

Abstract
The first coherent pathophysiological scheme for sickle cell disease (SCD) emerged in the sixties-seventies based on an extremely detailed description of the molecular mechanism by which HbS in its deoxy-form polymerises and forms long fibres within the red blood cell that deform it and make it fragile. This scheme explains the haemolytic anaemia, and the mechanistic aspects of the vaso-occlusive crises (VOCs), but, even though it constitutes the basic mechanism of the disease, it does not account for the processes that actually trigger VOCs. This paper reviews recent data which imply: red blood cell dehydration, its abnormal adhesion properties to the endothelium, the participation of inflammatory phenomenon and of a global activation of all the cells present in the vessel, and finally, abnormalities of the vascular tone and of nitric oxide metabolism. These data altogether have shed a new light on the pathophysiology of the first molecular disease i.e. sickle cell disease.
Keywords: Cell activation, cell adhesion, haemoglobin S, inflammation, ion channels, nitric oxide, sickle cell disease
Introduction
The discovery of haemoglobin S (HbS) by Linus Pauling and colleagues in 1949 was the first demonstration that the production of an abnormal protein could be the cause of a genetic disorder1. So was born the notion of “molecular disease” and sickle cell disease (SCD) was the first example. In 1956, Vernon Ingram identified the abnormality in the amino acid sequence of the β-globin chain (β6Glu→Val)2. During the sixties-seventies, the first coherent pathophysiological scheme based on the abnormal polymerization of deoxy-HbS was elaborated3. It explains the basic mechanisms of the vaso-occlusive events, hallmarks of the disease, the first of which being the classical vaso-occlusive painful crisis (VOC). It also accounts for the fragility of the red blood cells and thus for the haemolytic anaemia. But this initial scheme does not account for the initial events that trigger VOC. More recent data indicate a direct participation of the vascular endothelium, of multiple and complex cellular interactions, and of a global inflammation-mediated cell activation, in the initiation and propagation of the vaso-occlusive process4. The existence of (i) an enhanced adhesion of the sickle red blood cells (SS-RBCs) to the endothelial cells (ECs)5, not; (ii) a pro-inflammatory vascular environment6 as attested by circulating activated ECs7 and activated polymorphonuclear neutrophils8, (iii) signalling pathways in the red blood cell, susceptible to be modulated by stress, hypoxia, and by the inflammatory response and to influence the activation status of adhesion receptors9 and of ion transporters implicated in SS-RBC dehydration10,11 and finally, (iv) of a syndrome of complex endothelial dysfunction involving abnormalities of the metabolism of nitric oxide (NO)12, was brought into light.
The basic pathophysiological mechanism: haemoglobin S polymerization and red blood cell alterations
During the deoxygenation which follows the passage of RBCs in the microcirculation the Hb molecule undergoes a conformational change. In HbS, replacement of the hydrophilic glutamic acid at position 6 in the β-globin chain by the hydrophobic valine residue makes that this last one establishes hydrophobic interactions with other hydrophobic residues on the β-globin chain of another deoxy-HbS molecule (Fig. 1). A polymer forms and lengthens in helical fibres which, grouped together, stiffen, and induce the characteristic SS-RBC shape change, classically in the shape of a sickle3,13. This process needs a certain time to be primed, the so-called “delay time”, which is inversely proportional to the intracellular concentration of HbS.
Fig. 1.
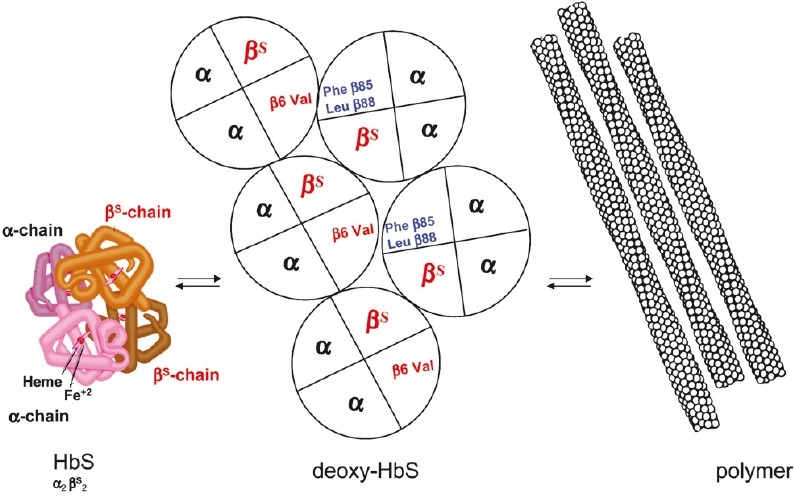
Basic pathophysiological mechanism of sickle cell disease: the polymerization of deoxy-HbS. The replacement of a glutamic acid by a valine residue at position 6 in the β-globin polypeptide chain characterizes the abnormal haemoglobin of SCD: HbS. At low oxygen pressure, deoxy-HbS polymerises and gets organised in long polymer fibres that deform, stiffen, and weaken the red blood cell (not shown). This process represents the basic mechanisms leading to haemolytic anaemia and to vaso-occlusive events in the microcirculation. [Source: Modified from Labie and Elion32].
The formation of these long polymer fibres triggers a cascade of several other cellular abnormalities which participate in the overall pathophysiological mechanism (Fig. 2). Dysregulation of cation homeostasis resulting from the activation of some ion channels, such as the K-Cl co-transport system and the Ca-dependent K-channel (Gardos channel) in particular, leads to a loss of potassium and cellular dehydration which, in turn, by increasing the intracellular Hb concentration, favours deoxy-HbS polymerization. Hb becomes denatured and hemichromes concentrate at the internal side of the membrane together with proteins of the cytoskeleton, in particular protein band 3. This process comes along with the loss of heme and with the liberation of Fe3+ which promotes the existence of an oxidizing microenvironment. The normal asymmetry of membrane phospholipids is disrupted with the exposure of anionic phosphatidylserine at the cell surface. Anti-band 3 IgGs accumulate on the protein band 3 aggregates, inducing erythrophagocytosis by macrophages14. Finally, all these membrane changes give rise to the production of microparticles.
Fig. 2.
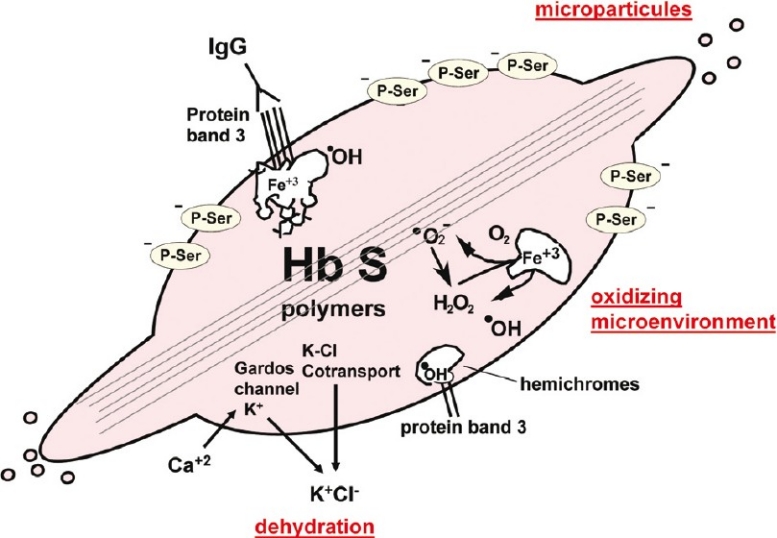
Membrane alterations in the sickle red blood cell. Formation of the deoxy-HbS polymer fibres triggers a whole series of changes of the red blood cell membrane. Ion channels are affected and their dysfunction is responsible for a cellular dehydration which, in a vicious circle, favours deoxy-HbS polymerization. Hemichromes are released and lead, in particular, to the formation of protein band 3 aggregates on which anti-band 3 IgGs accumulate. The liberation of heme and Fe3+ favours an oxidizing microenvironment. Exposure of anionic phosphatidylserines at the external side of the membrane creates a procoagulant surface. Finally, microparticles are released. [Source: Modified and completed from Elion and Labie33].
Stiffening and fragility of SS-RBC explain vaso-occlusion and haemolytic anaemia, respectively. However, if these mechanisms indeed constitute the pathophysiological bases of SCD, they do not explain what triggers VOCs. In basal conditions, the delay time, necessary for the polymerization of deoxy-HbS is longer than the time of passage of RBCs in the microcirculation. Recent data provide additional elements on various mechanisms that, by slowing down the blood flow in the microcirculation, are likely to precipitate VOCs.
Increased adhesion of sickle red blood cells to the endothelium
In the eighties and nineties the teams of R.P. Hebbel and of N. Mohandas showed the existence an increased adhesion of the SS-RBCs to the endothelium5,15. Unexpectedly, it turned out that rather than the distorted RBCs, the main actors of this abnormal adhesion process are a population of young RBCs, referred to as “stress reticulocytes”. These stress reticulocytes, coming out prematurely from the bone marrow because of the anaemic stress, express on their surface adhesion proteins that do normally maintain them in the marrow. Thus VOC seems to be composed of two consecutive steps. The first one involves adhesion of the stress reticulocytes to the endothelium of post-capillary veinules, slowing down the blood flow and thereby inducing and propagating sickling of mature SS-RBCs that are maintained for a longer time in a hypoxic environment. This first step leads to a second one which corresponds to the entrapment of irreversible sickle cells and to the complete occlusion of the micro-vessels4.
The first molecular partners identified as actors of these abnormal interactions on RBCs were the α4β1 integrin or very late antigen-4 (VLA-4) which directly binds to the vascular cell adhesion molecule-1 (VCAM-1) on the endothelial surface, and CD36 which interacts with another CD36 molecule on the endothelium through a molecular bridge formed by a molecule of plasmatic thrombospondin (TSP) (Fig. 3). Since then, the picture considerably complexified, with the identification of numerous other receptor/ligand couples on the RBCs, on one side, and on the endothelium on the other, of the involvement of various plasma proteins in addition to TSP, and with the description of an intricate network of probably co-operative and sometimes redundant interactions7,16. Clearly, the situation varies according to the vascular territories, for example VCAM-1 is specific of the endothelium of the microcirculation whereas von Willebrandt factor probably mediates abnormal cell-cell interactions in large vessels. The endothelium is altered as witnessed by circulating ECs17 and accordingly subendothelial structures are exposed that are also involved. For instance, the basal cell adhesion molecule (Lutheran blood group) Lu/BCAM antigen at the SS-RBC surface interacts with laminin in the subendothelial matrix9.
Fig. 3.
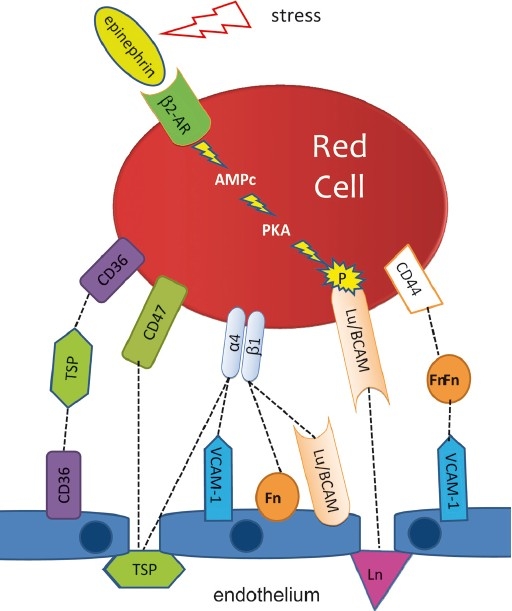
Adhesion of sickle red blood cells to the endothelium and cell activation. Simplified scheme of the main interactions involved in the abnormal adhesion of the sickle red blood cells to the endothelium. Locally, endothelial damage exposes sub-endothelial structures that also participate to the adhesion process. Some adhesion proteins are activated by extracellular stimuli. It is the case of the basal cell adhesion molecule (Lutheran blood group) (Lu/BCAM) that expresses its adhesion properties only when phosphorylated via the protein kinase A-dependent (PKA) pathway when the red blood cell is activated by epinephrine. 2-AR, type 2 adrenergic receptor; Fn, fibronectin; TSP, thrombospondin; Ln, laminin; α4β1, α4β1 integrin (or VLA-4). [Source: Modified from Elion et al34].
Even though these advances are remarkable, in particular the discovery of the major implication of stress reticulocytes, the SCD basic mechanism, namely deoxy-HbS polymerization, should never be forgotten. Even though other haemolytic anaemias come along with the presence of circulating stress reticulocytes as is the case, for example, of pyruvate kinase deficiency, none of these come along with VOCs. Thus, it is clear that, even though complex abnormal phenomena are at play, HbS is indeed the basic and the sole defect responsible of the SCD pathology, and in fine of the vaso-occlusive events.
All the cells in the blood vessel are implicated
Over the past years, it has become more and more clear that SS-RBCs and ECs are not the only actors of VOCs. For instance, plasma TSP is secreted by activated platelets. Polymorphonuclear neutrophils (PMN) also seem to be essential actors8,18. Hyperleukocytosis is almost constant in SCD patients, and a high PMN count is a pejorative element19. The presence of adherent leukocytes in post-capillary veinules suggests strongly that leukocytes, because of their cell volume, are major participants to the circulatory slowing down that initiates VOCs. In addition, it was shown that SS-RBCs are capable of abnormally interacting with leukocytes and particularly with PMNs.
All these interactions take place in a subintrant inflammatory context maintained by a set of additional mechanisms6. Phosphatidylserine abnormally exposed at the SS-RBC surface and tissue factor expressed by activated circulating ECs participate to a borderline activation of the coagulation system. The resulting production of thrombin, even at a minimal level, added to ischaemia-reperfusion injury is probably at the origin of these inflammatory phenomena20,21. These result in the production of proinflammatory cytokines that maintain a state of generalized cell activation. Furthermore, haemolysis leads to the liberation of free iron from the heme which is at the origin of an oxidative stress which in turn, via the activation of transcription factors such as nuclear factor-kappa β (NFκβ) and activator protein-1 (AP-1), participates into the endothelial expression of VCAM-1, inter-cellular adhesion molecule-1 (ICAM-1), and E-selectin, all proteins that are involved in the adhesion of stress reticulocytes and in leukocytes recruitment1. Finally, cell activation induces the production of microparticles. In addition to the microparticles of erythrocytic origin already mentioned, circulating microparticles of endothelial, platelet, and leukocytic origins are observed in SCD patients and are increased in number at the time of VOCs. Very significantly, these microparticles are not only passive witnesses of cell activation, but these are probably important actors through activating properties conferred to them by their reorganised membrane22.
Sickle red blood cells are activable and activated
For a long time considered as simple «haemoglobin bags», RBCs have been shown to express on their surface a variety of receptors susceptible to induce signalling pathways that modify their functional properties. This aspect is particularly relevant to the pathophysiology of SCD. Cytokines and hypoxia modulate signalling pathways involved in the regulation of ion transport and RBC hydration, the influence of which on HbS polymerization is already mentioned10,11,23,24. Some extracellular stimuli are also capable of activating adhesion mechanisms. Epinephrin for example, by inducing the activation of the protein kinase A-dependent (PKA) signalling pathway leads to the phosphorylation of Lu/BCAM, and this phosphorylation is indispensible for the expression of Lu/BCAM adhesion properties9,25 (Fig. 3).
All these data have brought a new vision of factors that participate in the initiation of the VOC. We now perceive better the precarious balance in which the SCD patient in steady state is and how this can tip over towards the initiation of a VOC for example, during an infection which increases inflammation, or because of a stress that activates adhesion proteins on the RBCs.
Haemolysis alters nitric oxide metabolism and vessel biology
For a long time, VOC has been the focus of researchers trying to decipher the intimate mechanisms of SCD pathophysiology and haemolysis was rather a neglected aspect left in the second rank. Still, over the last decade, the work of the group of M.T. Gladwin outlined the potential importance of haemolysis as one of the primary pathophysiological factor in SCD12,26. Regulation of the vascular tone depends on a subtle balance between mediators produced by the endothelium such as endothelin-1 (ET-1), with its vaso-constrictive action, and nitric oxide (NO), with its strong activity as a vaso-dilator. In SCD, plasma NO is low and ET-1 is increased, particularly during VOCs. Thus the normal balance is shifted towards a vaso-constrictive state that is also susceptible to slowing down the blood flow and leading to the precipitation and perpetuation of VOCs. Haemoglobin is the most powerful NO scavenger known. Free Hb destroys NO and generates free radicals one thousand times more quickly than Hb in the RBC. The originality of Gladwin's work was to connect haemolysis and the resulting liberation of Hb in plasma and a decrease of NO bioavailability in SCD. This depletion of NO is still majored by the fact that haemolysis also releases erythroid arginase in plasma where this enzyme degrades L-arginine, the substrate of the NO producing enzyme, i.e. the endothelial NO synthase (eNOS)27. Thus the mechanism of NO depletion is double: destruction of NO by free Hb and reduced NO production by depletion of its precursor.
Haemolysis-induced NO depletion is responsible for a set of abnormalities, the first of which being the essential loss of the vasodilator potential, incapable to counteract the vasoconstrictive action of ET-1, but also a facilitation of platelet activation and an endothelial dysfunction with an abnormal expression of adhesion molecules. From these data, the authors suggested to distinguish two sub-phenotypes in SCD, one referred to as “hyperviscosity – vaso-occlusion” and the other one “haemolysis – endothelial dysfunction”, each associated with different complications of the disease12. They proposed that the first one was preferentially associated with VOC, acute chest syndrome and femoral head osteonecrosis and the second one with a greater risk of developing arterial pulmonary hypertension (APHT), a complication with a severe prognosis and relatively underestimated until then in SCD, leg ulcer, priapism, and possibly stroke. These sub-phenotypes, however, are overlapping and simultaneously present in all the patients, but certain patients would express preferentially a sub-phenotype with regard to the other one and would develop preferentially the complications specifically associated to this sub-phenotype. This attractive hypothesis led to a whole series of new therapeutic hypotheses to restore NO bioavailability28. Various clinical assays to test these hypotheses were realized, none of which with very convincing results at the moment, but other assays are still in progress.
One should note however, that the relevance of Gladwin's hypothesis was recently challenged29,30. Certainly, NO scavenging by free Hb has been documented in some other haemolytic diseases as for example, paroxystic nocturnal haemoglobinuria (PNH). But SCD and PNH do differ on several important aspects: the level of free plasma Hb which is 10 times higher in PNH than in SCD, the acute character of intravascular haemolysis in PNH versus a most frequently chronic and primarily extravascular haemolysis in SCD, the erectile abnormalities: impotence in PHN versus priapism in SCD. Recent data also suggest that the APHT is a complication, certainly very severe, but rare in SCD, contradicting the initial data of Gladwin's group.
Thus, the relative importance of the abnormalities of NO metabolism in the pathophysiology of SCD still remains to be defined with more precision.
Conclusion
The first molecular disease to be described, SCD remains an unprecedented model. The elucidation of the precise molecular mechanisms of HbS polymerization represented, at the time, an absolutely remarkable achievement. Even though it still remains valid, this scheme does not explain, by itself only, factors triggering VOCs. In this domain, considerable progresses were realized over the last few years which gave of SCD a new vision. These factors are complex, still controversial for some of them, but clearly very intricate, sometimes redundant and probably complementary and synergistic. Clearly, the pathophysiological understanding of diseases in general is essential to the development of new therapeutic hypotheses to be tested. It is indeed the case for SCD for which the remarkable progress realized in terms of reduced morbidity, improved quality of life, and increased life expectancy is essentially the fact of early follow up after neonatal screening and early prevention of severe complications. A single drug, hydroxycarbamide (or hydroxyurea), has definitely been proven to be efficient at improving the patients’ quality of life, but its mode of action is still poorly defined. The development of new therapeutic approaches applicable in countries where SCD is endemic is an absolute necessity. Thus a lot is still to be made. The recent technological advances of biology now allow global approaches without preconceived idea. In science, the new “omic” technologies: genomics, transcriptomics, proteomics, metabolomics… now prevail. Simultaneouly, biocomputing, biomathematic, and modelling tools necessary for the interpretation of the huge mass of generated data are being rapidly developed. The so called “system biology” emerges, that encompasses the analysis of the functioning and dysfunctioning of the signalling pathway networks and of their interactions globally. Because relatively simple in its complexity, SCD could indeed continue to serve as a model for the application of these new approaches and concepts to the understanding of diseases in general31.
References
- 1.Pauling L, Itano H, Singer SJ, Wells IC. Sickle cell anemia: a molecular disease. Science. 1949;110:543–8. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 2.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anemia haemoglobin. Nature. 1956;178:792–4. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 3.Bunn H, Forget BG. Hemoglobin: Molecular, genetic and clinical aspects. Philadelphia, PA, USA: WB Saunders; 1986. [Google Scholar]
- 4.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–9. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 5.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11 Suppl):S83–6. [PubMed] [Google Scholar]
- 6.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106:337–8. doi: 10.1172/JCI10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebbel RP. Adhesion of sickle red cells to endothelium: Myths and future directions. Transfus Clin Biol. 2008;15:14–8. doi: 10.1016/j.tracli.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Chiang EY, Frenette PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am. 2005;9:771–94. doi: 10.1016/j.hoc.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 9.El Nemer W, Colin Y, Le Van Kim C. Role of Lu/BCAM glycoproteins in red cell diseases. Transfus Clin Biol. 2010;17:143–7. doi: 10.1016/j.tracli.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Rivera A, Jarolim P, Brugnara C. Modulation of Gardos channel activity by cytokines in sickle erythrocytes. Blood. 2002;99:357–603. doi: 10.1182/blood.v99.1.357. [DOI] [PubMed] [Google Scholar]
- 11.Merciris P, Giraud F. How do sickle cells become dehydrated? Hematol J. 2001;2:200–5. doi: 10.1038/sj.thj.6200107. [DOI] [PubMed] [Google Scholar]
- 12.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edelstein SJ, Telford JN, Crépeau RH. Structure of fibers of sickle cell hemoglobin. Proc Natl Acad Sci USA. 1973;70:1104–7. doi: 10.1073/pnas.70.4.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–60. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- 15.Mohandas N, Evans E. Adherence of sickle erythrocytes to vascular endothelial cells: requirement for both cell membrane changes and plasma factors. Blood. 1984;64:282–7. [PubMed] [Google Scholar]
- 16.Elion J, Brun M, Odievre MH, Lapoumeroulie CL, Krishnamoorthy R. Vaso-occlusion in sickle cell anemia: role of interactions between blood cells and endothelium. Hematol J. 2004;5:S195–8. doi: 10.1038/sj.thj.6200452. [DOI] [PubMed] [Google Scholar]
- 17.Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337:1584–90. doi: 10.1056/NEJM199711273372203. [DOI] [PubMed] [Google Scholar]
- 18.Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850–8. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease.Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 20.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–20. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–51. [PubMed] [Google Scholar]
- 22.Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–71. doi: 10.1016/j.blre.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Merciris P, Claussen WJ, Joiner CH, Giraud F. Regulation of K-Cl cotransport by Syk and Src protein tyrosine kinases in deoxygenated sickle cells. Pflugers Arch. 2003;446:232–8. doi: 10.1007/s00424-003-1025-z. [DOI] [PubMed] [Google Scholar]
- 24.Durpès MC, Nebor D, du Mesnil PC, Mougenel D, Decastel M, Elion J, et al. Effect of interleukin-8 and RANTES on the Gardos channel activity in sickle human red blood cells: role of the Duffy antigen receptor for chemokines. Blood Cells Mol Dis. 2010;44:219–23. doi: 10.1016/j.bcmd.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Hines PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, et al. Novel epinephrine and cyclic AMP-mediated activation of BCAM/Lu-dependent sickle (SS) RBC adhesion. Blood. 2003;101:3281–7. doi: 10.1182/blood-2001-12-0289. [DOI] [PubMed] [Google Scholar]
- 26.Reiter CD, Gladwin MT. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr Opin Hematol. 2003;10:99–107. doi: 10.1097/00062752-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack AK, Kato GJ. Sickle cell disease and nitric oxide: a paradigm shift? Int J Biochem Cell Biol. 2006;38:1237–43. doi: 10.1016/j.biocel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunn HF, Nathan DG, Dover GJ, Hebbel RP, Platt OS, Rosse WF, et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116:687–92. doi: 10.1182/blood-2010-02-268193. [DOI] [PubMed] [Google Scholar]
- 30.Hebbel RP. Reconstructing sickle cell disease: a data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol. 2011;86:123–54. doi: 10.1002/ajh.21952. [DOI] [PubMed] [Google Scholar]
- 31.Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. 2009;9:271–92. doi: 10.2174/1871529x10909040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labie D, Elion J. Molecular and cellular pathophysiology of sickle cell anemia. Pathol Biol (Paris) 1999;47:7–12. [PubMed] [Google Scholar]
- 33.Elion J, Labie D. Bases physiopathologiques moléculaires et cellulaires du traitement de la drépanocytose. Hématologie. 1996;2:499–510. [Google Scholar]
- 34.Elion J, Laurance S, Lapouméroulie C. Physiopathologie de la drépanocytose. Med Trop. 2010;70:454–8. [PubMed] [Google Scholar]