

SUMMARY
A20 is a potent anti-inflammatory protein that inhibits NF-κB, and A20 dysfunction is associated with autoimmunity and B-cell lymphoma. A20 harbors a deubiquitination enzyme domain and can employ multiple mechanisms to antagonize ubiquitination upstream of NEMO, a regulatory subunit of the IκB kinase complex (IKK). However, direct evidence of IKK inhibition by A20 is lacking, and the inhibitory mechanism remains poorly understood. Here we show that A20 can directly impair IKK activation without deubiquitination or impairment of ubiquitination enzymes. We find that polyubiquitin binding by A20, which is largely dependent on A20’s seventh zinc finger motif (ZnF7), induces specific binding to NEMO. Remarkably, this ubiquitin-induced recruitment of A20 to NEMO is sufficient to block IKK phosphorylation by its upstream kinase TAK1. Our results suggest a non-catalytic mechanism of IKK inhibition by A20 and a means by which polyubiquitin chains can specify a signaling outcome.
INTRODUCTION
NF-κB, a dimeric transcription factor consisting of the Rel family of proteins, controls genes involved in inflammation, immunity, and cell survival (Hayden and Ghosh, 2008). Under basal conditions, NF-κB is sequestered in the cytoplasm by inhibitor of NF-κB (IκB). Stimulation of cells with any of a multitude of agents, including inflammatory cytokines and toll-like receptor (TLR) ligands, induces IκB phosphorylation by the IκB kinase (IKK). Phosphorylated IκB is ubiquitinated, then degraded by the proteasome, liberating NF-κB to enter the nucleus and regulate gene expression.
Best known for its role in targeting protein degradation by the proteasome, ubiquitin also has non-degradative functions, including regulation of IKK (Chen and Sun, 2009). Indeed, binding of the TNF receptor (TNFR), IL-1 receptor (IL-1R), and many TLRs to their respective ligands results in activation of ubiquitin ligases. In the IL-1R and many TLR pathways, TNF receptor-associated factor 6 (TRAF6) is the major ubiquitin ligase. TRAF6 works with the dimeric ubiquitin-conjugating enzyme UBC13/UEV1A to synthesize polyubiquitin chains linked through Lys 63 (K63) of ubiquitin (Deng et al., 2000). Indeed, the E3 ligase activity of TRAF6, the catalytic activity of UBC13, and K63 of ubiquitin are required for IL-1β-induced IKK activation (Lamothe et al., 2008; Xu et al., 2009). In the TNFR pathway, numerous ubiquitin ligases are recruited, including TRAF2, cellular inibitor of apoptosis (cIAP) 1 and 2, and linear ubiquitin chain assembly complex (LUBAC) (Bianchi and Meier, 2009; Haas et al., 2009). The linkage of polyubiquitin chains in this pathway remains enigmatic, and it is unclear why so many ubiquitin ligases, and potentially different polyubiquitin linkages, are involved. Nevertheless, polyubiquitin chains clearly play an essential role in TNFα-induced IKK activation. In the TNFR pathway, receptor-interacting protein kinase 1 (RIP1) is a key ubiquitination substrate (Ea et al., 2006; Wu et al., 2006). In the IL-1R/TLR4 pathway, several proteins, including IL-1R associated kinase 1 (IRAK1) and TRAF6, are known to be ubiquitinated, but only unanchored polyubiquitin chains have been shown to directly activate TAK1 and IKK (Xia et al., 2009). The polyubiquitin chains in each pathway bind to the regulatory subunits of the TGFβ-activated kinase (TAK1) and IKK complexes, TAB2 and NEMO, respectively, and this binding leads to TAK1 and IKK activation (Ea et al., 2006; Kanayama et al., 2004; Laplantine et al., 2009; Wu et al., 2006). Activated TAK1 phosphorylates IKK, as well as MAP kinase kinases such as MKK6, promoting activation of IKK and MAP kinase signaling pathways (Wang et al., 2001).
A20 is a potent suppressor of the NF-κB signaling pathways, and A20 deficiency in mice results in excessive NF-κB activity and multiorgan inflammation (Boone et al., 2004; Lee et al., 2000). Recent evidence also implicates dysfunction of A20 as a risk factor for human disease. Polymorphisms in the A20 locus are associated with multiple autoimmune diseases including systemic lupus erythematosis, and A20 was recently identified as a tumor suppressor in B-cell lymphoma (Compagno et al., 2009; Kato et al., 2009; Musone et al., 2008; Schmitz et al., 2009; Vereecke et al., 2009). A20 has an N-terminal ovarian tumor (OTU) domain and seven C-terminal zinc finger (ZnF) motifs. The OTU domain can deubiquitinate RIP1, and the ZnF region can act as an E3 ligase to add K48 polyubiquitin chains to RIP1, promoting its proteasomal degradation (Wertz et al., 2004). A20 also promotes disassembly of ubiquitination complexes in the IL-1R and TNFR pathways, including TRAF6-UBC13; cIAP1/2-UBC13; and cIAP1/2-UBCH5, as well as proteasomal degradation of UBC13 and UBCH5 (Shembade et al., 2010). Both deubiquitination and disassembly of E2-E3 complexes require A20’s catalytic Cys 103 residue within the OTU domain. A20 has also been reported to block recruitment of the adaptor proteins TRADD and RIP1 to TNFR (He and Ting, 2002), and to promote lysosomal degradation of TRAF2 (Li et al., 2009). Although the relative contributions of deubiquitination, degradation of upstream signaling proteins, and disruption of ubiquitination complexes remain unclear, A20 appears to employ multiple mechanisms that could potentially reduce the amount of polyubiquitin chains available to interact with TAB2 and NEMO.
Yet certain data are difficult to reconcile with the proposed catalytic mechanisms. For example, in vitro, A20’s OTU domain readily disassembles K48 polyubiquitin chains, but only weakly disassembles K63 polyubiquitin chains (Komander and Barford, 2008; Lin et al., 2008). In addition, overexpression of A20 mutants lacking the catalytic Cys 103 residue inhibits NF-κB, suggesting a non-catalytic mechanism (Evans et al., 2004; Song et al., 1996). Furthermore, there is no direct evidence that A20 reduces the amount of polyubiquitin chains available to interact with TAB2 and NEMO.
We found evidence that A20 can inhibit IKK through a mechanism that cannot be accounted for by reducing the interaction of polyubiquitin chains with NEMO. We found that polyubiquitin binding, which is largely dependent on ZnF7, is required for IKK inhibition. Polyubiquitin chains non-covalently recruit A20 to NEMO, forming a complex involving specific NEMO-A20 interaction. Remarkably, when bound to the NEMO-polyubiquitin complex, A20 directly impairs phosphorylation of IKK by TAK1. Importantly, the catalytic Cys 103 residue of A20 is dispensable for this mechanism of inhibition. Our results therefore suggest a direct, non-catalytic mechanism of IKK inhibition by A20 and demonstrate that polyubiquitin chains can induce specific binding among proteins to which ubiquitin is not covalently attached.
RESULTS
Overexpressed A20 Impaired IKK Activation Without Reducing RIP1 Ubiquitination
As a tool to dissect the mechanism of IKK inhibition by A20, we stably overexpressed Flag-A20 in a HEK293 cell line that expresses IL-1R. In this cell line, IKK activation by TNFα was almost completely abolished, as evidenced by the lack of IκBα degradation and the failure of immunoprecipitated IKK complex to phosphorylate IκBα (Figure 1A). To test whether IKK inhibition in this cell line was caused by disruption of the TNFR complex, we stimulated cells with GST-TNFα, then pulled down the TNFR complex using glutathione-Sepharose and immunoblotted for TRADD and RIP1. Surprisingly, TRADD and RIP1, including a high molecular weight smear of RIP1 indicative of ubiquitination, were enriched on TNFR in A20-overexpressing cells compared to parental cells (Figure 1B). Notably, A20 was also recruited to this complex. We also immunoprecipitated NEMO from TNFα-treated cells and compared the abundance of ubiquitinated RIP1 that interacted with NEMO in A20-overexpressing vs. parental cells. Remarkably, NEMO co-immunoprecipitated more ubiquitinated RIP1 from the A20-overexpressing cells, and A20 was also present in this complex (Figure 1C). Thus, overexpressed A20 was recruited to the NEMO-RIP1 complex and inhibited IKK activation, apparently without deubiquitinating RIP1, causing degradation of RIP1, inhibiting ubiquitination of RIP1, or disrupting the interaction of NEMO with polyubiquitinated RIP1. Although a previous study showed A20-dependent deubiquitination and degradation of RIP1 (Wertz et al., 2004), perhaps this outcome was dependent on cell types and other experimental conditions.
Figure 1. A20 inhibits TNFα-induced IKK activation without antagonizing RIP1 ubiquitination.

(A) HEK293 cells stably overexpressing A20 and the parental (WT) HEK293 cells were stimulated with TNFα for the indicated time, then IKK complex was immunoprecipitated with a NEMO antibody for kinase assay using GST-IκBα N-terminus and γ-32P-ATP as substrates. Immunoprecipitates and cell lysates were immunoblotted with the indicated antibodies. N.S.: non-specific. (B) TNF receptor complexes were pulled down using glutathione Sepharose following stimulation of cells with GST-TNFα, and the proteins in the complexes were detected by immunoblotting with the indicated antibodies. (C) Similar to (B), except that a NEMO antibody was used for immunoprecipitation. (D) Parental HEK293 cells (WT) or HEK293 cells stably expressing A20 WT or A20 C103A with N-terminal TAP tag and C-terminal Flag tag were treated with TNFα for the indicated timepoints. Cell lysates were used for immunoblotting with the indicated antibodies.
To determine if the DUB activity of A20 is important for IKK inhibition, we established stable cell lines in which wild-type A20 (WT) and the catalytically inactive A20 (C103A) were overexpressed to a similar extent. In this case, the construct used for A20 expression contained an N-terminal tandem affinity purification (TAP) tag, consisting of Protein A; TEV Protease cleavage site; then calmodulin binding peptide, as well as a C-terminal Flag tag. Supporting the idea that IKK inhibition by A20 does not require deubiquitination, IκBα degradation did not occur when either A20 WT or C103A was overexpressed (Figure 1D). Notably, immunoblots for UBC13 and UBC5 showed no change in their abundance regardless of A20 expression level, suggesting that the IKK inhibition observed in these cells was not due to degradation of these E2 enzymes.
A20 Inhibits IKK Downstream of TRAF6 Through a Non-Catalytic Mechanism
To further dissect the mechanism of IKK inhibition by A20, we tested whether purified A20 protein could inhibit IKK activation by TRAF6 in a cell-free system (S100) (Deng et al., 2000). We purified A20 WT and C103A from the cell lines described above, in which A20 has an N-terminal TAP tag and C-terminal Flag tag, by two-step affinity purification (IgG-Sepharose followed by anti-Flag antibody Sepharose) (Figure 2A, left). Consistent with cell-based assays (Figure 1D), both A20 WT and C103A impaired IKK activation by TRAF6 in the cell-free system (Figure 2A, right).
Figure 2. Cys 103 is dispensable, while ZnF7-dependent polyubiquitin binding is important for IKK inhibition by A20.

(A) Left, silver stain of two-step affinity-purified A20. HSP70, 70kDa heat shock protein. Right, different amounts of purified A20 (WT and C103A) were incubated with HeLa S100, followed by addition of TRAF6 and ATP and further incubation for 1 hr. IKK activation in S100 was measured by immunoblotting for IκBα. (B) Domain architecture of A20, including relative locations of point mutations. OTU: ovarian tumor. ZnF: zinc finger. (C–E) Flag-A20 mutants were expressed in HEK293 cells and affinity purified. After incubation with HA-tagged polyubiquitin chains, A20 proteins were immunoprecipitated with a Flag antibody and ubiquitin chains were immunoblotted with an HA antibody. (F) IKK activation in S100 was measured as in (A). CC(AA): C779A/C782A. FG(AA): F770A/G771A. (G) MEF cells were infected with retrovirus expressing either GST or Flag-A20 WT, C103A, or F770A/G771A. The cells were stimulated with IL-1β, and then IKK activity was measured as in Figure 1A. Fold activation indicates activity normalized to the activity of unstimulated WT (A20+/+) cells (lane 1).
ZnF7-Dependent Polyubiquitin Binding is Important for IKK Inhibition
To elucidate this non-catalytic mechanism of IKK inhibition, we considered alternative biochemical properties of A20. A recent study found that A20 ZnF4 can bind to K63 polyubiquitin chains, and that this binding is important for NF-κB inhibition (Bosanac et al., 2010). Yet the potential contribution of other regions of A20 to polyubiquitin binding has not been addressed. To assess polyubiquitin binding, we mixed partially purified Flag-A20 with K63 polyubiquitin chains synthesized by TRAF6 and UBC13/UEV1A in which some of the ubiquitin has an HA tag, then performed Flag immunoprecipitation followed by HA immunoblotting. A mutant lacking the ovarian tumor domain (ΔOTU), but not one lacking the seven ZnF motifs (ΔZnF), bound to polyubiquitin chains as well as A20 WT (Figure 2B, C). To identify specific ZnFs involved in polyubiquitin binding, mutants lacking one or more ZnFs (ZnF 2–7, 3–7, 4–7, 5–7, 6–7, 1–6, 1–5, and 1–4) were tested. Interestingly, a substantial binding defect was observed in a mutant lacking only ZnF7 (Figure 2D, lane 3). ZnF4-7 and ZnF5-7, but not ZnF7 alone or ZnF1-5, were sufficient to bind polyUb, indicating that multiple ZnFs contribute to polyUb binding and neither ZnF4 nor ZnF7 was sufficient for this binding.
Co-crystallization of the ZnF motif of Rab5 guanine nucleotide exchange factor (RABEX-5) with ubiquitin revealed ZnF residues that interact with ubiquitin, including Tyr 26 and Gly 27 (Lee et al., 2006; Mattera et al., 2006; Penengo et al., 2006). We mutated the corresponding A20 ZnF7 residues Phe 770 and Gly 771, or the highly conserved Cys 779 and Cys 782, to Ala, and compared them to A20 WT or ΔZnF7. A20 ΔZnF7, C779A/C782A, and F770A/G771A had an equally severe impairment in polyubiquitin binding (Figure 2E). Importantly, each of these mutants was defective in IKK inhibition in S100 (Figure 2F), suggesting that ZnF7’s contribution to polyubiquitin binding is important for IKK inhibition in vitro.
Retrovirus-mediated expression of A20 WT or C103A in A20-deficient MEFs (A20−/−) limited IL-1β-induced IKK activation to a level similar to that observed in wild type (A20+/+) MEFs (Figure 2G, lanes 1–16; Figure S1A). By contrast, A20 F770A/G771A failed to inhibit IKK activation (Figure 2G, lanes 17–20). Similarly, for TNFα-induced IKK activation, expression of A20 WT or C103A largely rescued A20 function in A20−/− MEFs, while A20 F770A/G771A was defective (Figure S1B). Thus, in the first few minutes of ligand detection in MEF cells, Cys 103 is dispensable for limiting inflammatory cytokine-induced IKK activation, while ZnF7-dependent polyubiquitin binding is important.
To further test the roles of Cys 103 and polyUb binding for NF-κB downregulation in cells, we established stable cell lines wherein A20 WT, C103A, F770A/G771A (ZnF7*), or Y614A/F615A (ZnF4*) were expressed at similar levels in A20−/− MEFs (Figure 3A). The Y614A/F615A mutation was previously shown to impair Ub binding and NF-κB downregulation (Bosanac et al., 2010). We used RT-PCR to measure expression of two NF-κB target genes, interleukin-6 (IL-6) and cyclooxygenase-2 (COX-2) before or after stimulation of these cells for 3 hr with TNFα. Not surprisingly, A20−/− MEFs expressing GFP as a control had much higher levels of IL-6 and COX-2 mRNA than A20+/+ MEFs expressing GFP, and expression of A20 WT largely suppressed IL-6 and COX-2 expression (Figure 3B). Expression of IL-6 and COX-2 in cells with A20 C103A was higher than what was observed in cells with A20 WT, but substantially less than that in cells with GFP. This suggests that downregulation of NF-κ B by A20 is partially dependent on Cys 103. Importantly, IL-6 and COX-2 expression was significantly elevated in cells expressing either A20 ZnF7* or ZnF4*, confirming the key role of polyUb binding for suppressing NF-κB activity.
Figure 3. The roles of A20’s catalytic Cys 103 residue and ubiquitin binding in downregulation of NF-κB activity.

(A) Lysates from cells stably expressing A20 or GFP as a control were examined for A20 expression by immunoblotting. (B) Cells were untreated or treated with TNFα for 3 hr. RNA was extracted, and RT-PCR was performed to measure the relative expression of IL-6 or COX-2 as described in Experimental Procedures. Error bars indicate the standard error of the mean (SEM) among triplicates for each sample. Each graph is representative of three independent experiments. (C & D) Cells were treated for the indicated times with TNFα. Cell lysates were immunoblotted with the indicated antibodies; or immunoblotting was performed following immunoprecipitation with a NEMO antibody (D).
Importantly, no change was seen in the abundance of RIP1, UBC13, or UBC5 over the three-hour TNFα treatment regardless of whether A20 WT or mutants were expressed (Figure 3C). Thus, over this time period, A20 did not appear to affect the overall stability of these proteins. To see whether TNFα-induced RIP1 ubiquitination was affected by A20 in these cell lines, we immunoprecipitated NEMO from untreated or TNFα-treated cells and immunoblotted for RIP1. A20+/+ MEFs and A20−/− MEFs expressing A20 WT had ubiquitinated RIP1 conjugates with molecular weights slightly lower than those in A20−/− MEFs expressing GFP or A20 mutants (Figure 3D). This suggests that, in these cells, A20 WT is having some effect on RIP1 ubiquitination status. Nevertheless, A20 WT and C103A each appeared to impair TNFα-induced IKK phosphorylation as compared to A20−/− cells expressing GFP, whereas the ZnF7 and ZnF4 Ub-binding mutants did not (Figure 3D; compare lanes 8, 11, 14 & 17). Therefore RIP1 ubiquitination status appears to be not entirely predictive of IKK activation, i.e., A20 C103A impaired IKK activation even though it did not reduce RIP1 ubiquitination (lanes 10–12).
Polyubiquitin Chains Induce NEMO-A20 Interaction
A20 was previously found to interact with NEMO in a yeast two-hybrid screen and when both proteins were overexpressed in mammalian cells (Zhang et al., 2000). Given that A20 and NEMO are each recruited to polyubiquitin chains, we reasoned that their proximity might promote interaction with one another. Indeed, endogenous NEMO and A20 co-immunoprecipitated following IL-1β stimulation of HeLa cells with kinetics that correlated with IKK activity (Figure 4A). Similarly, TNFα stimulation of HEK293 cells induced interaction between NEMO and A20, concomitant with IKK activation (Figure S2A, lanes 1–4). This correlation suggested that IKK might promote NEMO-A20 interaction, which could explain how phosphorylation of A20 by IKK promotes NF-κB suppression (Hutti et al., 2007). However, to our surprise, the IKK inhibitor TPCA-1 enhanced TNFα-induced NEMO-A20 interaction (Figure S2A, lanes 5–8), suggesting that the kinase activity of IKK impairs NEMO-A20 interaction, while an alternative signal induces it. To identify this signal, NEMO-A20 interaction was examined in HeLa S100. The combination of TRAF6 and TPCA-1, or an alternative IKK inhibitor BMS-345541, induced NEMO-A20 interaction (Figures 4B and S2B). Addition of the deubiquitination enzyme vOTU (OTU domain from Crimean Congo Hemorrhagic Fever Virus L1 protein (Frias-Staheli et al., 2007)) abolished this interaction (Figure 4C, compare lanes 3 & 6). Depletion of UBC13 from S100 also abolished NEMO-A20 interaction, which was rescued by adding back recombinant His6-Ubc13 (Figure 4C, compare lanes 9 & 12). Thus, TRAF6/UBC13-synthesized polyubiquitin chains induce NEMO-A20 interaction. In support of this notion, A20 WT, but not the polyubiquitin-binding mutant F770A/G771A, co-immunoprecipitated with NEMO from S100 in a TRAF6-dependent manner (Figure 4D). NEMO’s interaction with A20 was selective, because under the same conditions it did not interact with the proteasomal subunit S5A, which also bound to polyubiquitin chains (Figure S2C, right panel).
Figure 4. Polyubiquitin chains induce NEMO-A20 interaction.

(A) HeLa cells were stimulated with IL-1β for the indicated time, then immunoprecipitation was performed with a NEMO antibody. Immunoprecipitate was used to measure IKK activity as in Figure 1A, or for immunoblotting with an A20 antibody. (B) S100 was incubated with ATP, −/+ TRAF6, −/+ the IKK inhibitor TPCA-1 (10 μM), for 1 hr. A NEMO antibody was used for immunoprecipitation. Proteins in the S100 and immunoprecipitate were detected by immunoblotting with the indicated antibodies. (C) As in (B), following depletion of UBC13 and/or addition of vOTU or (D) A20 WT or F770A/G771A was added to S100. Experiment was His6-UBC13. performed as in (B) but without TPCA-1. (E) shUb-Ub (WT) or shUb-Ub (K63R) cells were untreated or treated with 1 μg/ml tetracycline for 4 days. Cells were then treated with TPCA-1 (20 μM) for 3 hr to enhance NEMO-A20 association. Cells were stimulated with IL-1β for the indicated times, then a NEMO antibody was used for immunoprecipitation. Proteins in the S100 and immunoprecipitate were detected by immunoblotting with the indicated antibodies. (F) MEF stable cell lines were treated for the indicated times with TNFα. Cell lysates were used for immunoprecipitation with a NEMO antibody. Cell lysate and immunoprecipitate were immunoblotted with the indicated antibodies.
We next examined NEMO-A20 interaction in human osteosarcoma cells (U2OS) in which ubiquitin is knocked down by tetracycline-inducible shRNA, and replaced with tetracycline inducible RNAi-resistant WT or K63R Ub (Xu et al., 2009). IL-1β-induced interaction between NEMO and A20 was reduced in cells expressing the K63R mutant compared to cells expressing WT Ub (Figure 4E), indicating that K63 polyubiquitination provides a signal for NEMO-A20 interaction in cells.
In further support of the role of polyubiquitin in promoting signal-dependent interaction between A20 and NEMO in cells, we found that A20 WT, but not the ZnF7 or ZnF4 Ub-binding mutants, was co-immunoprecipitated with NEMO following TNFα stimulation of MEF cells stably expressing the A20 proteins (Figure 4F).
Polyubiquitin Chains Directly and Non-Covalently Induce Binding of A20 to NEMO
To determine if polyubiquitin chains directly promote NEMO-A20 interaction, we incubated highly purified K63 polyubiquitin chains, recombinant GST-NEMO, and A20, then performed GST pull down analyses. These polyubiquitin chains had been synthesized by recombinant E1, UBC13/UEV1A, and TRAF6, each of which was His6-tagged. The polyubiquitin chains were subsequently separated from the ubiquitination enzymes by nickel depletion and gel filtration (Figure S3A–C). NEMO and A20 did not detectably interact in the absence of polyubiquitin chains, but robustly interacted in their presence (Figure 5A). Importantly, A20 did not impair the interaction between NEMO and polyubiquitin chains, even when A20’s molar concentration was roughly 6-fold greater than that of NEMO (lane 8). This result suggests that non-covalent binding to polyubiquitin chains directly induces NEMO-A20 interaction. To confirm that the polyubiqutin chains inducing NEMO-A20 interaction were indeed “free,” or unanchored, we treated them with the deubiquitination enzyme Isopeptidase T (IsoT), which is specific for unanchored polyubiquitin chains (Reyes-Turcu et al., 2006). IsoT treatment destroyed the vast majority of polyubiquitin chains, and abolished the NEMO-A20 interaction (Figure 5B), supporting the idea that unanchored polyubiquitin chains can induce NEMO-A20 interaction. In contrast to long polyubiquitin chains, tetraubiquitin (Ub4) of K63-, K48-, or linear linkage did not induce NEMO-A20 interaction, nor did it affect NEMO-A20 interaction in the presence of long polyubiquitin chains (Figure S3E–G). It should be noted that, under the experimental conditions used here, NEMO bound to long polyubiquitin chains much more robustly than to Ub4 (Figure S3F). Therefore, the inability of Ub4 to affect NEMO-A20 binding may simply reflect the relatively low affinity between NEMO and short polyubiquitin chains.
Figure 5. Polyubiquitin chains directly and non-covalently induce specific binding between NEMO and A20.

(A) GST or GST-NEMO was incubated with affinity-purified A20 in the presence or absence of K63 polyubiquitin chains. After 15 min, 10% of input was withdrawn, and the remaining mixture was incubated with Glutathione Sepharose. Input and glutathione pull-down were immunoblotted using the indicated antibodies. (B) as in (A) except that, in lanes 6 & 7, polyUb was treated with isopeptidease T (IsoT). (C) As in (A), but including GST-NEMO ΔN. (D) Model of the polyubiquitin-NEMO-A20 complex. NT: N-terminus. NUB: NEMO ubiquitin binding. ZnF: zinc finger. Ub: ubiquitin.
In a yeast two-hybrid screen, it was found that A20’s interaction with NEMO required a region of NEMO encompassing residues 95–218, which includes its first coiled coil domain (Zhang et al., 2000). On the other hand, polyubiquitin chains bind to NEMO’s C-terminus, which contains two ubiquitin binding domains in tandem (Ea et al., 2006; Laplantine et al., 2009; Wu et al., 2006). This suggests that distinct regions of NEMO mediate interaction with A20 and polyubiquitin chains. To test this idea, we purified a fragment of NEMO lacking the first coiled coil domain but containing the ubiquitin-binding domains (251–419, or NEMO ΔN). NEMO ΔN did not bind A20 even in the presence of polyubiquitin chains (Figure 5C, lanes 7–9). In fact, A20 reduced the binding of NEMO ΔN to polyubiquitin chains (lane 9), suggesting that A20 and NEMO ΔN compete for polyubiquitin binding, whereas A20 and full-length NEMO cooperate to form a ternary complex in the presence of polyubiquitin. The formation of this complex involves not only the binding of NEMO and A20 to polyubiquitin chains, but also a domain within NEMO’s N-terminus, presumably because this domain interacts directly with A20 (Figure 5D).
If this complex involves direct interaction between NEMO and A20, it is expected that other polyubiquitin-binding proteins will not bind to NEMO under these conditions, assuming they lack an interface for binding to NEMO. Indeed, interaction was not detected between NEMO and recombinant TAB2 in the presence or absence of polyubiquitin chains; rather, TAB2 at high concentration appeared to compete with NEMO for binding to polyubiquitin chains (Figure S3D, lane 14). These results suggest that polyubiquitin chains can directly and non-covalently induce specific binding between NEMO and A20 (see Discussion).
A20 Prevents IKK Phosphorylation by TAK1
The signal-induced recruitment of A20 to NEMO suggests that A20 might directly regulate IKK. However, the activity of IKK complex immunoprecipitated from TNFα-stimulated cells was unaffected by A20 in vitro (Figure 6A). In fact, removal of polyubiquitin chains from NEMO-associated RIP1 by vOTU did not affect IKK activity, whereas dephosphorylation by lambda phosphatase abolished it (Figure 6B). Thus, IKK phosphorylation appears to be the primary determinant of its kinase activity.
Figure 6. A20 inhibits IKK phosphorylation by TAK1 through a direct, non-catalytic mechanism.

(A) HEK293 cells were untreated or treated with TNFα for 10 min, and a NEMO antibody was used for immunoprecipitation. Immunoprecipitate was incubated with affinity-purified A20, then washed and incubated with γ–32P-ATP and GST-IκBα N-terminus or immunoblotted with the indicated antibodies. (B) as in (A), but NEMO immunoprecipitate was treated with vOTU or lambda phosphatase. Immunoprecipitate was immunoblotted for RIP1, p-IKK, or IKKβ. Aliquots of the immunoprecipitates were also assayed for IKK activity by measuring IκBα phosphorylation. (C) Method to generate constitutively active TAK1 (TAK1-ca). (D) TAK1 or TAK1-ca was incubated with IKK complex (top) or His6-MKK6 (K82A) (bottom), −/+ polyubiquitin chains and ATP. Samples were immunoblotted for p-IKK or p-MKK6. (E) Top and middle: as in (D), except that A20 was included in the reactions. Bottom: MEKK1 was used to activate IKK. (F) as in (D) top, except that the reactions included A20 WT, C103A, or C103A/ΔZnF7.
We then tested whether A20 could prevent IKK phosphorylation, using an assay in which IKK complex is activated by TAK1 in the presence of unanchored K63 polyubiquitin chains (Xia et al., 2009). To uncouple the processes of TAK1 and IKK activation, TAK1 was first activated by polyubiquitin chains, which were subsequently destroyed using vOTU, then the TAK1 complex was affinity purified (Figure 6C). Phosphorylation of MKK6 by this constitutively active TAK1 (TAK1-ca) was no longer subject to regulation by polyubiquitin chains (Figure 6D, bottom). Interestingly, phosphorylation of IKK by TAK1-ca was significantly enhanced by polyubiquitin chains (Figure 6D, top; compare lanes 5 & 6), indicating that polyubiquitin chains are required not only for TAK1 activation, but also for the phosphorylation of IKK by active TAK1. Remarkably, A20 had no effect on phosphorylation of MKK6 by TAK1-ca, but impaired phosphorylation of IKK (Figure 6E, top and middle), indicating that A20 can impair IKK phosphorylation without affecting TAK1 activity per se. As a control, phosphorylation of IKK by the kinase MEKK1, which does not require ubiquitin, was unaffected by A20 (Figure 6E, bottom). Importantly, A20 C103A impaired IKK phosphorylation as potently as A20 WT, whereas further deletion of ZnF7 attenuated this inhibitory activity (Figure 6F). These results provide direct evidence that A20 inhibits IKK by preventing the phosphorylation of IKK by TAK1.
We have previously shown that purified IKK complex could be activated by polyubiquitin chains synthesized by TRAF6 and UBC5 through a mechanism involving the binding of NEMO to polyUb, which in turn induces IKK autophosphorylation (Xia et al., 2009). Interestingly, A20 blocked IKK autophosphorylation in a manner independent of Cys 103, and this inhibition was impaired by deletion of A20’s 7th ZnF (Figure S4A, B). Importantly, the A20:NEMO molar ratio required for IKK inhibition was roughly 1:1 (lanes 3–4). Yet, even at an A20:NEMO ratio of roughly 5:1, A20 did not impair the binding of NEMO to polyUb (Figure S4C). Therefore, formation of the polyubiquitin-NEMO-A20 complex is sufficient to impair IKK activation.
Notably, we found that A20 could also impair TAK1 activation by K63-linked polyubiquitin chains, as measured by TAK1 autophosphorylation and phosphorylation of MKK6 (Figure S4D). As was the case with IKK, impairment of TAK1 activation did not require deubiquitination, but was attenuated when ZnF7 was mutated. These results suggest that A20 can also limit the activation of TAK1 in a manner dependent on polyubiquitin binding, which may promote the interaction between A20 and TAB2.
Taken together, our results suggest that A20 can inhibit autophosphorylation of TAK1, phosphorylation of IKKβ by TAK1, as well as autophosphorylation of IKK, all of which involve polyubiquitin binding to TAB2 or NEMO. Through its ability to bind simultaneously to polyubiquitin chains and NEMO, and possibly TAB2, A20 limits the activation of IKK and TAK1.
DISCUSSION
Direct, Non-Catalytic Impairment of IKK Activation by A20
Our results suggest a mechanism by which A20 inhibits IKK without deubiquitination or impairment of ubiquitination enzymes. We propose that, within the first few minutes of ligand detection, A20 forms a complex with polyubiquitin chains and NEMO and non-catalytically limits the extent of IKK activation by TAK1 (Figure 7). Indeed, we present multiple lines of evidence that A20 inhibits IKK through a non-catalytic mechanism. First, when overexpressed, A20 inhibits TNFα-induced IKK activation without impairing ubiquitination of RIP1 or the interaction between ubiquitinated RIP1 and NEMO (Fig. 1). Second, in cell-free systems that recapitulate the IL-1R signaling pathway, the C103A mutant of A20 is as potent as A20 WT in inhibition of IKK (Fig. 2A; Fig. 6F). Finally, A20 C103A inhibits IKK activation when expressed in A20−/− MEFs (Fig. 2G; Fig. S1). Our measurements of NF-κB target gene expression suggest that the majority of NF-κB downregulation by A20 does not require the catalytic Cys 103 residue, although the C103A mutant was partially defective (Figure 3B). Of course, elucidation of the specific role of Cys 103 in mammalian physiology will likely require generation of a knockin mouse harboring mutation of this residue.
Figure 7. Model of the non-catalytic mechanism by which A20 limits IKK activation.
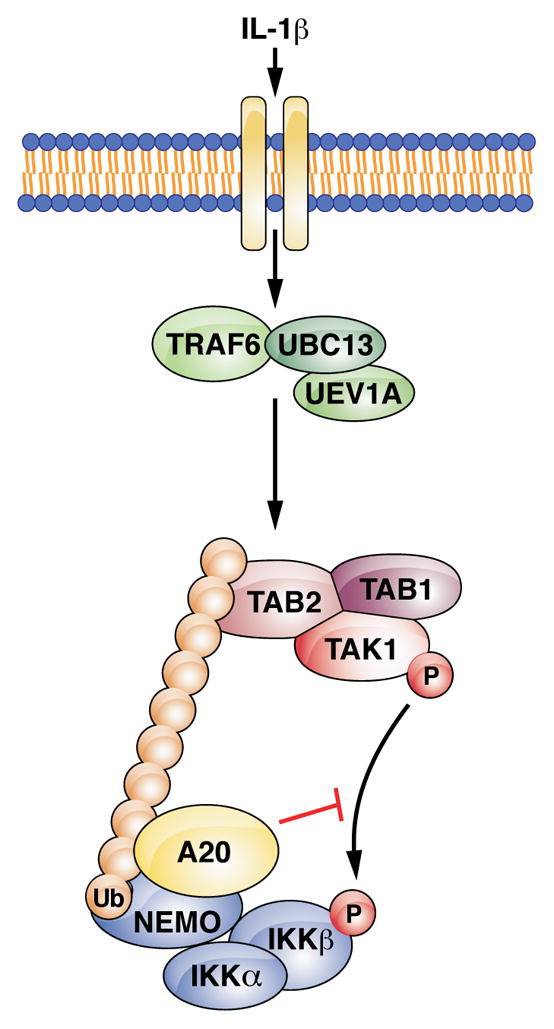
Binding of IL-1β to IL-1R activates the E3 ligase TRAF6, which works with UBC13 to synthesize K63-linked polyubiquitin chains. The TAK1 and IKK complexes are recruited through ubiquitin binding domains on TAB2 and NEMO, respectively, allowing IKK phosphorylation by TAK1. A20 is recruited to NEMO through bipartite binding to polyubiquitin chains and an N-terminal region of NEMO. Formation of this complex blocks IKK phosphorylation by TAK1, thereby inhibiting IKK.
Our results suggest that multiple A20 ZnFs contribute to polyubiquitin binding, including a prominent contribution by ZnF7 (Figure 2C). Consistent with a key role for ZnF7, mutations in this motif that impair polyubiquitin binding reduce A20’s ability to inhibit IKK in vitro and in reconstituted A20−/− MEFs (Figures 2E–G, S1), and to suppress NF-κB target gene expression (Figure 3B). It is noteworthy that among the mutations in the A20 gene (TNFAIP3) identified in B-cell lymphoma patients, the majority results in premature stop codons or frameshifts that preclude synthesis of some or all of the ZnFs (Compagno et al., 2009; Kato et al., 2009; Schmitz et al., 2009). Although their locations vary widely, almost all of these mutations occur prior to ZnF7. Our results suggest that each of these mutations causes a polyubiquitin-binding defect, which is sufficient to cause IKK dysregulation, although other biochemical properties of A20 affected by the mutations should also be considered.
Our results indicate that an outcome of polyubiquitin binding by A20 is its recruitment to NEMO. Binding between A20 and NEMO was initially reported more than a decade ago (Zhang et al., 2000), but the relevance of this interaction, and its potential regulation, have not been clearly defined. We found that A20 is recruited to NEMO by polyubiquitin chains, and when bound, can impair IKK activation. Thus, NEMO can receive both positive and negative IKK-regulatory signals. Remarkably, K63 polyubiquitin chains not only serve as a signal for the activation of TAK1 and IKK, but also as a signal for the recruitment of A20 to NEMO to limit IKK activation, suggesting a ‘built-in brake’ mechanism that tightly controls NF-κB activation.
Consistent with what was reported by Wertz et al. (2004) and Bosanac et al. (2010), we have found that A20’s C-terminal ZnF region can catalyze polyUb synthesis in the presence of E1 and UBC5, and this activity was impaired by mutation of ZnF4 (Figure S5, lanes 2 and 4). Interestingly, this activity was also impaired by mutation of ZnF7 (F770A/G771A) (lanes 2 and 3). This result suggests an unexpected role for ZnF7 Ub binding in A20’s E3 ligase activity. Because these mutations also impair A20’s recruitment to NEMO following TNFα detection (Figure 4F), it is difficult to delineate the cause of impaired NF-κB downregulation by the A20 ZnF7 and ZnF4 mutants in vivo (Figure 3B). Although we cannot rule out a role for A20 E3 ligase activity in downregulation of NF-κB in vivo, we did not detect an A20-dependent decrease in the stability of RIP1, UBC13, or UBC5 in the time periods we examined (Figure 3C). Given the ability of A20 to directly impair IKK phosphorylation when bound to NEMO in vitro, as well as the rapid recruitment of A20 to NEMO that correlates with IKK inhibition in cells, we think it is likely that direct, non-catalytic impairment of IKK activation plays an important role.
A previous study showed that A20 is recruited to E2–E3 ubiquitination enzymes such as UBC13 and TRAF6, leading to disruption of the E2–E3 interaction (Shembade et al., 2010). This recruitment, which is dependent on A20’s Cys 103 residue, appears to occur after IKK activity has declined from its peak, and may therefore serve to further reduce IKK activity towards its basal level and to desensitize the pathway to additional upstream stimulation.
In cells, recruitment of A20 to NEMO, TRAFs and RIP1 is facilitated by a multitude of A20-interacting proteins, including ABINs, TAX1BP1, ITCH, and RNF11 (Iha et al., 2008; Mauro et al., 2006; Shembade et al., 2007; Shembade et al., 2008; Shembade et al., 2009). We found evidence that IKK activity can limit the extent of A20 recruitment to NEMO. It seems plausible that IKK cooperates with the A20-regulatory proteins to regulate the timing and magnitude of A20 recruitment. In principle, it is logical to limit recruitment of A20, because if recruited in excess, A20 does not allow IKK activation (Figure 1A). Thus, IKK may enable its own activation by imposing a limit on the extent of A20 recruitment, and the function of the A20-interacting proteins might be to “release” some A20 from this limitation to allow its interaction with polyubiquitin chains, NEMO, and potentially other upstream factors. Further studies are required to elucidate the mechanism(s) by which IKK and the A20-interacting proteins regulate the function of A20. In any case, it appears that ubiquitin-mediated recruitment of A20 to NEMO is sufficient to impair IKK activation through a non-catalytic mechanism.
A Basis For Specificity In Ubiquitin-Mediated Signal Transduction
We have identified a complex between K63 polyubiquitin chains, A20, and NEMO. Remarkably, the ubiquitin binding domain of NEMO is insufficient for complex formation. Rather, the N-terminus of NEMO is required, presumably because it makes direct contact with A20. A ternary complex of a similar nature was previously described (Kang et al., 2007). In this case, one UIM domain of the proteasome subunit S5A binds to the ubiquitin-like domain of hHR23a, while the other UIM domain of S5A and the UBA domain of hHR23a bind to a K48-linked polyubiquitin chain. This interaction may facilitate delivery of K48 polyubiquitin chains into the proteasome. Our results suggest that this type of complex can also form in the context of signal transduction, leading to specific interaction among ubiquitin binding proteins. Importantly, this interaction can be achieved with polyubiquitin chains that are not attached to a target protein, such as those synthesized by TRAF6 and UBC13/UEV1A. Thus, unanchored polyubiquitin chains are sufficient to induce specific protein-protein interaction. This mode of interaction may help to account for the specific signaling outputs achieved by polyubiquitin chains in diverse cellular pathways.
EXPERIMENTAL PROCEDURES
Stimulation of Cells and Analyses by Immunoblotting, Immunoprecipitation, and Kinase Assays
HEK293 cells, HeLa cells, U2OS cells, or MEFs were treated with IL-1β (30 ng/ml) or GST-TNFα (1 μg/ml). Cells were lysed in Buffer A (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 20 mM β-glycerolphosphate, 1 mM DTT, 0.5% NP-40, and Complete protease inhibitor cocktail [Roche]). Cell lysates were centrifuged at 20,000 × g for 10–15 min, and supernatant was used for immunblotting or immunoprecipitation according to standard protocols.
For IKK activity assays, following immunoprecipitation of NEMO and several washes in Buffer A, immunoprecipitate was washed with Buffer B (20 mM Tris-HCl [pH 7.5], 1 mM DTT, 20 mM β-glycerolphosphate, 0.05% NP-40). The immunoprecipitate was incubated with GST-IκBα N-terminus (0.1 mg/ml), 10 μM ATP, and γ-32P-ATP at 30° for 30 min.
Cell-Free Assays of IKK Activity
HeLa S100 was prepared according to standard protocols. S100 was mixed with 5X Buffer C (100 mM Tris-HCl [pH 7.5], 2.5 mM DTT, 100 mM β-glycerophosphate, g of 12.5 mM ATP, and 25 mM MgCl2) for a final concentration of 1X buffer C and 1.2 μg of total protein per 1 μl. Flag-purified A20 was added as indicated, then His6-TRAF6 was added to ~100 nM final. After 30 min at 30°, reaction was analyzed by IκBα immunoblot.
For assays with purified TAK1 and IKK complexes, IKK complex was used at ~5 nM, TAK1 complex at ~1 nM, MKK6 (K82A) at ~1 μM, and two-step affinity-purified A20 at ~3–100 nM. Synthesis and purification of polyubiquitin chains, as well as purification of TAK1 and IKK complexes, were described previously (Xia et al., 2009). Components were mixed with Buffer C. After 30 min at 30°, reaction was analyzed by immunoblotting as indicated.
A20 Polyubiquitin Binding Assays
Constructs encoding N-terminally Flag-tagged A20, or mutants thereof, were transfected into HEK293 cells by calcium phosphate precipitation. Cells were lysed in Buffer D, and 20,000 × g supernatant was incubated for 4–6 hr with anti-FLAG (M2) agarose. Beads were washed with Buffer D (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM DTT, 0.5% NP-40), then with Buffer E (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM DTT, 0.05% NP-40, 5% glycerol). Protein was eluted with 0.2 mg/ml Flag peptide. This partially purified A20 was mixed with polyubiquitin chains synthesized by E1, UBC13/UEV1A, and TRAF6, in which some of the ubiquitin had an HA tag (Boston Biochem), in Buffer F (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.5 mM DTT, 0.2% NP-40, 5% glycerol, 0.1 mg/ml BSA) on ice for 15 min. 10% of reaction was analyzed by immunoblotting for HA. Remaining reaction was diluted 1:10 in Buffer F, then mixed with M2 agarose. Beads were rinsed with Buffer G (Buffer F without BSA), then with Buffer E. A20 was eluted with 0.2 mg/ml Flag peptide. Eluate was analyzed by immunoblotting for HA and Flag.
RT-PCR measurements of NF-κB target gene expression
RNA was extracted using Trizol, then chloroform using standard protocols. The iScript cDNA synthesis kit (BioRad) was used to create cDNA from 150 ng of RNA. Quantitative real time PCR was performed using Sybr® Green on a BioRad iCycler iQ™5 with the following primers: rp119 (AAATCGCCAATGCCAACTC; TCTTCCCTATGCCCATATGC); IL-6 (TCCATCCAGTTGCCTTCTTG; GGTCTGTTGGGAGTGGTATC); COX-2 (TCC TCA CAT CCC TGA GAA CC; AAG TGG TAA CCG CTC AGG TG). rp119 values were used as a standard for IL-6 and COX-2 values. Data were normalized to unstimulated WT MEFs.
Direct Binding Between NEMO, Polyubiquitin Chains, and A20
GST or GST-NEMO (~20 nM), two-step affinity-purified A20 (~10–120 nM) and purified K63 polyubiquitin chains (~25 nM assuming average molecular weight of 400 kDa) were mixed on ice for 15 min in Buffer F. 10% of the reaction was analyzed by immunoblotting as indicated. Remaining reaction was diluted 10-fold in Buffer F. Glutathione Sepharose 4B (GE Healthcare) was added and samples were rotated end-over-end at 4° for 1 hr. Beads were washed several times in Buffer G, then analyzed by immunoblotting as indicated.
Supplementary Material
Acknowledgments
We thank A. Ting (Mount Sinai School of Medicine) for retroviral constructs and protocol. We thank X. Chen, A. Adhikari, G. Pineda, M. Xu, and L.J. Sun in our lab for contributing reagents. We also thank J. Cabrera for assistance with preparation of figures. This work was supported by grants from NIH (RO1-GM63692) and the Welch Foundation (I-1389) to Z. J. C. B.S. is supported by an NIH pre-doctoral training grant (GM007062). Z.J.C. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bianchi K, Meier P. A tangled web of ubiquitin chains: breaking news in TNF-R1 signaling. Mol Cell. 2009;36:736–742. doi: 10.1016/j.molcel.2009.11.029. [DOI] [PubMed] [Google Scholar]
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, Phu L, Phung Q, Maurer B, Arnott D, et al. Ubiquitin Binding to A20 ZnF4 Is Required for Modulation of NF-kappaB Signaling. Mol Cell. 2010;40:548–557. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, Ploegh HL, Smith TS. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378:727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias-Staheli N, Giannakopoulos NV, Kikkert M, Taylor SL, Bridgen A, Paragas J, Richt JA, Rowland RR, Schmaljohn CS, Lenschow DJ, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol. 2002;22:6034–6045. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. IkappaB kinase beta phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-kappaB pathway. Mol Cell Biol. 2007;27:7451–7461. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, Starost MF, Yedavalli V, Heyninck K, Dikic I, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27:629–641. doi: 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen X, Lary JW, Cole JL, Walters KJ. Defining how ubiquitin receptors hHR23a and S5a bind polyubiquitin. J Mol Biol. 2007;369:168–176. doi: 10.1016/j.jmb.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85. doi: 10.1042/BJ20071399. [DOI] [PubMed] [Google Scholar]
- Lamothe B, Campos AD, Webster WK, Gopinathan A, Hur L, Darnay BG. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J Biol Chem. 2008;283:24871–24880. doi: 10.1074/jbc.M802749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28:2885–2895. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tsai YC, Mattera R, Smith WJ, Kostelansky MS, Weissman AM, Bonifacino JS, Hurley JH. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat Struct Mol Biol. 2006;13:264–271. doi: 10.1038/nsmb1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Soetandyo N, Wang Q, Ye Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim Biophys Acta. 2009;1793:346–353. doi: 10.1016/j.bbamcr.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Chung JY, Lamothe B, Rajashankar K, Lu M, Lo YC, Lam AY, Darnay BG, Wu H. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol. 2008;376:526–540. doi: 10.1016/j.jmb.2007.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Tsai YC, Weissman AM, Bonifacino JS. The Rab5 guanine nucleotide exchange factor Rabex-5 binds ubiquitin (Ub) and functions as a Ub ligase through an atypical Ub-interacting motif and a zinc finger domain. J Biol Chem. 2006;281:6874–6883. doi: 10.1074/jbc.M509939200. [DOI] [PubMed] [Google Scholar]
- Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, Shifrin N, Petri MA, Ilyas Kamboh M, Manzi S, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008 doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, Schneider TR. Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell. 2006;124:1183–1195. doi: 10.1016/j.cell.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD. The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell. 2006;124:1197–1208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
- Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I, Giefing M, Gesk S, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–989. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007;26:3910–3922. doi: 10.1038/sj.emboj.7601823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, Harhaj EW. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9:254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Parvatiyar K, Harhaj NS, Harhaj EW. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J. 2009;28:513–522. doi: 10.1038/emboj.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A. 1996;93:6721–6725. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Skaug B, Zeng W, Chen ZJ. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol Cell. 2009;36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.