

Abstract
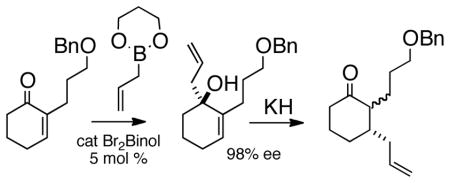
Enantioselective organocatalytic 1,2-allylation of a cyclic enone followed by anionic oxy-Cope rearrangement delivered the ketone as a mixture of diastereomers. This appears to be a general method for the net enantioselective conjugate allylation of cyclic enones.
Several procedures have been put forward in recent years1 for enantioselective conjugate addition to prochiral cyclic enones. To date, however, there has been only one report1b of enantioselective conjugate addition to an α-substituted cyclic enone such as 1a. It occurred to us that catalytic enantioselective 1,2-allylation2 followed by oxy-Cope rearrangement3 could offer a solution4,5 to this long-standing problem (Eq 1).
 |
(1) |
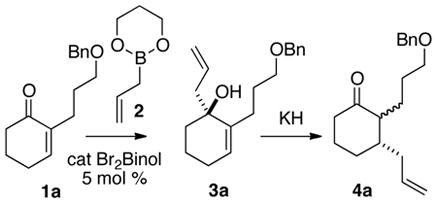
α-Iodo and α-alkyl6 cyclic enones (Table 1) are easily prepared.7 Of the several methods2 that have been put forward for the catalytic enantioselective allylation of ketones, we were attracted to that of Schaus,2g, 8 that employed the easily-prepared allylboronate 2 as the allyl donor and the commercially-available 3, 3′-dibromobinol as the enantioselective catalyst.
Table 1.
Enantioselective Allylation of Cyclic Enones.
| Enonea | Yieldb | Product | ee |
|---|---|---|---|
1
 1a |
84% |
 3a |
98% ee |
2
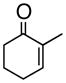 1bc,d |
46% |
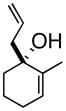 3b |
97% ee |
3
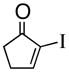 1c |
92% |
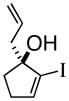 3c |
93% ee |
4
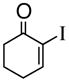 1d |
95% |
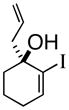 3d |
92% ee |
5
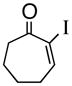 1e |
89% |
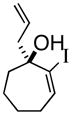 3e |
85% ee |
Additions were carried out using 5 mol % (S)-3,3′-dibromobinol.
Yields are for pure isolated substances.
Addition was effected using 5 mol % (R)-3,3′-dibromobinol.
Both the starting material and the product were volatile.
We initiated our studies with the enone 1a6a, f (Table 1). Following the updated Schaus protocol,8 stirring the enone with 2 in concentrated t-BuOH solution with a catalytic amount of (S)-3,3′-dibromo-1,1′-bi-2-naphthol at room temperature for 24 h, we found that the 1,2-addition proceeded smoothly. It was gratifying that 5 mol % of the organocatalyst was sufficient, and that > 90% of the catalyst could be recovered by extraction.
We found that this protocol worked equally well for 5, 6, and 7-membered rings, and with 2-alkyl and 2-iodo substitution. The enantiomeric excess for each of the 1, 2-allylations was established by chiral HPLC, except for 3b, the ee of which was secured by comparison of the optical rotation of the chromatographed and distilled product with that of the same substance prepared by methyl coupling of 3d.
For the oxy-Cope rearrangements (Table 2), we found it convenient to use KH in paraffin.9 With the alkyl enones, it was also necessary to include an equivalent of 18-crown-6.10 The oxy-Cope rearrangement of the aryl-substituted allylated alcohols (3dAr, 3cAr) proceeded efficiently without 18-crown-6, but the yields were slightly higher when it was included.
Table 2.
KH-Mediated oxy-Cope Rearrangement.
| Alcohola | Productb | Yieldc |
|---|---|---|
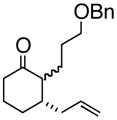
1
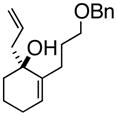 3a |
 4a |
70% |
2
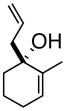 3bd |
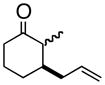 4b |
64% |
3
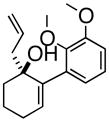 3dAre |
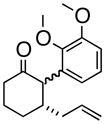 4c |
89% |
4
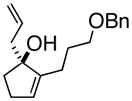 3cAlke |
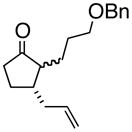 4d |
80% |
5
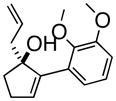 3cAre |
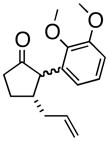 4e |
67% |
6
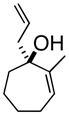 3eMef |
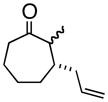 4f |
52% |
Oxy-Cope rearrangement was carried out with dicyclohexyl-18-crown-6 and KH in THF.
Products were a mixture of α-epimers.
Yields are for pure isolated substances.
t-BuOK was used for the rearrangement.
Prepared by Stille coupling of the corresponding iodoalkene.
Prepared by Kumada coupling of the corresponding alkene.
Of the substances reported here, only 4b had previously been reported, in racemic form and without characterization.11 We expect that the net catalytic enantioselective conjugate allylation of cyclic enones introduced here will have many applications in target-directed synthesis. The practicality of the Schaus organocatalytic allylation (room temperature in t-BuOH, 5 mol % catalyst, commercial and easily recoverable) is particularly noteworthy.
Experimental Section
General Procedures
1H NMR and 13C NMR spectra were recorded, as solutions in deuteriochloroform (CDCl3) unless otherwise indicated, at 400 MHz and 100 MHz, respectively. 13C multiplicities were determined with the aid of a JVERT pulse sequence, differentiating the signals for methyl and methine carbons as “d” from methylene and quaternary carbons as “u”. The infrared (IR) spectra were determined as neat oils. Rf values indicated refer to thin layer chromatography (TLC) on 2.5 × 10 cm, 250 μm analytical plates coated with silica gel GF and developed in the solvent system indicated. All glassware was oven dried and rinsed with dry solvent before use. THF was distilled from sodium metal/benzophenone ketyl under dry nitrogen. Toluene, dichloromethane and acetonitrile were distilled from calcium hydride under dry nitrogen. CH2Cl2 is dichloromethane, MTBE is methyl-tertbutyl ether and PE is petroleum ether. All reactions were conducted under N2 and stirred magnetically. We prepared KH in paraffin, but it is now commercially available.
(R)-1-(2-Propenyl)-2-(3-phenoxymethoxypropyl)-2-cyclohexenol (3a)
To a dry 10 mL round bottom flask was charged (S)-(−)-3,3′-dibromo-1,1′-bi-2-naphthol (73 mg, 0.165 mmol), followed by t-butyl alcohol (362 mg, 8.23 mmol) and allyl borane 2 (778 mg, 6.17 mmol). This suspension was stirred until all of the BINOL was dissolved. To this clear solution was charged enone 1a (1.00 g, 4.12 mmol), and the reaction was stirred at room temperature overnight. The reaction was concentrated directly to silica and chromatographed to give 3a (938 mg, 84% yield, 98% ee) as a yellow oil; enantioselective HPLC (3% i-PrOH/Hexanes, Chiralpak IA 4.6 mm × 250 mm, UV detection at 254 nm, 0.08 mL/min) tr= 6.74 (major) tr= 4.68 (minor) TLC Rf = 0.24 (MTBE:PE, 20:80) [α]D +23.3 (DCM, 20°C); IR (neat, cm−1) 3439 (s), 2928 (s), 2850 (m), 1668 (m), 1633.8 (s), 1442.1 (s), 1363.5 (s) 1H NMR (400 MHz, CDCl3) δ ppm 1.5–2.5 (m, 13H), 3.5 (m, 2H), 4.5 (s, 2H), 5.1 (m, 1H), 5.2 (m, 1H), 5.8 (m, 1H), 7.3–7.4 (m, 5H) 13C NMR (400 MHz, CDCl3) δ ppm u: 139.9, 138.0, 117.6, 77.0, 76.7, 76.3, 72.5, 71.4, 69.9, 43.5, 35.7, 28.4, 27.0, 25.3, 18.5; d: 133.9, 127.9, 127.3, 127.1, 124.9; HRMS calcd for C19H25O (M+ -OH) 269.1905, Found 269.1909.
(S)-1-(2-Propenyl)-2-methyl-2-cyclohexenol (3b)
Light yellow oil. [α]20D = −34.9° (c = 1.00, CH2Cl2); TLC: Rf (MTBE/PE, 1:4) = 0.46; 1H-NMR δ 5.78 (m, 1H), 5.64 (m, 1H), 5.2 (m, 2H), 2.40 (dt, J = 7.2, 1.2 Hz, 2H), 1.98 (m, 2H), 1.77 (m, 1H), 1.76 (s, 3H), 1.64 (m, 4H); 13C-NMR δ u: 137.0, 118.2, 71.6, 43.6, 35.7, 25.6, 19.1; d: 134.1, 126.6, 17.8; IR: 1639, 1440, 1174, 973, 912 cm−1; HRMS calcd for C10H15 (M – OH): 135.1174, obsd: 135.1173.
(R)-1-(2-Propenyl)-2-iodo-2-cyclopentenol (3c)
Yellow solid (mp 32–35 °C, 92% yield, 93% ee); TLC Rf = 0.29 (DCM:MTBE:PE 10:20:70); chiral HPLC (3% i-PrOH/Hexanes, Chirapak IA 4.6 mm × 250 mm, UV detection at 254 nm, 0.08 mL/min) tr= 8.420 (major), tr= 9.205 (minor): [α]D +52.4 (DCM, 20 °C); IR (neat, cm−1) 3400 (s), 3066 (m), 2918 (s), 2840 (m), 1638 (m), 1599 (m), 1427 (m), 1373 (m), 1309 (m) 1H NMR (400 MHz, CDCl3) δ ppm 1.9 (m, 2H), 2.2–2.6 (m, 5H), 5.1–5.2 (dd, 2H), 5.6–5.9 (m, 1H), 6.3 (s, 1H) 13C NMR (400 MHz, CDCl3) δ ppm u: 119, 134.0, 106.5, 86.0, 123.5, 44.5, 33.0, 32.8 d: 142.1, 132.7; HRMS calcd for C8H10I (M+-OH) 232.9827, Found 232.9826.
(R)-1-(2-Propenyl)-2-iodo-2-cyclohexenol (3d)
Clear oil (95% yield, 92% ee); TLC Rf = 0.29 (MTBE:PE 20:80) [α]D -34.9 (DCM, 20°C); chiral HPLC (2% i-PrOH/Hexanes, Chirapak IA 4.6 mm × 250 mm, UV detection at 254 nm, 0.08 mL/min) tr= 17.34 (major), tr= 16.80 (minor); IR (neat, cm−1) 3436 (s), 3074 (m), 2937 (m), 1638 (m), 1435 (s); 1H NMR (400 MHz, CDCl3) d ppm 1.6–1.8 (m, 2H), 1.8–2.2 (m, 5H), 2.4–2.5 (d, 2H), 5.1–5.2 (m, 2H), 5.7–5.9 (m, 1H), 6.6 (m, 1H); 13C NMR (400 MHz, CDCl3) d ppm u: 118.9, 111.7, 73.0, 47.1, 34.1, 29.6, 18.9; d: 141.9, 132.9; HRMS calcd for C9H12I (M+ -OH) 246.9992, Found 246.9984
(R)-1-(2-Propenyl)-2-iodo-2-cycloheptenol (3e)
Yellow oil (89% yield, 85% ee); TLC Rf = 0.50 (MTBE:PE, 20:80); [α]D -46 (DCM, 20°C); chiral HPLC (0.1% i-PrOH/Hexanes, Chiracel OJH 4.6 mm × 250 mm, UV detection at 254 nm, 0.08 mL/min) tr= 10.721 (major), tr= 12.399 (minor); IR (neat, cm−1) 3459 (b), 3076 (m), 2918 (s), 2859 (m), 1682 (m), 1643 (m), 1609 (m), 1442 (m), 1343 (m) 1H NMR (400 MHz, CDCl3) δ ppm 1.6–1.9,(m, 5H), 2.0–2.2 (m, 4H), 2.5 (m, 2H), 5.2(ds, 2H), 5.8–5.9 (m, 1H), 6.7 (t, 1H); 13C NMR (400 MHz, CDCl3) δ ppm 141.5, 132.5, 118.6, 118.0, 77.7, 44.3, 33.0, 28.5, 24.7, 20.7; HRMS calcd for C10H14I (M+-OH) 261.0141, Found 261.0198.
(1S, 2S)- 2-(3-Phenoxymethoxypropyl)- 3-(2-propenyl)-cyclohexanone (4a)
To a 100 mL round bottom flask was charged 3a (1.07 g, 3.77 mmol), followed by 18-crown-6 (996 mg, 3.77 mmol) and 50 mL of THF. The reaction was sparged with N2 for 10 minutes and KH(P) (452 mg 5.65 mmol) was added portion wise. The reaction was heated to reflux for 1 hour and then quenched with saturated aqueous ammonium chloride. The organic layer was partitioned between Et2O and water. The organic layer was dried (anhydrous Na2SO4) and concentrated. The residue was chromatographed giving 4a (749 mg, 70% yield) as a yellow oil. TLC Rf = 0.20 (DCM:MTBE:PE, 10:20:70); [α]D +18 (c = 0.02, CH2CL2, 20°C) IR (neat, cm−1) 3029 (m), 2931 (s), 1709 (m), 1639 (m), 1495 (s), 1453 (s), 1360 (s); major diastereomer: 1H NMR (400 MHz, CDCl3) δ ppm 1.49 – 2.45 (m, 14H) 3.5 (t, 2 H) 4.5 (s, 2 H) 5.1 (m, 2 H) 5.8 (m, 1 H) 7.21 - 7.4 (m, 5H); 13C NMR (400 MHz, CDCl3) δ ppm u: 213.3, 138.6, 117.0, 72.7, 70.3, 37.8, 28.7, 27.3, 26.2, 24.7, 24.1, 22.0 d: 135.7, 128.3, 127.5, 54.6, 41.9; HRMS calcd for C19H27O2 (M+H) 287.2011, Found 287.2002.
(1R, 2R)- 2-Methyl-3-(2-propenyl)-cyclohexanone (4b)
To a 500 mL round bottom flask was charged toluene (140 mL), 18-crown-6 (6.48 g, 24.5 mmol) and potassium tert-butoxide (5.49 g, 49 mmol). Ketone 3b (5.33 g, 35 mmol) was charged in 30 mL of toluene. The reaction was heated to 84 °C for 1 hour and was then quenched with saturated aqueous NH4Cl. The toluene/PE solution was then diluted with more PE and placed directly on a column for purification by silica gel chromatography to afford ketone 4b (2.162 g, 64% yield, mixture of trans/cis diastereomers) as a light yellow oil. [α]20D = +10.0° (c = 1.00, CH2Cl2, 2:1 mixture of trans:cis diastereomers); TLC: Rf (MTBE/PE, 1:4) = 0.67; 1H-NMR δ 5.85-5.73 (m, 1H, trans diastereomer), 5.75-5.63 (m, 1H, cis diastereomer), 5.11-5.03 (m, 2H, trans diastereomer), 5.03-4.98 (m, 2H, cis diastereomer), 2.66-2.00 (m, 6H), 1.93-1.41 (m, 4H), 1.05 (d, J = 12.8 Hz, 3H, trans diastereomer), 1.02 (d, J = 12.8 Hz, 3H, cis diastereomer); 13C-NMR (trans diastereomer) δ u: 213.4, 117.1, 41.5, 38.2, 30.3, 25.7; d: 135.5, 49.4, 45.2, 11.9; (cis diastereomer) δ u: 214.5, 116.3, 39.8, 33.7, 26.7, 23.7; d: 136.5, 48.7, 41.9, 11.5; IR: 1711, 1640, 1447, 1221, 999, 914 cm−1; HRMS calcd for C10H17O (M + H): 153.1279, obsd: 153.1283.
(1R, 2S)- 2-(2,3-Dimethoxyphenyl)- 3-(2-propenyl)-cyclohexanone (4c)
White solid, 89% yield, mp = 70–75 °C); TLC Rf = 0.5 (MTBE:PE 20:80); [α]20D – 64° (c = 0.2 CH2Cl2, 20° C); IR (neat, cm−1) 2934 (s), 2931 (s), 1710 (s), 1584 (m), 1475 (s), 1267 (s), 1H NMR (400 MHz, CDCl3) δ ppm 1.5 (m, 2H) 1.8 (m, 2 H) 2.0–2.2 (m, 4 H) 2.4 (m, 1H) 2.6 (m, 1 H), 3.6 (d, 1H, J=9.2 Hz), 3.7 (s, 3H), 3.9 (s, 3H), 4.9 (m, 2H), 5.6–5.8 (m, 1H), 6.6 (d, 1H, J= 8.2 Hz) 6.8 (d, 1H, J= 8.2 Hz) 7.1 (t, 1H, J= 8.2 Hz) 13C NMR (400 MHz, CDCl3) δ ppm u: 210.0, 152.6, 147.4, 131.6, 116.8, 41.8, 39.0, 30.6, 25.1 d: 135.8, 123.7, 121.7, 111.0, 60.4, 56.8, 55.6, 43.5; HRMS calcd for C17H23O3 (M+H) 275.1647, Found 275.1659.
(1S, 2S)- 2-(3-Phenoxymethoxypropyl)- 3-(2-propenyl)-cyclopentanone (4d)
Yellow oil (52% yield); TLC Rf = 0.35 (MTBE:PE, 20:80). [α]20D -72° (c = 0.1 CH2Cl2, 20° C); IR (neat, cm−1) 2909 (m), 2857 (s), 1735 (s), 1700 (s), 1638 (m), 1450 (m) 1H NMR (400 MHz, CDCl3) δ ppm 1.3–2.5 (m, 13H) 3.4–3.5 (m, 2H) 4.5 (m, 2H), 2.1 (m, 1H) 5.0–5.1 (m, 2H), 5.7–5.9 (m, 1H), 7.2–7.4 (m, 5H), 13C NMR (400 MHz, CDCl3) δ ppm u: 220.2, 138.1, 116.2, 72.4, 69.9, 38.1, 37.2, 27.3, 26.5, 21.6 d: 137.5, 127.9, 126.5, 126.0, 52.1, 41.9; HRMS calcd for C18H25O2 (M+H) 273.1854, Found 273.1850.
(1R, 2S)- 2-(2,3-Dimethoxyphenyl)- 3-(2-propenyl)-cyclopentanone (4e)
Yellow oil (67% yield); TLC Rf = 0.67 (MTBE:PE, 20:80);); [α]20D +38° (c = 0.1 CH2Cl2, 20° C) IR (neat, cm−1) 3073 (m), 2939 (s), 2836 (s), 1740 (s), 1640 (m), 1584 (m), 1478 (s), 1268 (s); 1H NMR (400 MHz, CDCl3) δ ppm 1.4–1.6 (m, 1H) 2.0 (m, 1H) 2.2–2.4 (m, 2H) 2.4–2.5 (m, 2H) 3.0 (d, 1H, J=10.8 Hz), 3.7 (s, 3H), 3.9 (s, 3H), 5.0 (m, 2H), 5.7 (m, 1H), 6.6 (d, 1H, J= 8.2 Hz), 6.8–6.9 (d, 1H, J= 8.2 Hz), 7.0 (t, 1H, J= 8.2 Hz); 13C NMR (400 MHz, CDCl3) δ ppm u: 218.4, 152.7, 146.9, 132.7, 116.6, 38.2, 38.0, 27.3 d: 135.7, 123.7, 122.8, 111.8, 59.8, 58.4, 55.7, 44.0; HRMS calcd for C16H20O3 (M+) 260.1412, Found 260.1401.
(1S, 2S)- 2-Methyl-3-(2-propenyl)-cycloheptanone (4f)
Yellow oil (52% yield) as a mixture of diastereomers. TLC Rf = 0.30 (MTBE/PE, 5:95); [α]20D -45.0° (c = 0.03; CH2Cl2, 20° C); IR (neat, cm−1) 3074 (m), 2927 (s), 2862 (s), 1700 (s), 1640 (m), 1451 (m), 1374 (m), 1H NMR (400 MHz, CDCl3) δ ppm 1.0–1.1 (d, 2H, J=8.2 Hz) 1.1–1.2 (d, 1H, J=8.2 Hz) 1.5–2.0 (m, 8H) 2.1 (m, 1H) 2.3–2.4 (m, 2H), 2.6 (m, 1H), 2.8–2.9 (m, 1H), 5.0 (m, 2H), 5.0–5.1 (m, 2H), 5.6–5.8 (m, 1H); major 13C NMR (400 MHz, CDCl3) δ ppm u: 215.7, 116.2, 43.3, 34.5, 32.3, 25.9, 24.2 d: 137.2, 49.3, 40.1, 12.9; HRMS calcd for C11H19O (M+H) 167.1436, Found 167.1435.
Supplementary Material
Acknowledgments
We thank Dr. John Dykins for mass spectrometric measurements, supported by the NSF (0541775), Dr. Shi Bai for NMR assistance (NSF CRIF:MU, CHE 0840401)and the NIH (GM42056) for financial support. We thank the Schaus group for sharing their results prior to publication.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Catalytic enantioselective conjugate allylation is effective with doubly-activated cycloalkenones: Shizuka M, Snapper ML. Angew Chem Int Ed. 2008;47:5049. doi: 10.1002/anie.200800628.Despite excellent progress with catalytic enantioselective conjugate addition, there has been only one report of addition to an α-alkyl or α-aryl cyclic enone: Vuagnoux-d’Augustin M, Alexakis A. Chem Eur J. 2007;13:9647. doi: 10.1002/chem.200701001.For other leading references, see Alexakis A, Li K. Angew Chem Int Ed. 2006;45:7600. doi: 10.1002/anie.200602417.Wencel J, Rix D, Jennequin T, Labat S, Crévisy C, Mauduit M. Tetrahedron: Asymm. 2008;19:1804.Malmgren M, Granander J, Amedjkouh M. Tetrahedron: Asymm. 2008;19:1934.Feringa BL, Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ. Chem Rev. 2008;108:2824. doi: 10.1021/cr068424k.Riguet E. Tetrahedron Lett. 2009;50:4283.Alexakis A, Palais L. Tetrahedron: Asymm. 2009;20:2866.
- 2.Several methods have been developed for the enantioselective 1,2-allylation of ketones: Tietze LF, Schiemann K, Wegner C. J Am Chem Soc. 1995;117:5851.Tagliavini E, Casolari S, D’Addario D. Org Lett. 1999;1:1061.Wada R, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910. doi: 10.1021/ja047200l.Walsh PJ, Kim JG, Waltz KM, Garcia IF, Kwiatkowski D. J Am Chem Soc. 2004;126:12580. doi: 10.1021/ja047758t.Chong JM, Wu TR, Shen L. Org Lett. 2004;6:2701. doi: 10.1021/ol0490882.Yamamoto H, Wadamoto M. J Am Chem Soc. 2005;127:14556. doi: 10.1021/ja0553351.Schaus SE, Lou S, Moquist PN. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308.Yamamoto H, Wadamoto M. Chem Asian J. 2007;2:692. doi: 10.1002/asia.200600413.Feng X, Zheng K, Qin B, Liu X. J Org Chem. 2007;72:8478. doi: 10.1021/jo701491r.Ishihara K, Hatano M. Synthesis. 2008:1647.x.
- 3.For reviews of the oxy-Cope rearrangement, see Durairaj K. Curr Sci. 1994;66:917.Paquette LA. Tetrahedron. 1997;53:13971.
- 4.For stoichiometric 1,2-allylation followed by oxy-Cope rearrangement, see Castle SL, Nielsen DK, Nielsen LL, Jones SB, Toll L, Asplund MC. J Org Chem. 2009;74:1187. doi: 10.1021/jo802370v.Li F, Tartakoff SS, Castle SL. J Am Chem Soc. 2009;131:6674. doi: 10.1021/ja9024403.
- 5.Conjugate addition/enolate trapping offers an alternative solution to this problem, but is limited to α-substituents that can be introduced by alkylation. For an overview, see Stork G, Rosen P, Goldman N, Coombs RV, Tsuji J. J Am Chem Soc. 1965;87:275.Taylor RJK. Synthesis. 1985:364.Coates RM, Jin AQ. J Org Chem. 1997;62:7475. doi: 10.1021/jo9706335.Feringa BL, Pineschi M, Arnold LA, Imbos R, de Vries AHM. Angew Chem Int Ed. 1997;36:2620.Alexakis A, Vuagnoux-d’Augustin M. Chem Eur J. 2007;13:9647. doi: 10.1002/chem.200701001.Alexakis A, Henon H, Mauduit M. Angew Chem Int Ed. 2008;47:9122. doi: 10.1002/anie.200803735.Jarugumilli GK, Cook SP. Org Lett. 2011;13:1904. doi: 10.1021/ol200059u.
- 6.For the preparation of α-alkyl cyclic enones, see Taber DF. J Org Chem. 1976;41:2649.Johnson CR, Braun MP. J Am Chem Soc. 1993;115:11014.Armitage MA, Lathbury DC, Mitchell MB. J Chem Soc, Perkin Trans 1. 1994;12:1551.Abbas AA, Kobayashi Y. Tetrahedron Lett. 2003;44:119.Takeishi K, Sugishima K, Sasaki K, Tanaka K. Chem Eur J. 2005;10:5681. doi: 10.1002/chem.200400679.Taber DF, Sheth R. J Org Chem. 2008;73:8030. doi: 10.1021/jo801767n.
- 7.For the preparation of α-aryl cyclic enones, see Bosse F, Tunoori AR, Niestro AJ, Gronwald O, Maier ME. Tetrahedron. 1996;52:9485.Prasad ASB, Knochel P. Tetrahedron. 1997;53:16711.Kim J, Kulawiec R. Tetrahedron Lett. 1998;39:3107.Felpin F. J Org Chem. 2005;70:8575. doi: 10.1021/jo051501b.To date, we have not been able to achieve high ee’s in the addition to such α-aryl cyclic enones.
- 8.For the preparation and use of the allyl boronate 2, see Schaus SE, Barnett DS, Moquist PN. Angew Chem Int Ed. 2009;48:8679. doi: 10.1002/anie.200904715.
- 9.For the preparation of KH in paraffin, see Taber DF, Nelson CG. J Org Chem. 2006;71:8973. doi: 10.1021/jo061420v.Huang H, Nelson CG, Taber DF. Tetrahedron Lett. 2010;51:3545.
- 10.For rate acceleration of the oxy-Cope rearrangement using 18-crown-6, see Evans D, Golob AM. J Am Chem Soc. 1975;97:4765.
- 11.Hayashi M, Mukaiyama T. Chem Lett. 1987:289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.