

Abstract
ATR-X syndrome is an X-linked mental retardation syndrome characterized by mental retardation, alpha thalassaemia and distinct facial features which include microcephaly, frontal hair upsweep, epicanthic folds, small triangular nose, midface hypoplasia and carp-shaped mouth. Here we report two brothers with clinical features of ATR-X syndrome, in whom a novel missense (C>T) mutation was identified in exon 31 of the ATRX gene.
Keywords: Alpha-thalassaemia, ATR-X, developmental delay, X-inactivation
Introduction
ATR-X syndrome, formally called alpha-thalassaemia X-linked mental retardation syndrome results from an abnormality in the ATRX gene. Clinical diagnosis is based on the presence of mental retardation in combination with alpha-thalassaemia and a characteristic and recognizable facial gestalt consisting of small head circumference, upsweep of the frontal hair, telecanthus, epicanthic folds or hypertelorism, small triangular nose, retracted columella, midface hypoplasia and tented upper lip which results in a consistently open mouth1. External genitalia abnormality is common (80%). Mental retardation is usually severe, with severe developmental delay and marked hypotonia. Alpha-thalassaemia is seen in 80 per cent of cases. The disease is rare. About 170 cases have been reported worldwide2. The estimated prevalence of the disease is <1-9/100,000 live born males3.
We report here two brothers with severe developmental delay, characteristic facial anomalies, and alpha-thalassaemia. Clinical diagnosis of ATR-X syndrome was not suspected until the second brother was evaluated for developmental delay. Presence of ATR-X syndrome was confirmed by mutation analysis of ATRX gene, which showed a novel substitution in exon 31 of the ATRX gene.
Case 1: Patient 1 was male, delivered at term by lower segment caesarian section with a breech presentation. He had delayed cry and his birth weight was 1.88 kg. Antenatally there was intra uterine growth retardation (IUGR) and a single umbilical artery was found. Facial dysmorphism was noted at birth. He had a preauricular sinus and undescended testes. He was evaluated at the age of 5 months with complaints of delayed head holding. Clinical examination showed subtle dysmorphism, severe hypotonia, brisk reflexes and hepatosplenomegaly. Metabolic screening by mass tandem spectrometry and gas chromatography mass spectrometry was carried out and found normal. Ophthalmic evaluation and EEG were normal. The cause of delay was not recognized then and at the age of five years he was re-evaluated for delay. He could neither recognize his parents, nor could he speak and was noted to have microcephaly (head circumference 42 cm,-4 SD) and severe hypotonia. Dysmorphic features consisted of a cowlick, bilateral epicanthic folds, depressed nasal bridge and bilateral camptodactyly of the upper limbs (Fig. 1). Mild hepatosplenomegaly was still present. Blood ammonia and lactate were normal. Karyotype was 46,XY. MRI (brain) suggested periventricular leucomalacia.
Fig. 1.
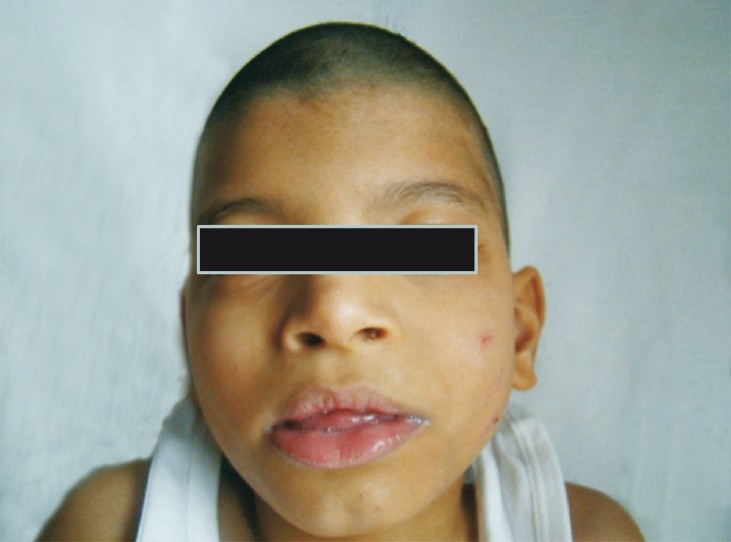
Facial features case 1.
Case 2: The younger brother of case 1(pedigree shown in Fig. 2) ) was evaluated for delayed development at 7½ months of age. He had a head lag and did not recognize his parents. He was unable to sit and was not able to follow objects. Speech was absent. There was no history of seizures. Head circumference was 41.1 cm (-1 SD). He had severe hypotonia. There was no organomegaly and deep tendon reflexes were normal. Facial features were similar to the elder sib consisting of a widow's peak or upsweep of the frontal hair, hypertelorism, low-set ears, flat nasal bridge, small nose, tented upper lip and everted lower lip (Fig. 3). He had multiple café-au-lait spots on his back, scrotum and left thigh. These spots were less than 6 in number and size was also less than 1 cm. His penis was small. MRI brain findings included a thin corpus callosum, mildly dilated ventricular system and white matter changes suggestive of periventricular leukomalacia. Fundus examination was normal. Complete blood count showed anaemia and low mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH). Peripheral smear suggested microcytosis, anisocytosis, tear drop cells. Serum iron and TIBC was normal. HPLC for haemoglobins was normal, with a slightly raised HbF level of 4.2 per cent corresponding to his age. Hb electrophoresis was done and showed three bands. Faint band in the region of HbA2, prominent band in the region of Hb A and fast moving band were suggestive of the presence of Hb H.
Fig. 2.
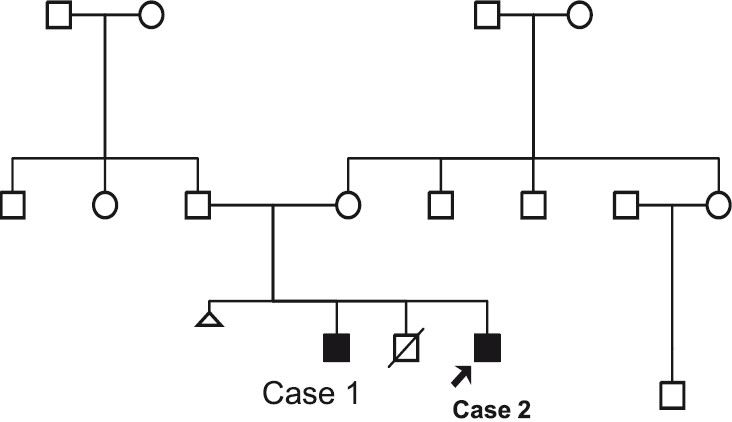
Three generation Pedigree.
Fig. 3.
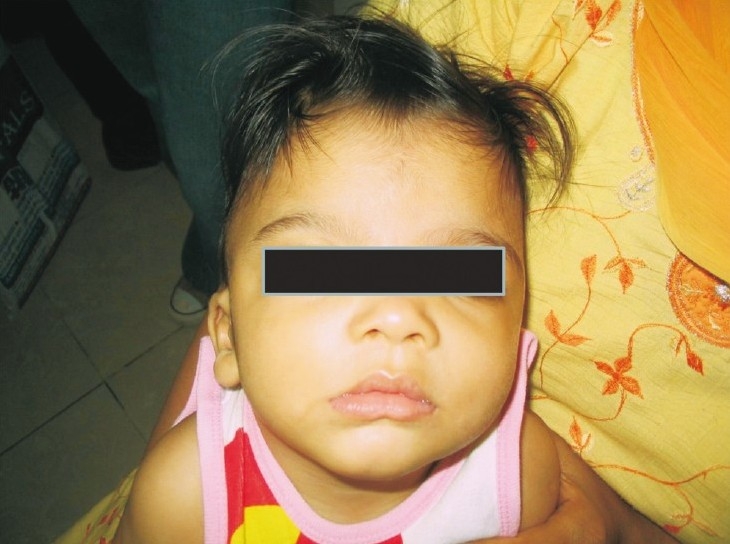
Facial features case 2.
Staining of red blood using brilliant cresyl blue was done which showed HbH inclusions in 25 per cent of cells examined. This suggested the diagnosis of alpha thalassaemia. Diagnosis of ATR-X was then suspected, mutation study was undertaken for confirmation.
Molecular study of ATRX gene: Deletion/duplication analysis of the alpha-thalassaemia gene was first performed using multiple ligation probe amplification (MLPA) using MRC Holland kit, the Netherlands. No deletions or duplications were found in both the brothers. ATRX gene analysis in younger child was then done by the methods described earlier using denaturating HPLC technique4. A hemizygous substitution in exon 31 c.6718C>T (p.L2240F) of the ATRX gene was found. Elder brother also harboured the same mutation. Mother was a carrier of the same mutation. X-inactivation study was done in mother, which showed that she had skewed X- inactivation.
Clinical features of both patients were consistent with ATR-X syndrome. Some facial features present in ATR-X syndrome are also found in Coffin-Lowry syndrome. However, the absence of genital anomalies, presence of pudgy tapering digits and the carrier manifestations in Coffin-Lowry distinguish it from ATR-X syndrome3. The facial features may have a similarity with Smith Lemli Opitz syndrome as was reported by Stanek et al2. Because of overlap of clinical features with other syndromes and history of perinatal asphyxia, the diagnosis of ATR-X syndrome in this case was not suspected until presentation of the second sibling. This highlights the importance of correct interpretation of dysmorphic features and combining clinical data with additional investigations and pedigree analysis.
The combination of mental retardation and alpha- thalassaemia could be confused with ATR-16 syndrome, which involves a gene deletion on chromosome 16. This can be differentiated from ATR-X by the absence of typical dysmorphic features. Most cases will have characteristic and recognizable facial gestalt during infancy which is probably due to facial hypotonia3. Definitive diagnostic criteria have not been established. The characteristic facial features seen in both cases are found in 94 per cent of the patients with ATR-X syndrome.
The occurrence of hepatosplenomegaly in patient 1 is usually not reported as a feature of ATR-X syndrome. In this case, it rather suggested the possibility of a metabolic disorder. As patient 2 did not have the same finding, we are unsure whether this should be included as a clinical feature. As per our knowledge, the café-au-lait spots observed in patient 2 have not been reported before in cases of ATR-X syndrome.
ATR-X is inherited in X-linked recessive manner. The disease is caused by mutation in ATRX gene located on Xq13.3. The gene is composed of 36 exons. The gene contains two major domains one PHD domain (Zinc finger domain) at N- terminus and the other helicase domain at the C- terminus. The gene is involved in the chromatin remodeling in combination with other chromatin associated proteins5. Alpha- thalassaemia is caused as the ATRX gene is involved in the regulation of the alpha-thalassaemia gene expression.
Although the causative mutations are scattered all over the gene, 43 per cent of mutations are seen in PHD- like domain and 41 per cent in helicase domains6. Missense mutations are more common than nonsense and frame shift mutations5. Our patient had mutation in exon 31, which is a novel mutation not reported earlier. Mutations in PHD domain are associated with severe psychomotor impairment and severe urogenital abnormality6. Our patients had mutation in helicase domain near C-terminus.
Carrier status for ATR-X syndrome can be identified by mutation detection, which is the most sensitive method. However, in case the mutation is unknown, X inactivation studies can be opted for. Unaffected carriers will display skewed X-inactivation, like the proband's mother in the present case. It should however, be noted that this is only a supportive test and a skewed X inactivation could also signify the presence of other X-linked syndrome or may simply be a variation of the normal pattern, which is found in about 10 per cent of women3. On the other hand, non-skewed X-inactivation could also result in phenotypic features of ATR-X syndrome in females and hence cause (moderate) mental retardation7.
Once a mutation is identified, prenatal diagnosis can be offered for future pregnancies and relevant family members could be tested for their carrier status. Management of patients with ATR-X is based on a multidisciplinary approach focusing on developmental skills and secondary problems related to the syndrome. The presence of anaemia due to alpha-thalassaemia does not require any treatment in general3.
In conclusion, this report described the presence of ATR-X syndrome in two brothers displaying many of the typical features associated with this syndrome. Clinical diagnosis is by dysmorphic facial features and mental retardation. Presence of Hb H inclusions supports the clinical diagnosis. A new c.6718C>T mutation has been identified in the family, which has further increased the number of causative mutations within the spectrum of ATR-X syndrome.
Acknowledgments
Authors thank Dr Catherine Badens, Département de génétique médicale, hôpital d’enfants-de-la-Timone, 264, rue Saint-Pierre, 13385 Marseille cedex 05, France for perfoming mutation studies in this family.
References
- 1.Gibbons RJ. ATR-X. In: Cassidy SB, Allanson JE, editors. Management of genetic diseases. 2nd ed. United States of America: John Wiley & Sons; 2005. pp. 77–86. [Google Scholar]
- 2.Jezela-Stanek A, Fishere C, Szarras-Czapnik M, Olczak-Kowalczyk D, Gibbons RJ, Słowikowska-Hilczer J, et al. X-linked alpha thalassemia/mental retardation syndrome: a case with gonnadal dysgenesis, caused by a novel mutation in ATRX gene. Clin Dysmorphol. 2009;18:168–71. doi: 10.1097/MCD.0b013e32832a9ea5. [DOI] [PubMed] [Google Scholar]
- 3.Gibbons R. Alpha thalassaemia-mental retardation, X-linked. Orphanet J Rare Dis. 2006;1:15. doi: 10.1186/1750-1172-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falco M, Luciano D, Sturnio M, Spalletta A, Scionti D, Lo Giudic M, et al. Denaturing HPLC-based assay for detection of ATRX gene mutations. Clin Chem. 2005;51:1314–5. doi: 10.1373/clinchem.2005.052407. [DOI] [PubMed] [Google Scholar]
- 5.Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP, et al. Mutations in chromatin associated protein ATR-X. Hum Mutat. 2008;29:796–802. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- 6.Badens C, Lacoste C, Phillip N, Martini N, Courrier S, Giuliano F, et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATR-X syndrome. Clin Genet. 2006;70:57–62. doi: 10.1111/j.1399-0004.2006.00641.x. [DOI] [PubMed] [Google Scholar]
- 7.Wada T, Sugie H, Fukushima Y, Saitoh S. Non-skewed X-inactivation may cause mental retardation in a female carrier of X-linked alpha-thalassemia/mental retardation syndrome (ATR-X): X-inactivation study of nine female carriers of ATR-X. Am J Med Genet A. 2005;138:18–20. doi: 10.1002/ajmg.a.30901. [DOI] [PubMed] [Google Scholar]