg/ml | 1:5 |
| M6 | 40 μg/ml (0.10%) | 40 μg/ml | 1:1 |
| M7 | 4 μg/ml (0.010%) | 20 μg/ml | 5:1 |
| M8 | 4 μg/ml (0.010%) | 40 μg/ml | 10:1 |
| M9 | 4 μg/ml (0.010%) | 100 μg/ml | 25:1 |
| M10 | 4 μg/ml (0.010%) | 200 μg/ml | 50:1 |
| M11 | 4 μg/ml (0.010%) | 400 μg/ml | 100:1 |
Selection of Peptides and Transitions for SRM
A total of 79 peptides from the six nonhuman standard proteins were preselected by in silico screening. The minimum length and maximum molecular weight of target tryptic peptide sequences were eight amino acid residues and 2400 Da, respectively. Most target peptides are without any missed cleavages with the exception of a few high responding peptides that contain partial sequences of KD or KK, which were shown to significantly inhibit trypsin activity (21). Similarly, 16 peptides for PSA/CRP (eight peptides for each protein) were preselected.
Initially, transitions of each selected peptide and its optimal collision energy were obtained from infusion experiments in which 500 fmol/μl solution of each standard protein digest in a 1:1 mixture of water and acetonitrile containing 0.1% formic acid were infused at a flow rate of 300 nl/min. The three most intense transitions of y-type ions were utilized for SRM quantification for each peptide. All of the peptides spiked in the mouse and human plasma digests were screened and validated by LC-SRM-MS, and at least two of the most responsive peptides per protein in the SRM mode were selected for the assessment of quantification. Moreover, six peptides, i.e. bradykinin fragments 1–7, kemptide, melittin, methionine enkephalin, renin substrate porcine, and [d-Ala2]-deltorphin II (Sigma), were used as quality control peptides (two optimized transitions per peptide) for monitoring the overall performance of the platform and for LC retention time markers. For quality control peptides without tryptic terminal, both b- and y-type transitions were considered. All of the preselected peptides and parameters of SRM screened peptides are summarized in supplemental Table 1.
For the 45 major human plasma proteins monitored by Kuzyk et al. (6), we selected 24 of the original 45 peptides without C-terminal inhibitory motifs. The other 21 peptides were found to contain inhibitory motifs (e.g. RK, KK, (D/E)K, or K(D/E)) for trypsin activity at their C termini (21–23), which could lead to ineffective 18O labeling. Therefore, these peptides were replaced by other peptides from the same proteins. A total of 42 peptides without such motifs for these 21 proteins were initially selected using the Skyline software tool (24) with a spectrum library based on previous LC-MS/MS data. All of the transitions were selected based on ion trap MS/MS spectra without further optimization because it has been reported the intensity order of transition is well correlated between ion trap CID and SRM (25). The predicted collision energies from Skyline were used for all peptides. After LC-SRM-MS screening, a total of 45 peptides for the 45 major plasma proteins (one peptide per protein) were selected for relative quantification in this study (see Table II).
Table II. SRM quantification of 45 human plasma proteins with known relative abundance ratios relative to the labeled reference.
| Protein | Peptide | Peptide replaced | Product ion | Slope ± CIa | y intercept ± CIa | R2 | Accuracy (Error %)b | Mean % CV |
|---|---|---|---|---|---|---|---|---|
| Afamin | DADPDTFFAK | y7+ | 1.013 ± 0.018 | 0.036 ± 0.086 | 0.999 | 3.45 | 3.41 | |
| Albumin, serum | LVNEVTEFAK | y5+ | 0.968 ± 0.032 | 0.053 ± 0.151 | 0.997 | 2.17 | 2.42 | |
| α1-Acid glycoprotein 1 | EQLGEFYEALDCLR | √ | y8+ | 0.870 ± 0.028 | 0.137 ± 0.130 | 0.997 | 6.82 | 2.39 |
| α1-Antichymotrypsin | EIGELYLPK | y2+ | 1.023 ± 0.007 | 0.025 ± 0.032 | 1.000 | 2.41 | 1.62 | |
| α1B-Glycoprotein | LETPDFQLFK | y8+ | 0.948 ± 0.028 | 0.141 ± 0.131 | 0.998 | 6.60 | 2.66 | |
| α2-Antiplasmin | LCQDLGPGAFR | √ | y6+ | 0.984 ± 0.029 | 0.091 ± 0.135 | 0.998 | 4.04 | 3.27 |
| α2-Macroglobulin | LLIYAVLPTGDVIGDSAK | y11+ | 0.985 ± 0.027 | 0.110 ± 0.129 | 0.998 | 8.36 | 2.04 | |
| Angiotensinogen | ALQDQLVLVAAK | y3+ | 0.982 ± 0.021 | 0.068 ± 0.097 | 0.999 | 4.66 | 3.52 | |
| Antithrombin-III | DDLYVSDAFHK | y6+ | 0.989 ± 0.054 | 0.040 ± 0.251 | 0.992 | 5.48 | 6.06 | |
| Apolipoprotein A-I | LLDNWDSVTSTFSK | √ | y8+ | 0.976 ± 0.012 | 0.058 ± 0.055 | 1.000 | 4.64 | 1.06 |
| Apolipoprotein A-II precursor | SPELQAEAK | y8+ | 0.947 ± 0.028 | 0.094 ± 0.131 | 0.998 | 6.16 | 3.43 | |
| Apolipoprotein A-IV | ALVQQMEQLR | √ | y6+ | 1.003 ± 0.013 | 0.033 ± 0.059 | 1.000 | 2.54 | 2.70 |
| Apolipoprotein B-100 | LTISEQNIQR | √ | y7+ | 0.993 ± 0.017 | 0.056 ± 0.078 | 0.999 | 4.03 | 4.76 |
| Apolipoprotein C-I lipoprotein | EWFSETFQK | √ | y7+ | 1.090 ± 0.037 | 0.060 ± 0.174 | 0.997 | 3.24 | 4.75 |
| Apolipoprotein C-III | DALSSVQESQVAQQAR | √ | y10+ | 1.025 ± 0.017 | 0.088 ± 0.081 | 0.999 | 5.36 | 2.97 |
| Apolipoprotein E | LGPLVEQGR | y7+ | 1.027 ± 0.022 | 0.017 ± 0.102 | 0.999 | 3.02 | 2.73 | |
| β2-Glycoprotein I | ATVVYQGER | y6+ | 0.999 ± 0.006 | 0.032 ± 0.029 | 1.000 | 3.33 | 1.43 | |
| Ceruloplasmin | EVGPTNADPVCLAK | √ | y6+ | 0.997 ± 0.026 | 0.037 ± 0.124 | 0.998 | 2.97 | 1.68 |
| Clusterin | ELDESLQVAER | y3+ | 0.978 ± 0.022 | 0.050 ± 0.104 | 0.999 | 4.76 | 3.22 | |
| Coagulation factor XIIa HC | VVGGLVALR | y7+ | 0.969 ± 0.017 | 0.066 ± 0.081 | 0.999 | 6.17 | 1.93 | |
| Complement C3 | AVLYNYR | √ | y5+ | 0.987 ± 0.008 | 0.03 ± 0.039 | 1.000 | 2.69 | 1.32 |
| Complement C4 β chain | VGDTLNLNLR | y3+ | 0.986 ± 0.009 | 0.022 ± 0.040 | 1.000 | 2.45 | 2.1 | |
| Complement C4 γ chain | VEYGFQVK | √ | y6+ | 1.024 ± 0.015 | 0.001 ± 0.071 | 1.000 | 3.69 | 2.02 |
| Complement component C9 | VVEESELAR | √ | y7+ | 1.014 ± 0.023 | 0.075 ± 0.108 | 0.999 | 5.49 | 2.61 |
| Complement factor B | LEDSVTYHCSR | √ | y6+ | 0.984 ± 0.048 | 0.002 ± 0.223 | 0.994 | 3.44 | 3.94 |
| Complement factor H | EIMENYNIALR | √ | y7+ | 0.897 ± 0.035 | 0.115 ± 0.163 | 0.996 | 3.24 | 3.62 |
| Fibrinogen α chain | VQHIQLLQK | √ | y7+ | 0.963 ± 0.018 | 0.057 ± 0.086 | 0.999 | 3.96 | 2.23 |
| Fibrinogen β chain | EDGGGWWYNR | √ | y4+ | 1.000 ± 0.013 | 0.047 ± 0.062 | 1.000 | 5.67 | 1.32 |
| Fibrinogen γ chain | DNCCILDER | √ | y4+ | 1.022 ± 0.018 | 0.014 ± 0.086 | 0.999 | 4.35 | 1.76 |
| Gelsolin, isoform 1 | TGAQELLR | y4+ | 0.984 ± 0.011 | 0.028 ± 0.043 | 1.000 | 0.88 | 3.01 | |
| Haptoglobin β chain | VGYVSGWGR | y6+ | 1.049 ± 0.006 | −0.004 ± 0.030 | 1.000 | 1.05 | 0.78 | |
| Hemopexin | NFPSPVDAAFR | y7+ | 0.989 ± 0.013 | 0.030 ± 0.060 | 1.000 | 2.98 | 2.01 | |
| Heparin cofactor II | TLEAQLTPR | y6+ | 1.021 ± 0.022 | 0.031 ± 0.102 | 0.999 | 2.04 | 2.30 | |
| Inter-α-trypsin inhibitor HC | AAISGENAGLVR | y9+ | 1.016 ± 0.024 | 0.043 ± 0.111 | 0.999 | 3.40 | 2.07 | |
| Kininogen-1 | YFIDFVAR | √ | y6+ | 0.965 ± 0.010 | 0.065 ± 0.048 | 1.000 | 3.74 | 1.40 |
| l-selectinc | AEIEYLEK | y5+ | 0.980 ± 0.141 | 0.731 ± 0.571 | 0.943 | - | 22.2 | |
| Plasma retinol-binding protein | YWGVASFLQK | y6+ | 1.021 ± 0.013 | 0.018 ± 0.059 | 1.000 | 2.97 | 4.17 | |
| Plasminogen | WELCDIPR | √ | y6+ | 1.000 ± 0.012 | 0.038 ± 0.057 | 1.000 | 3.31 | 2.32 |
| Prothrombin | YGFYTHVFR | √ | y5+ | 1.064 ± 0.039 | 0.015 ± 0.185 | 0.996 | 3.03 | 4.49 |
| Serum amyloid P-component | VGEYSLYIGR | y6+ | 0.972 ± 0.012 | 0.063 ± 0.058 | 1.000 | 1.81 | 2.16 | |
| Transferrin | EGYYGYTGAFR | √ | y7+ | 1.012 ± 0.009 | 0.037 ± 0.043 | 1.000 | 2.86 | 0.94 |
| Transthyretin | CPLMVK | √ | y5+ | 0.994 ± 0.027 | 0.022 ± 0.110 | 0.998 | 1.90 | 3.32 |
| Vitamin d-binding protein | THLPEVFLSK | y3+ | 0.951 ± 0.014 | 0.014 ± 0.064 | 0.999 | 4.00 | 2.66 | |
| Vitronectin | FEDGVLDPDYPR | y5+ | 0.961 ± 0.027 | 0.097 ± 0.127 | 0.998 | 4.28 | 3.95 | |
| Zinc-α2-glycoprotein | EIPAWVPFDPAAQITK | y10+ | 0.946 ± 0.017 | 0.064 ± 0.081 | 0.999 | 3.12 | 2.79 | |
| Averagec | 0.991 ± 0.021 | 0.050 ± 0.096 | 0.999 | 3.79 | 2.67 |
a CI, 95% confidence interval.
b Accuracy was calculated based on the percentage of error relative to the known relative abundance ratios.
c l-Selectin was excluded in calculation of the average values.
LC-SRM-MS
Peptide samples were analyzed using either an Agilent 1100 LC system (Agilent Technologies) or ACQUITY UPLC (Waters) coupled on-line to a triple quadrupole mass spectrometer (TSQ Vantage; Thermo Fisher Scientific). The capillary analytical column was prepared by slurry-packing 3-μm Jupiter C18 bonded particles (Phenomenex, Torrence, CA) into a 25-cm-long, 75-μm-inner diameter fused silica capillary (Polymicro Technologies, Phoenix, AZ) and connected via a Valco 100-μm-inner diameter stainless steel union to a chemically etched fused silica emitter (20-μm inner diameter) produced in-house (26). The mobile phases consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). One-μl aliquots of each sample containing ∼0.5 μg/μl peptides were injected onto the analytical column. Following 100% mobile phase A for 20 min, the samples were separated using a 40-min linear gradient of 10–50% mobile phase B at a flow rate of 400 nl/min. The inlet capillary of the mass spectrometer was maintained at 240 °C with an electrospray ionization voltage of 2.2 kV. All of the samples were analyzed in triplicate. A fixed dwell time of 10 ms and a scan window of 0.002 m/z were employed, and the maximum number of transitions was less than 150 to ensure a cycle time of less than 2 s for mouse plasma samples. For monitoring the 45 human plasma proteins with total 270 transitions, scheduled SRM was used with cycle time fixed as ∼1 s, whereas dwell time varied according to the number of transitions in each scheduled segment from 11 to 20 ms. For LC-SRM-MS quantification of PSA and CRP in human plasma, the dwell time was increased to 40 ms to keep at a fixed cycle time (∼1 s) with the reduced number of transitions. Both Q1 and Q3 resolution were set to a peak width of 0.7 Da at FWHM unless otherwise noted.
Data Analysis
Data sets for calibration mixtures were analyzed by Pinpoint software (Thermo Fisher Scientific) for peak detection and integration. For some low concentration points, the data were analyzed using Thermo Xcalibur 2.0.7, specifically, with Qual Browser, for peak detection. Data sets for quantifying 45 human plasma proteins were analyzed by Skyline 0.7.0. Peak smoothing was not applied to determine the peak areas in all analyses. All subsequent data analyses were performed in MS Excel to generate standard deviations and calibration curves. The calibration curves were generated by plotting the averaged peak area ratio of light/heavy from technical triplicate analyses versus the known concentration ratio of light/heavy with linear regression for all data points. The error bars in each plot represent standard deviations from triplicate analyses.
RESULTS
Overall Concept and Experimental Workflow
Fig. 1A illustrates the concept of using the 18O-labeled proteome reference as GIS for relative quantification of individual biological or clinical samples using SRM. The 18O-labeled reference sample can be generated by pooling aliquots from several or all individual digested samples followed by post-digestion 18O labeling (20). The 18O-labeled reference is then spiked back into individual samples prior to LC-SRM-MS analysis. All tryptic peptides present in the individual samples should have their 18O-labeled counterparts as internal standards; thus the reference is considered as GIS. We should note that only the fully 18O-labeled form (two 18O incorporation on peptide C terminus) is used as the heavy isotope-labeled internal standard. Quantification of relative peptide and protein abundances across different samples is based on the isotopic peak area ratios (light/heavy, 16O/18O) of each SRM transition pair (18). Fig. 1B shows the workflow for the proof of concept experiments. An 18O-labeled mouse plasma reference was prepared by labeling a plasma sample spiked with the six standard proteins. Unlabeled standard protein digests were spiked into the labeled reference to make a series of calibration mixtures with known 16O/18O concentration ratios (Table I). The accuracy, reproducibility, and linear dynamic range of 16O/18O-based quantification were assessed by LC-SRM-MS analyses of the calibration mixtures.
Fig. 1.

The use of 18O-labeled proteome reference as GIS for SRM quantification. A, overall conceptual workflow. A reference sample is generated by pooling aliquots from digested individual biological samples and 18O-labeled. The labeled proteome reference as GIS is spiked into each sample prior to LC-SRM-MS analysis. B, a workflow for proof of concept experiments.
18O Heavy Isotope Incorporation
The usage of stable isotope-labeled peptides as internal standards requires good stability of the labeled peptides and a high percentage of heavy isotope incorporation with minimum presence of the unlabeled form in the labeled standards. The stability of 18O-labeled peptides mainly depends on the quenching of residual trypsin activity to prevent trypsin-catalyzed back-exchange. We recently reported complete quenching of residual trypsin activity via boiling (20), and the stability of labeled peptides was demonstrated even after 1 week of storage at room temperature (20).
The potential presence of unlabeled form within labeled internal standards presents a limitation for the dynamic range of quantification in stable isotope dilution studies. To evaluate this issue in the 18O-labeled reference, the percentage of heavy isotope incorporation was measured by LC-SRM-MS for 18O and 16O transitions. The percentage of heavy isotope incorporation is defined as heavy/(heavy + light), where the heavy denotes the fully labeled 18O2 form as internal standards, and the light denotes the unlabeled form 16O2. The partially labeled form with only one 18O was not included in this calculation because only the fully labeled peptide form with two 18O is utilized as internal standard for SRM monitoring. Based on triplicate analyses, 14 of 16 peptides exhibited a percentage of heavy isotope incorporation of >99.5%, indicating the presence of <0.5% unlabeled form in heavy labeled peptides. The other two peptides, TGPNLHGLFGR from bovine cytochrome c and TPEVDDEALEK from bovine β-lactoglobulin, had incorporation percentages of ∼99.0 and 97.8%, respectively (supplemental Table 2).
Fig. 2 exemplifies the high percentage of heavy isotope incorporation for one peptide (VLVLDTDYKK) that was confirmed by both LC-MS and LC-SRM. The presence of <0.5% unlabeled form within the labeled peptide, although higher than those in typically 13C/15N-labeled synthetic peptide standards, is sufficiently low for providing a good dynamic range for quantification. Although ∼6% of the labeled peptides are from single 18O-labeled form when using 97% enriched 18O-water, the single 18O-labeled form only contributes to a small percentage (1–2%) to the signal of double 18O-labeled form because of the isotopic overlap between the two forms. In SRM-based quantification, only the unlabeled (light) and double 18O-labeled (heavy) transitions are monitored. Although the measured heavy transition intensity does include the small percentage of contribution from the single 18O-labeled form, the total intensity of heavy transition should be identical in all biological samples spiked with the same level of labeled reference, thus serving well as internal standards. Also <0.5% of the unlabeled 16O2-form versus the 18O2-form was observed consistently in both orbitrap-MS and SRM-MS, indicating sufficient resolution of SRM-MS for monitoring the light and heavy transitions.
Fig. 2.

18O heavy isotope incorporation. 18O-Labeled VLVLDTDYKK2+ from β-lactoglobulin detected by LTQ-Orbitrap and triple quadrupole instrument. A, LTQ-Orbitrap MS spectrum. B, extracted ion chromatogram of fully labeled (18O2), singly labeled (18O1), and unlabeled (16O2) peak from LTQ-Orbitrap. C, SRM extracted ion chromatogram of fully 18O2-labeled transitions (solid lines) and light unlabeled transitions (dashed lines in zoom-in view) from LC-TSQ. Green, y6; blue, y7; red, y8 ion, respectively. The intensities of the light transitions are near the noise level.
Peptide Detection Selectivity
One of the challenges associated with analyzing complex samples is the potential matrix interference with the analyte of interest. To examine the selectivity of SRM detection of target peptides in a complex mixture, we applied the following criteria: 1) coelution of 16O- and 18O-labeled peptides, 2) consistent peak area ratios of the three transitions for a given 16O- and 18O-labeled peptide pair, 3) consistent response in relative intensity of transitions between LC-SRM-MS and infusion analysis of the standard proteins, and 4) confirmed retention time with increased response following spiking unlabeled peptides from the standard protein digest. All 16 peptides were selectively detected by LC-SRM based on multiple pairs of transitions. The consistent light/heavy transitions were shown in supplemental Fig. 1 (27). Fig. 3A exemplifies coelution and consistent response of 16O- and 18O-labeled transitions of LFTGHPETLEK3+. Moreover, the consistent relative intensities of three transitions between LC-SRM-MS detection in mouse plasma and infusion MS2 spectra of the standard protein (Fig. 3, B and C) support the selective detection of this peptide.
Fig. 3.

An example of SRM detection of a selected peptide. A, the six 16O- and 18O-labeled transitions of peptide LFTGHPETLEK3+ in 1:5 molar ratio (16O:18O) in mouse plasma detected in accordance with expected ratio. Solid lines, 18O transitions; dashed lines, 16O transitions. B, relative intensities of transitions of the unlabeled peptide detected by LC-SRM-MS. C, relative intensities of transitions of the unlabeled peptide detected by infusion MS/MS of standard protein digest.
Cross-interference between 16O and 18O-Labeled Transitions
The potential for signal cross-interference between unlabeled, singly labeled (18O1), and doubly labeled (18O2) in SRM-based measurements initially raised a concern. To examine such potential cross-interference in SRM mode, the 16O/18O peak area ratios from an approximate 1:4 (mass ratio) mixture of unlabeled sample and 18O-labeled reference were analyzed by LC-SRM-MS with Q1 resolution settings varying from 0.5–3.0 Da peak width at FWHM and Q3 fixed at 0.7 Da peak width; samples were analyzed in triplicate for each Q1 setting. In Fig. 4, the light/heavy area ratios of all four peptides were consistent with ratio variations <7% when the Q1 resolution increased from 0.5 to 3.0 Da. This result indicated that the unit resolution of Q3 was sufficient to differentiate the product ions originating from the unlabeled and labeled forms of a given peptide, even when the Q1 filter allowed unlabeled and labeled peptides to pass simultaneously at low Q1 resolution settings (2–3 Da peak width). The selectivity for differentiating y-type transitions is attributed to the 4 Da mass differences between 16O and 18O transitions for the relatively small sizes of singly charged product ions. Some discrepancies of the abundance ratios from 0.16 to 0.35 were observed for the four peptides, which were anticipated for mixing an unlabeled and labeled sample at a given mass ratio because the preparation of the 18O-labeled reference required extra lyophilizing and labeling steps. These steps resulted in slightly different recoveries and labeling efficiencies for different peptides compared with the unlabeled sample. In the case shown in Fig. 4, although the overall quantity of unlabeled versus labeled was 1:4 based on BCA protein assay, the observed ratios ranging from 0.16 to 0.35 were within this expectation. Despite this discrepancy, it does not prevent the labeled reference from being used as effective internal standards for relative quantification across different samples when it is spiked at the same level.
Fig. 4.

Effect of Q1 window resolution on 16O/18O area ratio. The 16O/18O area ratios of four peptides are relatively consistent with <7% variations despite the increases of Q1 resolution from 0.5 to 3.0 Da.
Another concern is that the fifth isotope of unlabeled 16O-form overlaps with the monoisotope of fully 18O-labeled form, which will affect the final 16O/18O area ratios, especially when the unlabeled peptide abundance was significantly greater (i.e. >20-fold) than the 18O-labeled internal standard. For this reason, the abundance ratios (16O/18O) need to be corrected by the following equations modified from previously published methods (28, 29).
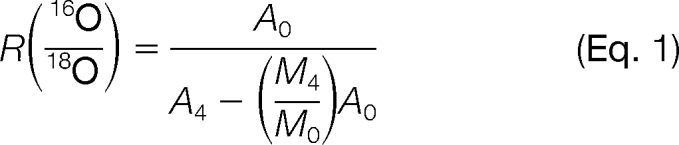 |
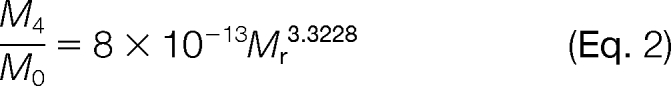 |
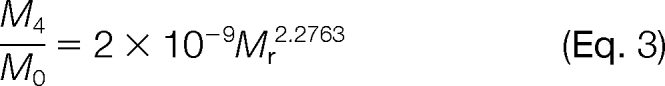 |
A0 and A4 are the measured peak areas for the monoisotopic peaks of an unlabeled transition and double 18O-labeled transition, respectively. M0 and M4 are the predicted relative abundances for the monoisotopic and fifth isotopic peak, respectively, of a given transition. Equation 2 was applied to all transitions except for sulfur-containing (e.g. methionine) transitions, in which case Equation 3 was applied. M4/M0 ratios (Equations 2 and 3) were estimated by plotting theoretical isotopic distribution ratios versus the molecular weight of transitions represented by Mr (supplemental Fig. 2). The application of these equations provided improved quantification accuracy in situations where the unlabeled peptide concentrations are significantly greater than the 18O-labeled internal standards (supplemental Fig. 2C).
Dynamic Range and Reproducibility of Relative Quantification
To assess the dynamic range of relative quantification based on isotopic peak area ratios, 11 calibration mixtures (Table I) with known 16O/18O concentration ratios ranging from 0.01 to 100 were prepared by spiking equal amounts of each of the unlabeled six standard protein digests into two 18O-labeled references. All 16 peptides were monitored by SRM to assess the accuracy, reproducibility, and linear dynamic range of quantification using the 18O-labeled GIS. All six standard proteins were detected at their lowest spike-in level (400 ng/ml) based on at least one peptide/protein, suggesting that the limit of detection is below the μg/ml level in nondepleted plasma, which is comparable with previous reports (1, 6). Eight peptides exhibited a linear response in relative 16O/18O concentration ratios from 0.01 to 100. The other peptides had a linear response range of in relative 16O/18O concentration ratios from 0.04 to 100 (supplemental Fig. 3) because of lower SRM signal intensities or interference for the unlabeled peptides at lower spike-in levels. Fig. 5 shows representative calibration curves for LFTGHPETLEK3+ and VEADIAGHGQEVLIR3+, two peptides from equine skeletal muscle myoglobin. The measured peak area ratios have a linear correlation with known concentration ratios from 0.01 to 100.
Fig. 5.

Calibration curves of selected peptides from standard proteins. Calibration curves of two peptides from equine skeletal muscle myoglobin in both natural and log scale: LFTGHPETLEK3+ (A) and VEADIAGHGQEVLIR3+ (B). The precision of quantification for both 16O/18O ion-pair (blue error bars) and label-free approach (red error bars) is shown. The error bar means standard deviation. A smaller error bar (marked as an asterisk in B) of label-free approach in the high end is due to the saturated peak intensity of the unlabeled transition. 95% confidence intervals for the slope and y intercept are indicated in the linear equations derived from the ion-pair data. The concentration ranges of all samples are included in Table I.
The reproducibility or precision of the GIS-based relative quantification was evaluated among the replicates and compared with that of label-free quantification. To compare the label-free data with the GIS-based 16O/18O pair ratio data, the label-free 16O abundances for peptides measured in each LC-SRM-MS analysis were converted into a similar relative abundance format by dividing the 16O-peak areas from individual analyses by a common denominator, the averaged 18O-peak area across the triplicate analyses. The converted relative abundance data from label-free 16O peak areas is different from the 16O/18O isotopic pair data because the 16O/18O pair data were measured by coeluting isotopic pair within the same LC-MS analysis, whereas the label-free relative abundance data were normalized against the averaged 18O abundance derived from the triplicate analyses. Following this conversion, 16O/18O pair ratio data and label-free 16O intensity data were in similar formats that allowed for direct comparison of the reproducibility. Compared with the label-free approach, the 16O/18O pair ratio exhibits significantly better precision in terms of coefficient of variance for all concentration levels. For example, the averaged coefficient of variance value for LFTGHPETLEK (Fig. 5A) is 8.6% for the pair ratio compared with 20.8% for the label-free approach, and that for VEADIAGHGQEVLIR (Fig. 5B) is 9.2% for the pair ratio versus 14.0% for the label-free approach. Although the observed coefficient of variance values from label-free approach are within acceptable range, it should be noted that all of these data are from back-to-back technical replicates. The reproducibility may become significantly worse for label-free approach in the case of large scale studies where instrument variations over time are anticipated.
Relative Quantification of 45 Major Human Plasma Proteins
To further illustrate the benefits of using the 18O-labeled proteome reference in preverification of biomarkers without the need for individual synthetic peptides, 45 major proteins in human plasma with SRM assays previously reported (1, 6) were selected for targeted quantification. Although all of the previously reported peptides for SRM monitoring were without internal missed cleavage, a number of the proteins (21 proteins) were based on peptide sequences with C-terminal motifs that had inhibitory effect for trypsin activity (e.g. RK, KK, (D/E)K, or K(D/E)). It was observed that these types of peptides within plasma digest were not effectively labeled by post-digestion 18O labeling. The reason is that the reduced trypsin activity leads to incomplete digestion and the production of both the short and long form (containing miscleavage) of peptides with the same N-terminal sequence. In post-digestion 18O labeling, further cleavage of the long form peptides resulting in only a single 18O incorporation, leading to an overall incomplete labeling of the short form peptides. However, in the standard proteins individually digested, this abnormal abundance of single 18O-labeled form was not observed, presumably because of a relatively complete digestion in individually digested proteins. To address this issue, we selected new peptides without such C-terminal motifs for each of these 21 proteins (Table II) and finalized SRM parameters for all 45 proteins (supplemental Skyline file). To select these peptides, the 18O-labeled reference was initially screened for 42 peptides using SRM, and finally 21 peptides with good labeling efficiencies were selected for quantification.
The final 45 peptides with total 270 transitions were scheduled and analyzed in triplicate with five standard mixtures with 1:10, 1:3, 1:1, 3:1, and 10:1 peptide mass ratios of the unlabeled versus the labeled reference. All relative protein abundance ratios except for l-selectin were accurately quantified with good linearity, accuracy, and precision (see Table II and supplemental Table 5). In the case of l-selectin, an interference peak for the heavy transition was observed, leading to less accurate quantification. The accurate quantification results of several representative proteins were shown in Fig. 6.
Fig. 6.

Accurate relative quantification of human plasma proteins. The detected peak area ratios accurately match with the known concentration ratios. 95% confidence intervals for the slope and y intercept are indicated in the linear equations. The four panels represent four selected proteins.
Quantification of Protein Concentrations
If purified protein standards are available, the 18O-labeled reference can be utilized to quantify unknown protein concentrations in a given sample by applying a standard addition method. To illustrate this utility, we prepared a human female plasma sample with spiked PSA for testing the quantification of PSA and endogenous CRP concentrations. The additions of standard proteins to plasma were performed prior to protein digestion so that all of the samples were carried out with the same protocol. One plasma sample spiked with 2 μg/ml of intact PSA and CRP was labeled as the 18O-labeled reference. The labeled reference was spiked into three unlabeled standard addition samples and the hypothetical endogenous sample with 1:1 mass ratio. These four samples with 18O-labeled reference were analyzed in technical triplicates. Fig. 7 shows the standard addition curves for the best responding peptide SVILLGR and AFVFPK for the quantification of PSA and CRP concentrations, respectively, in the hypothetical endogenous female plasma sample with PSA spiked at 0.5 μg/ml. Based on the standard addition method, the concentrations of PSA and CRP were 0.55 ± 0.05 and 1.07 ± 0.13 μg/ml, respectively, which agree well with the original spiked value of PSA (∼10% error) and the previously reported concentration of CRP (30, 31). These results demonstrate that an 18O-labeled reference can be successfully coupled with a standard addition method for quantifying protein concentrations in a given sample.
Fig. 7.

Quantification of PSA and CRP concentrations in plasma using the GIS coupled with the standard addition method. Standard addition curves of SVILLGR2+ (379.25 → 345.22) from spiked PSA and AFVFPK2+ (354.71 → 490.30) from endogenous CRP in human female plasma. The standard deviations are shown as error bars at all data points.
DISCUSSION
In typical biomarker discovery efforts applying shotgun proteomics or transcriptomics, hundreds of proteins or genes of potential biological interest can often be identified. It is an important step to quantitatively screen a relatively large number of these initial candidates across many biological or clinical samples to identify protein candidates that show statistically significant changes between different conditions for further verification. However, quantitative measurements of a large number of candidate proteins present a significant challenge for current SRM-based strategies because of the relatively high cost associated with the application of isotope-labeled synthetic peptides as internal standards. The concept of employing an 18O-labeled proteome reference as GIS is ideally suited for SRM-based quantitative screening for a broad list of candidate protein biomarkers following the initial discovery phase for identification of higher quality candidates that exhibit significant changes relevant to disease or treatment. Because essentially every possible tryptic peptide will have its own heavy internal standard, all candidate proteins of interest in principle can be quantitatively screened by SRM without the need for individually labeled synthetic peptides. Recent advances in informatics software tool and resources such as Skyline software (24), the global proteome machine database (32), and MRMatlas (33) have enabled the selection of target peptides and transitions for any given candidate proteins based on previously acquired MS/MS data sets as well as the prediction of collisional energies for SRM monitoring. Such informatics capabilities facilitate the broad utility of the GIS concept for monitoring candidate proteins without the need for optimizing the individual SRM assays using synthetic peptides in the initial quantitative screening experiments. An additional step utilizing either synthetic peptides or purified standard proteins can be applied to further verify the selectivity of SRM detection or the identity of the peptide detected; however, after the initial screening using GIS, the number of synthetic peptides or protein standards needed for this further verification step will be significantly reduced.
In this proof of concept study, we demonstrated that the 18O-labeled proteome reference can be effectively utilized as GIS for targeted SRM-based relative quantification. Following LC-SRM-MS analysis of a series of calibration mixtures, a dynamic range for relative quantification from 0.01 to 100 in light/heavy concentration ratios (Table I and Fig. 5) along with high reproducibility (<10% coefficient of variance) was observed. Accurate relative quantification of 45 major human plasma proteins (∼4% error relative to known relative abundance ratios) was also demonstrated by mixing the unlabeled plasma digest with the labeled reference with two orders of dynamic range of concentrations relative to the reference. All of the target peptides and transitions were selected based on either previously reported SRM assays or existing MS/MS spectra, illustrating the utility of the 18O-labeled reference for broad SRM screening. A standard addition method with purified protein standards further allowed us to demonstrate the quantification of spiked intact PSA and endogenous CRP concentrations in human female plasma.
There are several distinct advantages to utilizing an 18O-labeled reference as GIS for SRM-based quantification. Compared with other labeling approaches, 18O labeling is a simple and effective approach for labeling peptides in any types of biological samples, including tissues and body fluids. Moreover, labeling need only be performed on one reference sample instead of labeling each biological sample. To be effective, it is important to quench residual trypsin activity in each unlabeled tryptic digest by boiling to prevent oxygen back-exchange of spiked labeled reference (20). The preparation of the 18O-labeled reference is also straightforward without much additional cost. Another advantage is that an 18O-labeled pooled reference provides internal standards at balanced median concentrations for a given biological study, which is important for achieving accurate relative quantification (6). It is often difficult to determine the optimal concentration level for spiking synthetic peptides in complex biological samples because the target analyte concentrations can vary widely from one to another and are generally unknown.
Concerns related to the use of 18O-labeled reference include its stability and the potential issue of partial labeling. The stability of 18O-labeled peptides was well demonstrated in a previous study where the abundance ratios between the unlabeled and labeled forms were unchanged even after 1 week of storage at room temperature (20). In another study, reproducible results were obtained for repeated analyses of 40 human plasma samples spiked with an 18O-labeled reference with at least 3 months apart between the two batches of analyses, again illustrating the stability of labeled reference (19).
Regarding the issue of partial labeling, it is observed that the signal intensity of single 18O1-labeled form is 6–10% of double 18O2-labeled form (Fig. 2) for most peptides. In the case of traditional “pair-wise” comparisons, both the singly and doubly labeled forms need to be taken into consideration for quantification. However, in this work only the double 18O2-labeled form is utilized as internal standard across multiple samples for SRM-based relative quantification. The SRM mode provides sufficient resolution to allow the utility of double 18O-labeled forms as internal standards without the interference from the single 18O-labeled form (Fig. 4) when only singly charged y-type product ions are being monitored. The single 18O-labeled form does have a small percentage (1–2%) of contribution to the total signal of double 18O-labeled form monitored by SRM; however, it should be emphasized that as the internal standards being monitored, the total signal of any double 18O-labeled forms are consistent across all biological samples spiked with the same level of labeled reference regardless of the contribution from the singly 18O-labeled form. The negligible unlabeled portion (<0.5%) compared with the fully 18O-labeled portion for most peptides demonstrates that the labeled reference is well suited as an internal standard. It is possible that some peptides may not be efficiently labeled; therefore, it is necessary to screen all selected peptides for their light/heavy ratios by analyzing the labeled reference only to finalize the peptides for SRM. We also note that because of the nature of 18O labeling on the C-terminal, only y ions can be selected for SRM measurements, which may limit sensitivity in cases where b ions are most prevalent.
In conclusion, the balanced concentrations of internal standards afforded by an 18O-labeled proteome reference should benefit reproducibility, accuracy, and dynamic range for SRM-based quantitative proteomics in biomarker preverification or verification studies without the need for individually labeled peptide standards. Further verification of the candidate detection selectivity and quantification of candidate protein concentrations can be performed by applying unlabeled standard proteins or peptides with standard addition methods. The strategy is applicable to any types of cells, tissues, or biofluids and is well suited for accurate and quantitative screening of a relatively large number of biomarker candidates from the initial discovery phase across many biological or clinical samples. The 18O labeling approach should also be useful for generating heavy-labeled peptides from protein standards for quantitative studies.
Footnotes
* This work was supported in part by the National Institutes of Health Director's New Innovator Award Program 1-DP2OD006668-01 (to W.-J. Q.), the National Institutes of Health National Center for Research Resources RR18522 (to R. D. S.), Korea Research Foundation Grant KRF-2008-357-C00093 (to J-.S. K.), and Department of Energy Contract DE-AC05-76RLO-1830 (to Pacific Northwest National Laboratory). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
 This article contains supplemental Figs. S1–S3, Tables S1–S5, and Skyline files.
This article contains supplemental Figs. S1–S3, Tables S1–S5, and Skyline files.
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- GIS
- global internal standards
- PSA
- prostate-specific antigen
- CRP
- C-reactive protein.
REFERENCES
- 1. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 2. Rifai N., Gillette M. A., Carr S. A. (2006) Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 3. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stahl-Zeng J., Lange V., Ossola R., Eckhardt K., Krek W., Aebersold R., Domon B. (2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 5. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 8, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hossain M., Kaleta D. T., Robinson E. W., Liu T., Zhao R., Page J. S., Kelly R. T., Moore R. J., Tang K., Camp D. G., 2nd, Qian W. J., Smith R. D. (2011) Enhanced sensitivity for selected reaction monitoring-mass spectrometry-based targeted proteomics using a dual-stage electrodynamic ion funnel interface. Mol. Cell. Proteomics 10.1074/mcp.M000062-MCP201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lange V., Picotti P., Domon B., Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodriguez H., Rivers R., Kinsinger C., Mesri M., Hiltke T., Rahbar A., Boja E. (2010) Reconstructing the pipeline by introducing multiplexed multiple reaction monitoring mass spectrometry for cancer biomarker verification: An NCI-CPTC initiative perspective. Proteomics Clin. Appl. 4, 904–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu C. C., MacCoss M. J., Howell K. E., Matthews D. E., Yates J. R., 3rd (2004) Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem. 76, 4951–4959 [DOI] [PubMed] [Google Scholar]
- 13. Ishihama Y., Sato T., Tabata T., Miyamoto N., Sagane K., Nagasu T., Oda Y. (2005) Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23, 617–621 [DOI] [PubMed] [Google Scholar]
- 14. Beynon R. J., Doherty M. K., Pratt J. M., Gaskell S. J. (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589 [DOI] [PubMed] [Google Scholar]
- 15. Rivers J., Simpson D. M., Robertson D. H., Gaskell S. J., Beynon R. J. (2007) Absolute multiplexed quantitative analysis of protein expression during muscle development using QconCAT. Mol. Cell. Proteomics 6, 1416–1427 [DOI] [PubMed] [Google Scholar]
- 16. Zhao Y., Jia W., Sun W., Jin W., Guo L., Wei J., Ying W., Zhang Y., Xie Y., Jiang Y., He F., Qian X. (2010) Combination of improved O-18 incorporation and multiple reaction monitoring: A universal strategy for absolute quantitative verification of serum candidate biomarkers of liver cancer. J. Proteome Res. 9, 3319–3327 [DOI] [PubMed] [Google Scholar]
- 17. Petyuk V. A., Qian W. J., Chin M. H., Wang H., Livesay E. A., Monroe M. E., Adkins J. N., Jaitly N., Anderson D. J., Camp D. G., 2nd, Smith D. J., Smith R. D. (2007) Spatial mapping of protein abundances in the mouse brain by voxelation integrated with high-throughput liquid chromatography-mass spectrometry. Genome Res. 17, 328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian W. J., Liu T., Petyuk V. A., Gritsenko M. A., Petritis B. O., Polpitiya A. D., Kaushal A., Xiao W., Finnerty C. C., Jeschke M. G., Jaitly N., Monroe M. E., Moore R. J., Moldawer L. L., Davis R. W., Tompkins R. G., Herndon D. N., Camp D. G., Smith R. D. (2009) Large-scale multiplexed quantitative discovery proteomics enabled by the use of an (18)O-labeled “universal” reference sample. J. Proteome Res. 8, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qian W. J., Petritis B. O., Kaushal A., Finnerty C. C., Jeschke M. G., Monroe M. E., Moore R. J., Schepmoes A. A., Xiao W., Moldawer L. L., Davis R. W., Tompkins R. G., Herndon D. N., Camp D. G., 2nd, Smith R. D. (2010) Plasma proteome response to severe burn injury revealed by O-18-labeled “universal” reference-based quantitative proteomics. J. Proteome Res. 9, 4779–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petritis B. O., Qian W. J., Camp D. G., 2nd, Smith R. D. (2009) A simple procedure for effective quenching of trypsin activity and prevention of 18O-labeling back-exchange. J. Proteome Res. 8, 2157–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riviere L. R., Tempst P. (2001) Enzymatic digestion of proteins in solution, Curr. Protoc. Protein Sci. 11.1.1–11.1.19 [DOI] [PubMed] [Google Scholar]
- 22. Yen C. Y., Russell S., Mendoza A. M., Meyer-Arendt K., Sun S., Cios K. J., Ahn N. G., Resing K. A. (2006) Improving sensitivity in shotgun proteomics using a peptide-centric database with reduced complexity: Protease cleavage and SCX elution rules from data mining of MS/MS spectra. Anal. Chem. 78, 1071–1084 [DOI] [PubMed] [Google Scholar]
- 23. Siepen J. A., Keevil E. J., Knight D., Hubbard S. J. (2007) Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics. J. Proteome Res. 6, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherwood C. A., Eastham A., Lee L. W., Risler J., Vitek O., Martin D. B. (2009) Correlation between y-type ions observed in ion trap and triple quadrupole mass spectrometers. J. Proteome Res. 8, 4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelly R. T., Page J. S., Luo Q., Moore R. J., Orton D. J., Tang K., Smith R. D. (2006) Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal. Chem. 78, 7796–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abbatiello S. E., Mani D. R., Keshishian H., Carr S. A. (2010) Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 56, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson K. L., Muddiman D. C. (2004) A method for calculating 16O/18O peptide ion ratios for the relative quantification of proteomes. J. Am. Soc. Mass Spectrom. 15, 437–445 [DOI] [PubMed] [Google Scholar]
- 29. Qian W. J., Monroe M. E., Liu T., Jacobs J. M., Anderson G. A., Shen Y., Moore R. J., Anderson D. J., Zhang R., Calvano S. E., Lowry S. F., Xiao W., Moldawer L. L., Davis R. W., Tompkins R. G., Camp D. G., 2nd, Smith R. D. (2005) Quantitative proteome analysis of human plasma following in vivo lipopolysaccharide administration using 16O/18O labeling and the accurate mass and time tag approach. Mol. Cell. Proteomics 4, 700–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rost N. S., Wolf P. A., Kase C. S., Kelly-Hayes M., Silbershatz H., Massaro J. M., D'Agostino R. B., Franzblau C., Wilson P. W. (2001) Plasma concentration of C-reactive protein and risk of ischemic stroke and transient ischemic attack: The Framingham Study. Stroke 32, 2575–2579 [DOI] [PubMed] [Google Scholar]
- 31. Albert M. A., Glynn R. J., Ridker P. M. (2003) Plasma concentration of C-reactive protein and the calculated Framingham Coronary Heart Disease Risk Score. Circulation 108, 161–165 [DOI] [PubMed] [Google Scholar]
- 32. Craig R., Cortens J. P., Beavis R. C. (2004) Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 3, 1234–1242 [DOI] [PubMed] [Google Scholar]
- 33. Picotti P., Lam H., Campbell D., Deutsch E. W., Mirzaei H., Ranish J., Domon B., Aebersold R. (2008) A database of mass spectrometric assays for the yeast proteome. Nat. Methods 5, 913–914 [DOI] [PMC free article] [PubMed] [Google Scholar]