

Abstract
The thalassaemias and sickle cell disease are the commonest monogenic disorders in India. There are an estimated 7500 - 12,000 babies with β-thalassaemia major born every year in the country. While the overall prevalence of carriers in different States varies from 1.5 to 4 per cent, recent work has shown considerable variations in frequencies even within States. Thus, micromapping would help to determine the true burden of the disease. Although screening in antenatal clinics is being done at many centres, only 15-20 per cent of pregnant women register in antenatal clinics in public hospitals in the first trimester of pregnancy. There are only a handful of centres in major cities in this vast country where prenatal diagnosis is done. There is considerable molecular heterogeneity with 64 mutations identified, of which 6 to 7 common mutations account for 80-90 per cent of mutant alleles. First trimester foetal diagnosis is done by chorionic villus sampling (CVS) and DNA analysis using reverse dot blot hybridization, amplification refractory mutation system (ARMS) and DNA sequencing. Second trimester diagnosis is done by cordocentesis and foetal blood analysis on HPLC at a few centres. Our experience on prenatal diagnosis of haemoglobinopathies in 2221 pregnancies has shown that >90 per cent of couples were referred for prenatal diagnosis of β-thalassaemia after having one or more affected children while about 35 per cent of couples were referred for prenatal diagnosis of sickle cell disorders prospectively. There is a clear need for more data from India on non-invasive approaches for prenatal diagnosis.
Keywords: Haemoglobinopathies, India, invasive and non-invasive approaches, prenatal diagnosis
Introduction
The inherited disorders of haemoglobin are the most common monogenic disorders globally. Around 7 per cent of the population worldwide are carriers with more than 3,00,000 severely affected babies born every year1.
Prenatal diagnosis is an integral component of a community control programme for haemoglobinopathies. Estimating the disease burden, generating awareness in the population, screening to identify carriers and couples at - risk and genetic counselling are prerequisites for a successful prevention programme. The remarkable success of such programmes in the 1970s in Cyprus, Italy, Greece and the UK led to the development of control programmes in many other countries2–6.
The extent of the problem in India
β-thalassaemia has been reported in most of the communities that have been screened so far in India. While the overall prevalence varies from 1.5 to 4 per cent in different States, communities like Sindhis, Punjabis, Lohanas, Kutchi Bhanushalis, Jains and Bohris have a higher prevalence (4-17%)7–12. Different reports have estimated that 7500-12,000 β-thalassaemia major babies would be born in India each year12–14. It has also been shown recently by micromapping at the district level in two States, Maharashtra and Gujarat in western India that the prevalence of β-thalassaemia trait in different districts within these States is variable (0 - 9.5%). Based on these estimates there would be around 1000 births of β-thalassaemia major babies each year in these two States alone15. Thus, such data should be obtained from different states to know the true burden of the disease and for planning and executing control programmes.
Haemoglobin S (Hb S) is prevalent in central India and among the tribal belts in western, eastern and southern India, the carrier rates varying from 1-40 per cent16–18. It has been estimated that over 5000 babies with sickle cell disease would be born each year19.
Haemoglobin E is widespread in the north eastern States in Assam, Mizoram, Manipur, Arunachal Pradesh and Tripura, the prevalence of Hb E trait being highest (64%) among the Bodo-Kacharis in Assam and going up to 30-40 per cent in some other populations in this region20–22. In eastern India the prevalence of Hb E trait varies from 3-10 per cent in West Bengal8,23. Both Hb E and Hb S when co-inherited with β-thalassaemia result in a disorder of variable clinical severity24–26.
These inherited haemoglobin disorders cause considerable pain and suffering to the patients and their families and are a major drain on health resources in the country.
The need for accurate identification of carries and couples at risk
Classical β-thalassaemia carriers have typically reduced red cell indices [mean corpuscular volume (MCV)<80 fl, mean corpuscular haemoglobin (MCH)<27 pg] with high RBC counts and elevated HbA2 levels (4-8%)27. However, a few β-thalassaemia heterozygotes fail to manifest these classical haematological features and are termed as silent carriers or normal HbA2 β-thalassaemia carriers. Individuals with borderline HbA2 levels (3.3 to 3.9%) need to be evaluated carefully to avoid a misdiagnosis. Mutations in the promoter region of the β-globin gene like -86 (C→G), -87 (C→G), -88 (C→T), -101 (C→T) as well as the capsite +1 (A→C) and the poly A (T→C) mutation can lead to borderline Hb A2 levels28. Among these, the capsite +1 (A→C) mutation is seen in 1-2.5 per cent of β-thalassaemia heterozygotes in India29–31. Other factors like associated α-thalassaemia or δ-thalassaemia and iron deficiency anaemia may also contribute in reduction of HbA2 levels28,32,33.
It is also important to identify carriers of δβ-thalassaemia as compound heterozygotes of δβ-thalassaemia and β-thalassaemia can lead to a severe disorder. δβ-thalassaemia carriers are characterized by a modest elevation in Hb F levels (5-20%) with reduced or normal HbA2 levels and hypochromic and microcytic red cells. This phenotype partially overlaps with that of carriers of hereditary persistence of foetal haemoglobin (HPFH) and genotyping of high Hb F determinants is required as these are not infrequent in India. The common determinant of δβ-thalassaemia in India is the Asian Indian deletional inversion Gγ(Aγδβ)°thalassaemia34.
The most appropriate time to screen
Screening in antenatal clinics is the best way to identify couples at immediate risk of having an affected child. However, experiences in India have shown that only 15-20 per cent of pregnant women come to antenatal clinics in public hospitals in the first trimester of pregnancy when prenatal diagnosis should ideally be done11,35. This emphasizes the need for generating awareness in the population for early registration in antenatal clinics as well as among obstetricians to ask for a screening for β-thalassaemia and other haemoglobinopathies along with other investigations which are done routinely. The other target groups for screening in India include high school children, college or university students, high risk communities and extended family members of affected children11,12,36. The latter cascade screening approach appears to be a practical way of identifying a larger number of β-thalassaemia carriers in a cost-effective way in India. The large size of families as well as the system of living in joint families rather than nuclear ones is an advantage in this approach.
Genetic counselling
Genetic counselling plays a major role in a prevention programme. The Indian population comprises several ethnic groups with considerable cultural and religious diversity and this has to be kept in mind while counselling families to avoid social stigmatization37. The influence of elders in the family also weighs heavily and has to be dealt with appropriately. While earlier, parents with a thalassaemia major child kept to themselves, today their relatives and extended families are coming forward to get screened38. There is only one centre in Lucknow in north India which offers a formal course for genetic counsellors and there is a need for more such courses throughout the country.
Counsellors should be aware that couples at risk of having a child with β-thalassaemia major, sickle cell disease, Hb S β-thalassaemia, Hb E β-thalassaemia, δβ - β-thalassaemia, Hb Lepore β-thalassaemia and Hb SD disease should be given the option of prenatal diagnosis to avoid the birth of a child with a severe disorder. However, couples at risk of having a child with Hb D disease, Hb D β-thalassaemia and Hb E disease do not require prenatal diagnosis as these disorders are mild.
In Sardinia, identification of the maximum number of carriers followed by effective genetic counselling helped to reduce the birth rate of β-thalassaemia major babies from 1:250 to 1:400039.
Prenatal diagnosis
The first initiatives in India
Facilities for prenatal diagnosis became available in India in the mid 1980s40. Until then, although prenatal diagnosis was offered by a few centres, foetal samples were sent to the UK and other countries for analysis. Foetal blood sampling by foetoscopy done between 18 and 22 wk gestation and diagnosis by globin chain synthesis were done for the next 4 to 5 years at 2 centres in Mumbai40,41.
Chorionic villus sampling and DNA analysis in the first trimester
In the 1990s first trimester foetal diagnosis by chorionic villus sampling (CVS) and DNA analysis was established at 4-5 centres in the north in Delhi42, in the west in Mumbai41,43,44 and in the south in Vellore45. These services then expanded to other cities like Lucknow and Chandigarh in the north46,47, and Kolkata in the east48. However, these services are still limited to major cities where couples are referred to or CVS samples are sent from surrounding areas.
Molecular analysis
β-thalassaemia is extremely heterogeneous with more than 200 mutations described worldwide49. In India, about 64 mutations have been characterized by studies done at different centres30,31,49–51 (Table I). Six to seven mutations [IVS 1-5 (G→C), 619 bp deletion, IVS 1-1 (G→T), Codon 8/9 (+G), Codons 41/42 (-CTTT), Codon 15 (G→A), Codon 30 (G→C)] are common accounting for 85-95 per cent of mutant alleles. However, regional differences in their frequencies have been noted30,31,50,51 (Fig. 1). The prevalence of IVS 1 -5 (G→C), the most common mutation in India varies from 15-88 per cent in different States. Codon 15 (G→A) is the second most frequent mutation in Maharashtra and Karnataka and Codon 5 (-CT) is the third most common mutation in Gujarat. The -88 (C→T) and the Cap site +1 (A→C) mutations are more common in the northern region30,31,50. The 619 bp deletion is the most common mutation among the immigrant population from Pakistan.
Table I.
Spectrum of β-thalassaemia mutations in India

Fig. 1.
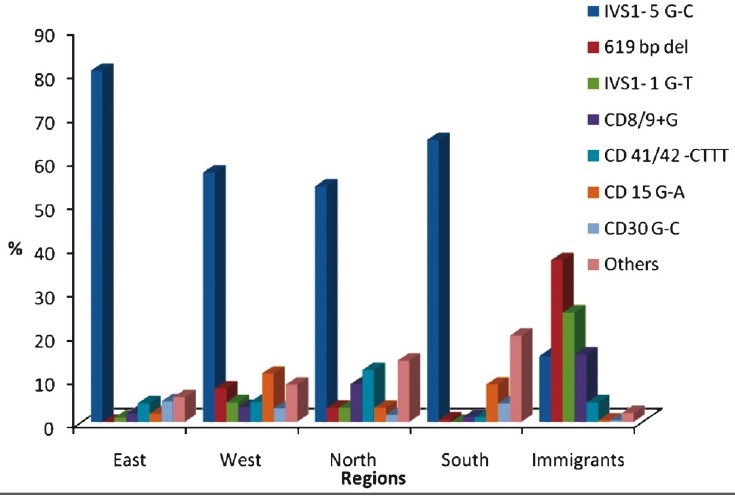
Regional distribution of β-thalassaemia mutation in India.
This knowledge on the distribution of mutations in different regions and in people of different ethnic backgrounds has facilitated prenatal diagnosis using molecular techniques like covalent reverse dot blot hybridization (CRDB), amplification refractory mutation system (ARMS), denaturing gradient gel electrophoresis (DGGE), and DNA sequencing43,44,52.
Foetal blood analysis in the second trimester
Most of the prenatal diagnosis programmes in the Mediterranean countries started with second trimester foetal blood analysis but they were able to switch over to first trimester diagnosis in a short span5,39.
In India, second trimester diagnosis is still done as many couples at risk are identified late during pregnancy. Foetal blood sampling is done by cordocentesis at 18 to 20 wk gestation and after confirming that there is no maternal contamination in the foetal sample by foetal cell staining using the Kleihauer-Betke method, it is analysed by HPLC on the Variant Hemoglobin Testing System (Bio Rad Laboratories, Hercules, USA). The Hb A levels in foetuses affected with β-thalassaemia major have ranged from 0 to 0.5 per cent and these were distinguishable from heterozygous babies where the Hb A levels were >1.3 per cent in different studies. However, there was some overlap in Hb A levels between heterozygotes and normals53–55. Sickle cell disease and Hb E thalassaemia have also been diagnosed in this way. On the other hand, experience in Thailand showed that while β°thalassaemia homozygotes and Hb E -β°thalassaemia compound heterozygotes could be diagnosed by HPLC analysis of foetal blood, β++ thalassaemia homozygotes may be misdiagnosed as heterozygotes56.
Amniotic fluid cells have not been used extensively in India for prenatal diagnosis of haemoglobinopathies.
Experience at National Institute of Immunohaematology (NIIH), Mumbai
Both first and second trimester prenatal diagnosis for the β-thalassaemias and sickle cell disorders are done at National Institute of Immunohaematology, Mumbai, and over the last 25 years 2,221 pregnancies at risk have been investigated (Table II). While majority of the couples were at risk of having children with β-thalassaemia major, a significant number of couples at risk of having children with sickle cell disorders have been referred for prenatal diagnosis in the last 4 to 5 years. Our experience in western India has shown that there are still very few couples (<10%) who come prospectively in the first pregnancy for prenatal diagnosis of the β-thalassaemias while 25 -35 per cent of couples come prospectively for prenatal diagnosis of sickle cell disorders. This is a reflection of the greater awareness in regions particularly in Maharashtra and Gujarat where sickle cell disorders are common. Realizing the need for more centres for prenatal diagnosis in the country, the Indian Council of Medical Research, New Delhi, is trying to establish regional centres with the help of NIIH, Mumbai, in Nagpur in Maharashtra, Valsad and Surat in Gujarat, Bangalore in Karnataka, Kolkata in West Bengal and Ludhiana in Punjab mainly in medical colleges.
Table II.
Summary of prenatal diagnosis of haemoglobinopathies done from 1986 to 2010 at NIIH, Mumbai
Non-invasive prenatal diagnosis from maternal blood
Current methods for obtaining foetal genetic material like chorionic villus sampling, amniocentesis and cordocentesis are invasive procedures which are associated with a small risk of foetal loss even when done by experienced hands57. This has led to many studies on the possibility of accessing foetal cells from the maternal circulation for non-invasive diagnosis.
Isolation of foetal cells from maternal blood
Trophoblasts, lymphocytes and nucleated erythrocytes (NRBCs) were the 3 cell types used as a source of foetal DNA. Foetal NRBCs were most widely used as they are mononuclear, are specific to the ongoing pregnancy and can be detected in the first trimester. These express several antigens like transferrin receptor and produce foetal haemoglobin chains like zeta (ζ), epsilon (ε) and gamma (γ) which are useful markers58,59. However, their numbers in the maternal circulation during pregnancy are only a few. It is estimated that around 2 μl of foetal blood crosses into the maternal circulation per day in the first and second trimesters and 1/50,000 cells are of foetal origin. Thus enrichment of foetal NRBCs was done by density gradient centrifugation using percoll or ficoll and isolation of the cells by fluorescence activated cell sorting (FACS) or magnetic activated cell sorting (MACS) using different monoclonal antibodies or pick up of single NRBCs after staining by microdissection60.
A few reports were published on the non-invasive prenatal diagnosis of sickle cell anaemia and β-thalassaemia using foetal NRBCs and PCR based protocols61–63. The number of pregnancies studied were few and accurate diagnosis was not possible in every case.
Our experience
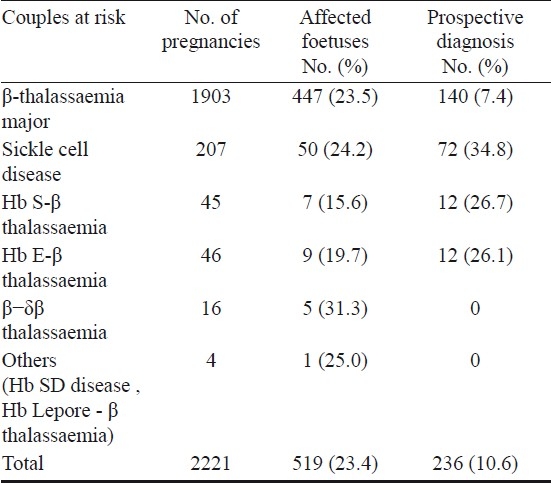
We attempted non-invasive prenatal diagnosis of β-thalassaemia major, sickle cell disorders and Hb E β-thalassaemia by isolating foetal NRBCs from 7 ml of maternal blood at different periods of gestation in 100 cases. As shown schematically in Fig. 2, after percoll density gradient centrifugation for enrichment of foetal NRBCs, a combination of 3 monoclonal antibodies CD 45, Glycophorin A(GpA) and anti-Hb F were used for flow sorting. The CD45 negative cells were first selected and from this population the cells which were dual positive for Gycophorin A and Hb F were sorted. Foetal NRBCs could be isolated between 9 and 22 wk gestation although maximum number of cells were obtained between 10 and 14 wk gestation. DNA was isolated from the sorted foetal NRBCs and a nested ARMS PCR approach was developed for the detection of 12 different mutations. The foetal genotype was correctly determined in 84 of the 100 cases (84%). In 10 cases, the diagnosis was incorrect due to maternal contamination in the flow sorted cells and in 6 cases this was due to non amplification of alleles64. In another 30 pregnancies substituting Glycophorin A by CD 71 increased the accuracy of diagnosis to 90 per cent. Thus, the problems of maternal contamination and allele dropouts remain.
Fig. 2.
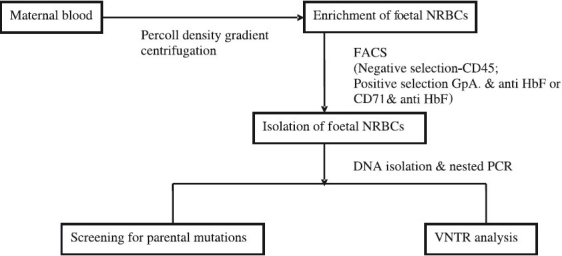
Schematic representation of the approach used for non-invasive prenatal diagnosis using foetal nucleated red blood cells (NRBCs) from maternal blood.
Circulatory foetal DNA in maternal plasma
The discovery of cell free foetal DNA in maternal plasma by Lo et al65 opened up new avenues for non-invasive prenatal diagnosis. However, for the diagnosis of monogenic disorders like the thalassaemias, this approach is still a challenge as foetal DNA represents only 5 per cent of the DNA in maternal plasma66, although recently it has been shown to be present in higher concentrations (10-20%)67. It was shown that foetal DNA fragments are smaller in size (<313 bp) than maternal DNA fragments (>1 kb) and cell free foetal DNA in maternal plasma can be enriched by size fractionation on agarose gels68. Most of the reports so far have been based on detection or exclusion of paternal β-thalassaemia mutations when the maternal and paternal mutations are different or on the efficacy of detection of informative single nucleotide polymorphisms (SNPs) linked to the paternal mutant or normal allele. The diagnostic methods used have included real time allele specific PCR, allele specific arrayed primer extension (AS-APEX) technology, conventional PCR - DGGE and MALDI-TOF mass spectrometry69–76.
The potential disadvantage of most of these studies is that only paternally inherited alleles can be studied and invasive procedures may still be required in many cases when the paternal mutation is present. Besides, the technology is sophisticated and would not be available at many diagnostic laboratories.
Our experience with cell free foetal DNA analysis
Fig. 3 shows the approach used by us for prenatal diagnosis using cell free foetal DNA from maternal plama. In 30 couples where the paternal and maternal mutations were different, we extracted DNA from maternal plasma. After size separation on agarose gels a fragment of about 300 bp was excised and used for looking for the presence or absence of the paternal mutation by semi- nested PCR. An accuracy of 80 per cent was achieved (unpublished data).
Fig. 3.
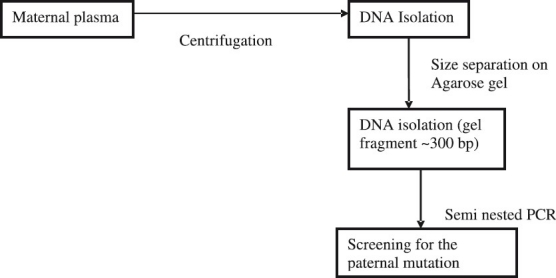
Schematic representation of the approach used for non-invasive prenatal diagnosis using circulating cell free foetal DNA from maternal plasma.
The future
It has been predicted that within the next 10 years, the complete foetal genome from maternal plasma would be successfully sequenced. The main technology for non-invasive prenatal diagnosis even for monogenic disorders could be based on single molecule analysis methods. Digital PCR and massively parallel sequencing would be the technology used77.
Acknowledgments
Authors thank Drs D. Mohanty and K. Ghosh for their support, and are grateful to all their collaborating obstetricians and sonologists and their technical staff.
References
- 1.Weatherall DJ. The inherited disorders of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–6. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao A. Results of programs for antenatal detection of thalassemia in reducing the incidence of the disorder. Blood Rev. 1987;1:169–76. doi: 10.1016/0268-960x(87)90032-4. [DOI] [PubMed] [Google Scholar]
- 3.Angastiniotis MA, Hadjiminas MG. Prevention of thalassemia in Cyprus. Lancet. 1991;1:369–70. doi: 10.1016/s0140-6736(81)91682-2. [DOI] [PubMed] [Google Scholar]
- 4.Petrou M, Modell B. Prenatal screening for hemoglobin disorders. Prenat Diagn. 1995;15:1275–95. doi: 10.1002/pd.1970151308. [DOI] [PubMed] [Google Scholar]
- 5.Loukopoulos D. Current status of thalassaemia and the sickle cell disorders in Greece. Semin Hematol. 1996;33:76–86. [PubMed] [Google Scholar]
- 6.Management of hemoglobin disorders, Report of Joint WHO-TIF meeting, Nicosia, Cyprus, 16-18 November, 2007. Geneva: WHO; 2008. World Health Organization. [Google Scholar]
- 7.Mehta BC, Dave VB, Joshi SR, Baxi AJ, Bhatia HM, Patel JC. Study of hematological and genetical characteristics of Kutchi Bhanushali Community. Indian J Med Res. 1972;60:305–11. [PubMed] [Google Scholar]
- 8.Sukumaran PK. Abnormal hemglobins in India. In: Sen NN, Basu AK, editors. Trends in hematology. Calcutta: Sree Saraswati Press; 1975. pp. 225–61. [Google Scholar]
- 9.Chouhan DM, Chouhan V. Epidemiology: Symposium on thalassemia. Indian J Hematol Blood Transf. 1992;10:1–6. [Google Scholar]
- 10.Jawahirani A, Mamtani M, Das K, Rughwani V, Kulkarni H. Prevalence of beta thalassemia in sub-castes of Indian Sindhis: Results from a two phase survey. Public Health. 2007;121:193–8. doi: 10.1016/j.puhe.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 11.Mohanty D, Colah R, Gorakshakar A, editors. Report of the Jai Vigyan S & T Mission Project on community control of thalassaemia syndromes- Awareness, screening, genetic counselling and prevention. A National Multicentric Task Force Study of Indian Council of Medical Research, New Delhi; 2008. New Delhi: ICMR; 2008. [Google Scholar]
- 12.Madan N, Sharma S, Sood SK, Colah R, Bhatia HM. Frequency of β-thalassemia and other hemoglobinopathies in northern and western India. Indian J Hum Genet. 2010;16:16–25. doi: 10.4103/0971-6866.64941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modell B, Petrou M. The problem of hemoglobinopathies in India. Indian J Haematol. 1983;1:1–5. [Google Scholar]
- 14.Choudhry VP, Upadhyay A. Thalassemia screening and control programme. In: Ghosh K, Colah R, editors. Control and management of thalassaemia and other hemoglobinopathies in the Indian subcontinent - Synoptic views. Mumbai: National Institute of Immunohaematology; 2008. pp. 36–44. [Google Scholar]
- 15.Colah R, Gorakshakar A, Phanasgaonkar S, D’Souza E, Nadkarni A, Surve R, et al. Epidemiology of β-thalassemia in western India:Mapping the frequencies and mutations in sub-regions of Maharashtra and Gujarat. Br J Haematol. 2010;149:739–47. doi: 10.1111/j.1365-2141.2010.08131.x. [DOI] [PubMed] [Google Scholar]
- 16.Bhatia HM, Rao VR. Mumbai: Institute of Immunohaematology; 1986. Genetic atlas of the Indian tribes. [Google Scholar]
- 17.Balgir RS. Genetic epidemiology of the three predominant abnormal hemoglobins in India. J Assoc Physicians India. 1996;44:25–8. [PubMed] [Google Scholar]
- 18.Mohanty D, Mukherjee MB. Sickle cell disease in India. Curr Opin Haematol. 2002;9:117–22. doi: 10.1097/00062752-200203000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Verma IC, Bijarnia S. The burden of genetic disorders in India and a framework for community control. Community Genet. 2002;5:192–6. doi: 10.1159/000066335. [DOI] [PubMed] [Google Scholar]
- 20.Deka R, Reddy AP, Mukherjee BN, Das BM, Banerjee S, Roy M, et al. Hemoglobin E distribution in 10 endogamous population groups of Assam, India. Hum Hered. 1988;38:261–6. doi: 10.1159/000153796. [DOI] [PubMed] [Google Scholar]
- 21.De M, Halder A, Podder S, Sen R, Chakraborty S, Sengupta B, et al. Anemia and hemoglobinopathies in tribal population of eastern and north eastern India. Hematology. 2006;11:371–3. doi: 10.1080/10245330600840180. [DOI] [PubMed] [Google Scholar]
- 22.Sharma SK, Mahanta J. Prevalence of hemoglobin variants in malaria endemic north east India. J Biol Sci. 2009;9:288–91. [Google Scholar]
- 23.Das MK, Dey B, Roy M, Mukherjee BM. High prevalence of Hb E in 3 populations of the Malda district, West Bengal, India. Hum Hered. 1991;41:84–8. doi: 10.1159/000153983. [DOI] [PubMed] [Google Scholar]
- 24.Nadkarni A, Ghosh K, Gorakshakar A, Colah R, Moahtny D. Variable clnical severity of Hb E β-thalassemia among Indians. J Assoc Physicians India. 1999;47:966–8. [PubMed] [Google Scholar]
- 25.Panigrahi I, Agwaral S, Gupta J, Singhal P, Pradhan M. Hemoglobin E beta thalassemia: Factors affecting phenotype. Indian Pediatr. 2005;42:357–62. [PubMed] [Google Scholar]
- 26.Kulozik AE, Bail S, Kar BC, Serjeant BE, Serjeant GR. Sickle cell -β+ thalassemia in Orissa state, India. Br J Haematol. 1991;77:215–20. doi: 10.1111/j.1365-2141.1991.tb07980.x. [DOI] [PubMed] [Google Scholar]
- 27.Weatherall DJ, Clegg JB. The thalassemia syndromes. 4th ed. Oxford: Blackwell Scientific Publications; 2001. [Google Scholar]
- 28.Giambano A, Passarello C, Vinciguerra M, Li Muli R, Teresi P, Anzà M, et al. Significance of borderline HbA2 values in an Italian population with a high prevalence of β-thalassemia. Haematologica. 2008;93:1380–4. doi: 10.3324/haematol.12840. [DOI] [PubMed] [Google Scholar]
- 29.Garewal G, Das R, Awasthi A, Ahluwalia J, Marwaha RK. The clinical significance of the spectrum of interactions of Cap + 1 (A→C), a silent β globin gene mutation with other β thalassemia mutations and globin gene modifiers in north Indians. Eur J Haematol. 2007;79:417–21. doi: 10.1111/j.1600-0609.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 30.Colah R, Gorakshakar A, Nadkarni A, Phanasgaonkar S, Surve R, Sawant P, et al. Regional heterogeneity of beta thalassemia mutations in the multi ethnic Indian population. Blood Cells Mol Dis. 2009;42:241–6. doi: 10.1016/j.bcmd.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Sinha S, Black ML, Agarwal S, Colah R, Das R, Ryan K, et al. Profiling β-thalassemia mutations in India at state and regional levels: implications for genetic education, screening and counseling programmes. Hugo J. 2009;3:51–62. doi: 10.1007/s11568-010-9132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panigrahi I, Rafeeq PHA, Choudhry VP, Saxena R. High frequency of deletional α thalassemia in β thalassemia trait: Implications for genetic counselling. Am J Hematol. 2004;76:297–99. doi: 10.1002/ajh.20083. [DOI] [PubMed] [Google Scholar]
- 33.Bouva M, Harteveld C, van Delft P, Giordano P. Known and new delta globin gene mutations and their diagnostic significance. Haematologica. 2006;91:129–32. [PubMed] [Google Scholar]
- 34.Nadkarni A, Wadia M, Gorakshakar A, Kiyama R, Colah RB, Mohanty D. Molecular charaterization of δβ thalassemia and hereditary persistence of fetal hemoglobin in the Indian population. Hemoglobin. 2008;32:425–33. doi: 10.1080/03630260802341687. [DOI] [PubMed] [Google Scholar]
- 35.Colah R, Surve R, Wadia M, Solanki, Mayekar P, Thomas M, et al. Carrier screening for β-thalassemia during pregnancy in India: A 7 year evaluation. Genet Test. 2008;12:181–5. doi: 10.1089/gte.2007.0066. [DOI] [PubMed] [Google Scholar]
- 36.Gorakshakar AC, Colah RB. Cascade screening for β-thalassemia: A practical approach for identifying and counseling carriers in India. Indian J Community Med. 2009;34:354–6. doi: 10.4103/0970-0218.58399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangnani B, Sukumaran PK, Mahadik C, Yagnik H, Telang S, Vaz F, et al. Thalassemia in Bombay: The role of medical genetics in developing countries. Bull World Health Organ. 1990;68:75–81. [PMC free article] [PubMed] [Google Scholar]
- 38.Saxena A, Phadke SR. Feasibility of thalassemia control by extended family screening in Indian context. J Health Popul Nutr. 2002;20:31–5. [PubMed] [Google Scholar]
- 39.Cao A, Rosatelli MC, Galanello R. Control of β-thalassemia by carrier screening, genetic counseling and prenatal diagnosis- the Sardinian experience. Ciba Found Symp. 1996;197:131–51. doi: 10.1002/9780470514887.ch8. [DOI] [PubMed] [Google Scholar]
- 40.Colah RB, Dastur AE, Aras A, Pawar A, Mudera V, Gorakshakar AC. Prenatal diagnosis of thalassemia in India - The first experience. J Obstet Gynaecol India. 1991;41:733–8. [Google Scholar]
- 41.Thakur (Mahadik) C, Vaz F, Banerjee M, Kapadia C, Natrajan PG, Yagnik H, et al. Prenatal diagnosis of β-thalassemia and other hemoglobinopathies in India. Prenat Diagn. 2000;20:194–201. [PubMed] [Google Scholar]
- 42.Saxena R, Jain PK, Thomas E, Verma IC. Prenatal diagnosis of β-thalassemia: Experience in a developing country. Prenat Diagn. 1998;18:1–7. [PubMed] [Google Scholar]
- 43.Gorakshakar AC, Lulla CP, Nadkarni AH, Pawar AR, Desai SN, Colah RB, et al. Prenatal diagnosis of beta thalassemia using denaturing gradient gel electrophoresis among Indians. Hemoglobin. 1997;21:421–35. doi: 10.3109/03630269708993128. [DOI] [PubMed] [Google Scholar]
- 44.Colah R, Gorakshakar A, Lu C, Nadkarni A, Desai S, Pawar A, et al. Application of covalent reverse dot-blot hybridization for rapid prenatal diagnosis of the common Indian thalassemia syndromes. Indian J Hemat Blood Transf. 1997;15:10–3. [Google Scholar]
- 45.Muralitharan S, Srivastava A, Shaji RV, Mathai M, Srivastava VM, Dennison D, et al. Prenatal diagnosis of β-thalassemia mutations using the reverse dot blot technique. Natl Med J India. 1996;9:70–1. [PubMed] [Google Scholar]
- 46.Agarwal S, Gupta A, Gupta UR, Sarwai S, Phadke S, Agarwal SS. Prenatal diagnosis in beta thalassemia: An Indian experience. Fetal Diagn Ther. 2003;18:328–32. doi: 10.1159/000071975. [DOI] [PubMed] [Google Scholar]
- 47.Garewal G, Das R, Jaur J, Marwaha RK, Gupta I. Establishment of prenatal diagnosis for β-thalassemia; a step towards control in a developing country. Ann Hum Biol. 2005;32:138–44. doi: 10.1080/03014460500075019. [DOI] [PubMed] [Google Scholar]
- 48.Bandyopadhyay A, Bandyopadhyay S, Basak J, Mondal BC, Sarkar AA, Majumdar S, et al. Profile of β-thalassemia in eastern India and its prenatal diagnosis. Prenat Diagn. 2004;24:992–6. doi: 10.1002/pd.1049. [DOI] [PubMed] [Google Scholar]
- 49. [accessed on October 5, 2010]. Globin Gene Server: http://globin.cse.psu.edu/
- 50.Edison ES, Shaji RV, Devi SG, Moses A, Viswabandhya A, Mathews V, et al. Analysis of β-globin mutations in the Indian population: presence of rare and novel mutations and regionwise heterogeneity. Clin Genet. 2008;73:331–7. doi: 10.1111/j.1399-0004.2008.00973.x. [DOI] [PubMed] [Google Scholar]
- 51.Kumar R, Tamhankar PM, Panigrahi I, Dalal A, Agarwal S. A novel β-globin mutation (HBB; c: 107 A>G; or codon 35 β(A→G) at alpha -beta chain interfaces. Ann Hematol. 2009;88:1269–71. doi: 10.1007/s00277-009-0760-4. [DOI] [PubMed] [Google Scholar]
- 52.Old JM, Varawalla NY, Weatherall DJ. The rapid detection and prenatal diagnosis of β-thalassemia in the Asian Indian and Cyproit populations in the UK. Lancet. 1990;336:834–7. doi: 10.1016/0140-6736(90)92338-i. [DOI] [PubMed] [Google Scholar]
- 53.Rao VB, Natrajan PG, Lulla CP, Bandodkar SB. Rapid mid-trimester prenatal diagnosis of beta-thalassaemia and other haemoglobinopathies using a non- radioactive anion exchange HPLC technique - an Indian experience. Prenat Diagn. 1997;17:725–31. doi: 10.1002/(sici)1097-0223(199708)17:8<725::aid-pd134>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 54.Wadia MR, Phanasgaokar SP, Nadkarni AH, Surve RR, Gorakshakar AC, Colah RB, et al. Usefulness of automated chromatography for rapid fetal blood analysis for second trimester prenatal diagnosis of beta-thalassemia. Prenat Diagn. 2002;22:153–7. doi: 10.1002/pd.251. [DOI] [PubMed] [Google Scholar]
- 55.Rao S, Saxena R, Deka D, Kabra M. Use of HbA estimation by CE-HPLC for prenatal diagnosis of beta-thalassemia; experience from a tertiary care centre in north India: a brief report. Hematology. 2009;14:122–4. doi: 10.1179/102453309X385269. [DOI] [PubMed] [Google Scholar]
- 56.Winichagoon P, Sriphanich R, Sae-Mgo WB, Chowthaworm J, Tantisirin P, Kanokpongsakdi S, et al. Application of automated HPLC in prenatal diagnosis of thalassemia. Lab Hematol. 2002;8:29–35. [Google Scholar]
- 57.Holzgreve W. Will ultrasound screening and ultrasound guided procedures be replaced by non-invasive techniques for the diagnosis of fetal chromosome anomalies? Ultrasound Obstet Gynecol. 1997;9:217–9. doi: 10.1046/j.1469-0705.1997.09040217.x. [DOI] [PubMed] [Google Scholar]
- 58.Steele CD, Wapner RJ, Smith JB, Haynes MK, Jackson LG. Prenatal diagnosis using fetal cells isolated from maternal peripheral blood.Clin. Obstet Gynecol. 1996;39:801–13. doi: 10.1097/00003081-199612000-00009. [DOI] [PubMed] [Google Scholar]
- 59.Mesker WE, Ouwerkerk-vn Velzen MC, Oosterwijk JC, Bernini LF, Golbus MS, Kanhai HH, et al. Two colour immunocytochemical staining of gamma and epsilon type hemoglobin in fetal red cells. Prenat Diagn. 1998;18:1131–7. doi: 10.1002/(sici)1097-0223(199811)18:11<1131::aid-pd426>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 60.Takabayashi H, Kuwabara S, Ukita T, Ikawa K, Yamafuji K, Igarashi T. Development of non-invasive fetal DNA diagnosis from maternal blood. Prenat Diagn. 1995;15:74–7. doi: 10.1002/pd.1970150116. [DOI] [PubMed] [Google Scholar]
- 61.Cheung MC, Goldberg JD, Kan YW. Prenatal diagnosis of sickle cell anemia and thalassemia by analysis of fetal cells in maternal blood. Nat Genet. 1996;14:264–8. doi: 10.1038/ng1196-264. [DOI] [PubMed] [Google Scholar]
- 62.Di Naro E, Ghezzi F, Vitucci A, Tannoia N, Campanale D, D’ Addario V, et al. Prenatal diagnosis of β-thalassemia using fetal erythroblasts enriched from maternal blood by a novel gradient. Mol Hum Reprod. 2000;6:571–4. doi: 10.1093/molehr/6.6.571. [DOI] [PubMed] [Google Scholar]
- 63.Kolialexi A, Vrettou C, Traeger-Synodinos J, Burgemeister R, Papantoniou N, Kanavakis E, et al. Non invasive prenatal diagnosis of β-thalassemia using individual fetal erythroblasts isolated from maternal blood after enrichment. Prenat Diagn. 2007;27:1228–32. doi: 10.1002/pd.1881. [DOI] [PubMed] [Google Scholar]
- 64.D’Souza E, Sawant PM, Nadkarni AH, Gorakshakar A, Mohanty D, Ghosh K, et al. Evaluation of the use of monoclonal antibodies and nested PCR for non-invasive prenatal diagnosis of hemoglobinopathies in India. Am J Clin Pathol. 2008;130:202–9. doi: 10.1309/1WDCPGTAJJ6A938V. [DOI] [PubMed] [Google Scholar]
- 65.Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 66.Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, et al. Quantitaive analysis of fetal DNA in maternal plasma and serum: implications for non invasive prenatal diagnosis. Am J Hum Genet. 1998;62:768–75. doi: 10.1086/301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lun FMF, Chiu RWK, Allen Chan KC, Lau TK, Leung TY, Dennis Lo YM. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–72. doi: 10.1373/clinchem.2008.111385. [DOI] [PubMed] [Google Scholar]
- 68.Li Y, Zimmermann B, Rusterholz C, Kang A, Holzgrave W, Hahn S. Size separation of circulating DNA in maternal plasma permits ready detection of fetal DNA polymorphisms. Clin Chem. 2004;50:1002–11. doi: 10.1373/clinchem.2003.029835. [DOI] [PubMed] [Google Scholar]
- 69.Chiu RW, Lau TK, Leung TK, Chow KC, Chui DH, Lo YM. Prenatal exclusion of beta thalassemia major by examination of maternal plasma. Lancet. 2002;360:998–1000. doi: 10.1016/s0140-6736(02)11086-5. [DOI] [PubMed] [Google Scholar]
- 70.Papasavva T, Kalakoutis G, Kalikas I, Neokli E, Papacharalambous S, Kyrri A, et al. Non-invasive prenatal diagnostic assay for the detection of beta thalassemia. Ann NY Acad Sci USA. 2006;1075:148–53. doi: 10.1196/annals.1368.020. [DOI] [PubMed] [Google Scholar]
- 71.Tungwiwat W, Fucharoen G, Fucharoen S, Ratanasiri T, Sanchaisuriya K, Sae-Ung N. Application of maternal plasma DNA analysis for noninvasive prenatal diagnosis of Hb E beta thalassemia. Transl Res. 2007;150:319–25. doi: 10.1016/j.trsl.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 72.Lazaros L, Hatzi E, Bouba I, Makrydimas G, Dalkalitsis N, Stefos T, et al. Non invasive first trimester detection of paternal beta globin gene mutations and polymorphisms as predictors of thalassemia risk at chorionic villus sampling. Eur J Obstet Gynecol Repord Biol. 2008;140:17–20. doi: 10.1016/j.ejogrb.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Di Naro E, Vitucci A, Grill S, Ahong XY, Holzgreve W, et al. Size fractionation of cell free DNA in maternal plasma improves the detection of a paternally inherited beta thalassemia point mutation by MALDI - TOF mass spectrometry. Fetal Diagn Ther. 2009;25:246–9. doi: 10.1159/000223442. [DOI] [PubMed] [Google Scholar]
- 74.Papasavva T, Kalikas I, Kyrri A, Kleanthous M. Arrayed primer extension for the noninvasive prenatal diagnosis of beta thalassemia based on detection of single nucleotide polymorphism. Ann N Y Acad Sci USA. 2008;1137:302–8. doi: 10.1196/annals.1448.029. [DOI] [PubMed] [Google Scholar]
- 75.Li Y, Di Naro E, Vitucci A, Zimmermann B, Holzgreve W, Hahn S. Detection of paternally inherited fetal point mutations for beta thalassemia using size fractionated cell free DNA in maternal plasma. J Am Med Assoc. 2005;293:843–9. doi: 10.1001/jama.293.7.843. [DOI] [PubMed] [Google Scholar]
- 76.Chan K, Yam I, Leung KY, Tang M, Chan TK, Chan V. Detection of paternal alleles in maternal plasma for non-invasive prenatal diagnosis in beta thalassemia: a feasibility study in southern China. Eur J Obstet Gynecol Repord Biol. 2010;150:28–33. doi: 10.1016/j.ejogrb.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 77.Lo YMD. Non invasive prenatal diagnosis in 2020. Prenat Diagn. 2010;30:702–3. doi: 10.1002/pd.2516. [DOI] [PubMed] [Google Scholar]