

Abstract
Proteolytic cleavage of tau at glutamic acid 391 (E391) is linked to the pathogenesis of Alzheimer disease (AD). This C-terminal truncated tau species exists in neurofibrillary tangles and abnormal neurites in the brains of AD patients and may potentiate tau polymerization. We generated a mouse model that expresses human tau truncated at E391 to begin to elucidate the role of this C-terminal truncated tau species in the development of tau pathology. Our results show that truncated but otherwise wild type human tau is sufficient to drive pre-tangle pathological changes in tau, including accumulation of insoluble tau, somatodendritic redistribution, formation of pathological conformations, and dual phosphorylation of tau at sites associated with AD pathology. In addition, these mice exhibit atypical neuritic tau immunoreactivity, including abnormal neuritic processes and dystrophic neurites. These results suggest that changes in tau proteolysis can initiate tauopathy.
Keywords: Aggregation, Alzheimer disease, tau, Transgenic mice, Truncation
INTRODUCTION
Tau is a normal, highly abundant protein in brain that is also part of the neuropathology of a number of neurodegenerative diseases including Alzheimer disease (AD) and frontotemporal lobar degeneration with tau inclusions. Diseases with pathological deposits of tau protein are collectively referred to as tauopathies; tau aggregates in these disorders form abnormal fibrillar tangles in neurons, and in some cases in oligodendrocytes and astrocytes (1–4). Tau is a microtubule-binding protein that promotes microtubule assembly and stabilizes formed microtubules. In tauopathies, fibrillar tau is found in low level aggregates, or as mature tangles, both of which may contribute to cellular dysfunction (5). Human tau is encoded by a single gene (MAPT) that is alternatively spliced to generate 6 tau isoforms that are distinguishable by the exclusion or inclusion of a repeat region of exon 10, referred to as 3-repeat (3R) and 4-repeat (4R) tau, respectively (6).
Proteolytic cleavage of tau at the C-terminus is linked to the pathogenesis of AD (7). Truncated tau terminating at glutamic acid 391 (E391) is a component of the paired helical filaments core, present in neurofibrillary tangles (NFTs) and abnormal neurites in AD patient brains (8, 9) and is positively correlated with the severity of dementia (10). In addition, the appearance of E391-truncated tau precedes that of tangle formation (8, 11). This C-terminal truncation greatly increases the rate of tau polymerization in vitro compared to that of full-length tau; thus, tau truncation may play a role in seeding tangle formation (11, 12).
We generated a human 4R-tau truncation construct of E391 and introduced this transgene into mice to elucidate the role of this C-terminal truncated tau species in the development of tau pathology. We found that a moderate level expression of truncated but otherwise wild type human tau is sufficient to drive pathological changes in tau.
MATERIALS AND METHODS
Antibodies
The 3R- and 4R-specific tau antibodies were developed by de Silva et al (13). The 3R-specific mouse monoclonal antibody (RD3, Upstate Cell Signaling, Lake Placid, NY) is raised against a synthetic peptide corresponding to amino acids 209–224 of human tau (numbering based on the 0N3R isoform). The 4R-specific mouse monoclonal antibody (RD4, Upstate Cell Signaling) is raised against a synthetic peptide corresponding to amino acids 275–291 of human tau (numbering based on 2N4R tau). T14 is a mouse monoclonal antibody that recognizes human tau residues 83–120 (based on 2N4R tau numbering) (14) but not mouse tau. T49 is a mouse monoclonal antibody that is specific for rodent tau and does not recognize human tau (15). Rabbit polyclonal 17025 is a pan-tau antibody recognizing total mouse and human tau raised against full-length recombinant tau. T14, T49 and 17025 were provided by Dr. Virginia Lee (University of Pennsylvania, Philadelphia, PA). AT8 (Pierce Biotechnology, Rockford, IL) is a phosphorylation-dependent mouse monoclonal antibody that recognizes paired helical filament-tau phosphorylated on dual sites Ser202 and Thr205. Two other monoclonal anti-phospho-tau antibodies related to tau pathology were also used: AT180 (phospho-Thr 231; Pierce Biotechnology) and CP13 (phospho-Ser 202; provided by Peter Davies, Albert Einstein College of Medicine, Bronx, NY). Alz50 is a phosphorylation-independent mouse monoclonal antibody that recognizes amino acids 5–15 and 312–322 of tau and is specific for a pathological tau conformation (16). MC-1 is also a conformation-specific mouse monoclonal antibody similar to Alz-50 that recognizes amino acids 7–9 and 313–322 of tau (17, 18). Alz-50 and MC-1 were both provided by Peter Davies. A third conformation-specific mouse monoclonal antibody, Tau2 (Sigma, St. Louis, MO), recognizes a phosphorylation-independent epitope (corresponding to amino acid 95–108 of bovine tau) that is pathologically modified as tau protein is phosphorylated to form NFTs (19). The anti-Actin monoclonal antibody (Sigma) recognizes a C-terminal Actin epitope from many species.
Construction of Transgenic Mice
The cDNA encoding the most abundant brain isoform (4R1N) truncated at position 391 (11) was cloned into the unique XHO I site in a mouse neuron-specific expression vector containing pThy1.2 (20). Transgenic mice were generated by pronuclear microinjection of the THY1.2::TauΔ391 transgene at the University of Washington Transgenic Resources Program as a fee-for-service project. Founders were identified by PCR analysis of tail biopsies, as described below. Founder mice were intercrossed with C57BL/6 mice to establish lines. Hemizygous mice were used as study subjects.
Genotyping
Mice were genotyped using DNA prepared from tail clips for live mice. The presence of the THY1.2::TauE391 transgene was detected by PCR analysis using the following primers:
cDNA trunc-F AAGATCGGCTCCACTGAGAA
cDNA trunc-R GGACGTGGGTGATATTGTCC
yielding a 316-bp product for the hemizygous E391 transgenic mice and no product for wild type control animals.
Immunoblot Analysis
We conducted a sequential extraction of tau protein using buffers of increasing solubilizing strength as previously described (21). Mouse brains from 3 non-transgenic (NTG), 6 3608, and 7 3610 transgenic mice were isolated and snap-frozen in liquid N2 prior to extraction. Mouse brain hemispheres were homogenized in high salt re-assembly buffer (RAB-High Salt [0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4, 0.75 M NaCl, 0.02 M NaF, 0.5 mM PMSF, 0.1% protease inhibitor cocktail, pH 7.0]) and ultra-centrifuged at 50,000 × gravity yielding the soluble fraction (supernatant) and an insoluble pellet. Next, myelin was floated by resuspending the pellet in RAB/1M sucrose and centrifuging as above. To extract detergent soluble tau, the RAB-insoluble material was re-extracted with an ionic and non-ionic detergent containing RIPA buffer (50 mM Tris, 150 mM NaCl, 1% NP40, 5 mM EDTA, 0.5% DOC, 0.1% SDS, 0.5 mM PMSF, 0.1% protease inhibitor cocktail, pH 8.0) and centrifuged as above yielding abnormal tau in the supernatant. Finally, the detergent-insoluble pellet was re-extracted with 70% formic acid (FA) to solubilize detergent-insoluble tau. Total protein fractions containing ~15 mg of protein per lane were boiled for 5 minutes and loaded onto 10% pre-cast SDS-PAGE gels (BioRad, Hercules, CA). Subsequent RIPA and FA-extracted fractions were normalized according to the levels of total protein in the total fraction. For semiquantitative immunoblotting, we detected mouse and human tau using the pan tau antibody 17025 at a dilution of 1:3000, as described previously (22). Densitometry measurements were performed using Adobe Photoshop to determine total tau levels in non-transgenic and transgenic mice. We also used human specific tau antibody T14 at 1:1000, mouse specific tau antibody T49 at 1:2000, and anti-actin antibody at 1:1000.
Immunohistochemistry
For all genotypes, 1-year-old and 2-year-old mice (n = 3 each) were examined by immunohistochemistry; the images presented are representative of each genotype. No discernible differences were detected between 1-year-old and 2-year-old mice with any antibody. Mice were anesthetized and fixed by transcardial perfusion with 4% paraformaldehyde. Brains were removed, paraffin-embedded, and coronal sections from the hippocampus were cut at 10-μm thickness and stored at 4°C until use. Sections were immunostained with RD4 (1:80), RD3 (1:800), T14 (1:500), AT8 (1:250), Alz50 (1:25), MC-1 (1:75), and Tau2 (1:500). Sections were deparaffinized and rehydrated through alcohols and an antigen retrieval step consisting of heat pretreatment by microwave (AT8, Alz50, MC-1, Tau2 and RD3) or autoclave (RD4) in DakoCytomation Target Retrieval Solution (Vector Laboratories, Burlingame, CA) was used. Antibody T14 did not require antigen retrieval. Sections were treated for endogenous peroxidases with 3% H2O2 in PBS (pH 7.4), blocked in 5% non-fat milk in PBS, incubated with primary antibody overnight at 4°C, followed by biotinylated secondary antibody for 45 minutes at room temperature. Finally, sections were incubated in an avidin-biotin complex (Vectastain Elite ABC kit, Vector Laboratories) and the reaction product was visualized with 0.05% diaminobenzidine/0.01% H2O2 in PBS. Negative controls with secondary antibody alone did not immunostain tissue sections (data not shown).
Photomicrography and Figure Preparation
Photomicrographs were taken with a digital camera and imported into Adobe Photoshop for mounting. To optimize visualization of staining, photomicrographs were modified (when necessary) by adjusting brightness and contrast.
RESULTS
Accumulation of Truncated tau in the Insoluble Fraction
To test the hypothesis that truncated tau promotes tau aggregation in vivo, we generated transgenic mice expressing tau truncated at position E391 (E391 mice) using the Thy1.2 neuron specific promoter (Fig. 1A). This construct expresses the 4R1N tau isoform, which is the most abundant isoform in human brain. The construct contains sequences encoded by alternatively spliced exons 2 and 10 and has 4 microtubule binding repeats. Two founder mouse lines (E391-3610 and E391-3608) expressing E391 truncated tau were generated. To measure total tau levels, whole 1-year-old mouse brains were homogenized and immunoblotted with a pan-tau-specific antibody, demonstrating total tau levels ~1.9-fold greater than normal endogenous mouse tau in E391-3608 mice and ~2.6-fold greater in E391-3610 mice (Fig. 1B). We homogenized brain hemispheres from NTG mice and the 2 transgenic founder lines in RAB, a high salt buffer, yielding the soluble tau fraction. There were roughly similar levels of total tau among animals in this fraction. Although the large amount of total tau made it difficult to distinguish soluble human tau from endogenous mouse tau, these similar levels indicated that expression of the human transgene was relatively low compared to that of endogenous mouse tau. We re-extracted material insoluble in RAB with RIPA, an ionic and non-ionic detergent containing buffer, yielding the detergent soluble fraction. Subsequently, we recovered detergent-insoluble material by extraction with FA. NTG animals exhibited nearly all tau in the soluble fraction (RAB), with no detergent-insoluble truncated tau detected. In contrast, E391-3608 and E391-3610 mice exhibited marked accumulation of truncated tau species in the detergent soluble (RIPA) and insoluble (FA) fractions, with higher expression exhibited by mouse line E391-3610 (Fig. 1C). FA fractions contained abundant truncated human E391 tau as detected by T14 human tau-specific antibody, but virtually no endogenous mouse tau was detected by T49, the mouse tau-specific antibody (Fig. 1D).
Figure 1.

Human E391 truncated tau expression in transgenic mice. (A) Diagram of the normal 4R1N tau isoform (410 amino acids long) and the neuronal truncated human E391 tau-expressing transgene. The E391 truncation is denoted according to the naming convention for the longest tau isoform. (B) Immunoblot analysis of total tau levels (pan-tau antibody 17025) in the brains of non-transgenic (NTG, n = 3) and E391 transgenic mice (3608, n = 6; 3610, n = 7). Densitometry measurements demonstrate that total tau levels are ~1.9 ± 0.1 and 2.6 ± 0.2 above endogenous mouse tau in E391-3608 and 3610 mice, respectively. All groups are significantly different from each other by one way ANOVA (Kurskal-Wallis test, p < 0.05) and error bars are SEM. (C) To measure detergent-insoluble tau species, mouse brains were subjected to a sequential extraction using buffers of increasing solubilizing strength and immunoblots were probed with the pan-tau antibody 17025. NTG animals exhibit nearly all tau in the soluble fraction (RAB) with no detergent-insoluble truncated tau detected. E391 transgenic mice exhibit accumulation of truncated tau species in the detergent soluble (RIPA) and insoluble (FA) fractions. (D) FA fractions contain abundant truncated human E391 tau as detected by T14 human specific tau antibody (FA-human), but virtually no endogenous mouse tau, as detected by T49 mouse specific tau antibody (FA-mouse).
E391 Mice Exhibit Re-localization of tau and Atypical Neuritic tau Immunoreactivity
In newborn mouse brain, 3R-tau is the predominant isoform, whereas in adult mice the predominant isoform changes to 4R-tau (23–25). We examined isoform-specific expression of tau in the brains of aged (1 and 2 year old) NTG mice and transgenic mice expressing E391-truncated human 4R-tau by immunohistochemistry using antibodies specific for 3R and 4R tau. As expected, 3R-tau was undetectable in all mouse lines used with the exception of occasional neurons in the subgranular zone of the dentate gyrus, as previously reported (26, 27 and data not shown). We next used a panel of tau-specific antibodies to compare tau distribution and pathology in NTG and E391 transgenic mice. All tau immunoreactivity was specific for 4R-tau since 3R-tau was not expressed in the mouse lines studied. Using an antibody specific for 4R-tau (RD4), we demonstrated that aged NTG mice exhibit a normal axonal distribution of tau, with high levels of expression in CA3 mossy fibers, hilus, inner molecular layer of the dentate gyrus, and somewhat lower levels in the stratum radiatum and oriens (Fig. 2A). All hippocampal CA pyramidal neurons were negative. In the cortex, tau staining was primarily localized to the neuropil (Figs. 2A, 3A). Neuronal cell bodies were never positively stained with antibodies to 4R-tau in the cortex or hippocampus of NTG mice. This axonal distribution in NTG mice was similar to previous reports (28, 29). In addition, we detected tau immunostaining in small, non-neuronal cells throughout the brain, in both white and gray matter; these were likely glial cells (Figs. 2A, 3A).
Figure 2.

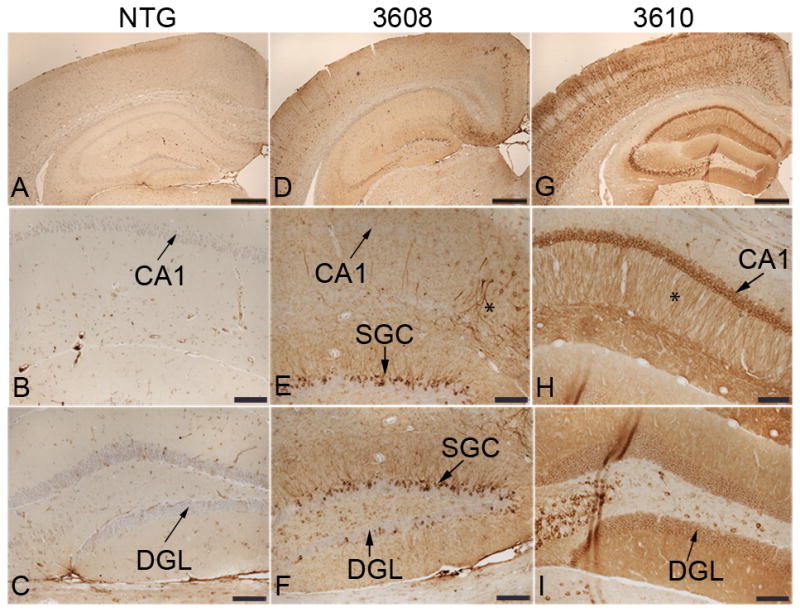
Truncated tau is distributed in a somatodendritic pattern in the hippocampus and cortex of E391 transgenic mice. (A–N) Coronal paraffin sections were stained with the 4R-tau-specific antibody RD4, which recognizes both mouse and human 4R-tau (A–C, G–J) or with the human specific tau antibody T14 (D–F, K–N). Non-transgenic (NTG) mice exhibit a normal axonal distribution of 4R-tau in the hippocampus and cortex (A). E391-3608 mice (lower expressers) exhibit somatodendritic distribution of 4R-tau throughout the cortex, in hippocampal superficial granule cells (SGC) of the dentate gyrus, and in CA1 apical dendrites and cell bodies (B, E, G, I, K, M). E391-3610 mice (higher expressers) exhibit somatodendritic distribution of 4R-tau throughout the cortex, in hippocampal CA3 pyramidal neurons, CA1 apical dendrites and cell bodies and dentate granule cells (C, F, H, J, L, N). Inset in C is a higher power of boxed region. DGL, dentate granule layer; DML, dentate molecular layer; iDML, inner dentate molecular layer; * CA1 apical dendrites. Scale bars: A–F, 500 μm; G–N, 100 μm; inset in C, 50 μm.
Figure 3.
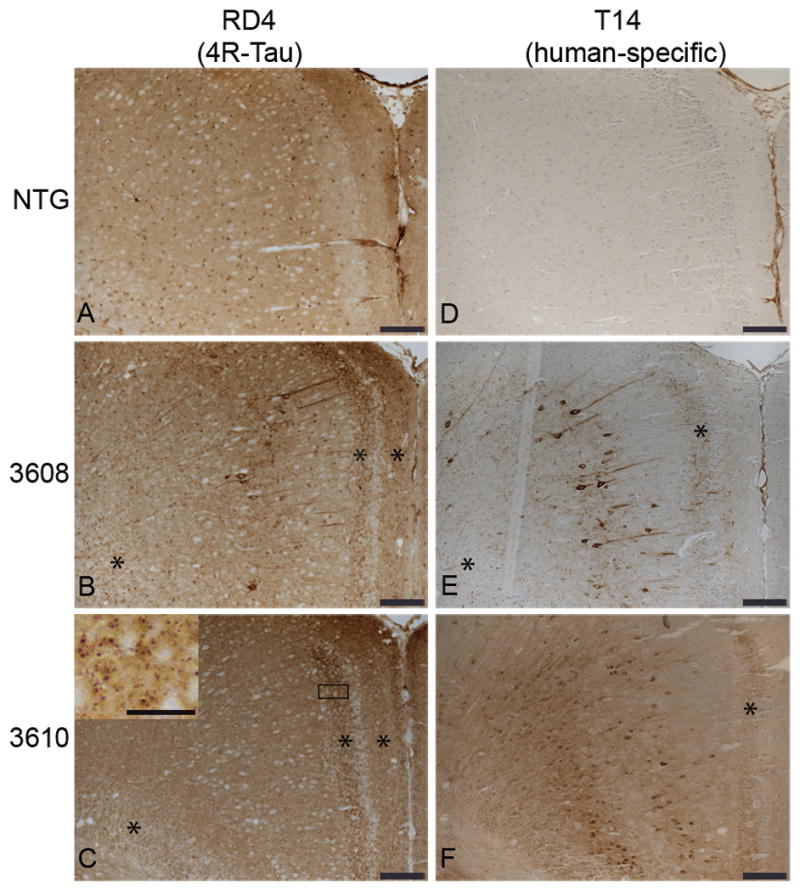
Somatodendritic distribution of 4R-tau in the cingulate gyrus (CG) of E391 transgenic mice. (A–F) Coronal paraffin sections were stained with the 4R-tau specific antibody RD4, which recognizes both mouse and human 4R-tau (A–C) or with the human specific tau antibody T14 (D–F). Non-transgenic (NTG) mice exhibit a normal axonal distribution of 4R-tau in the CG (A). Somatodendritic distribution of 4R-tau in the CG is detectable with the 4R-tau specific (B) and human tau specific (E) antibodies in E391-3608 mice (lower expressers). Only the human specific tau antibody demonstrates somatodendritic localization in the CG in E391-3610 mice (higher expressers) (C, F). *Granular dot-like neuritic processes in CG and supracallosal regions. Inset in (C) is a higher power of boxed region showing granular dot-like neuritic processes. Scale bars: A–F, 100 μm; inset in C, 25 μm.
In the line expressing higher levels of E391-truncated 4R-tau (E391-3610), we observed a similar axonal distribution of tau. In addition, tau immunostaining was prevalent in CA3 and CA1 pyramidal neurons and CA1 apical dendrites, as well as granule cells of the dentate gyrus (Fig. 2C, H, J). CA2 pyramidal neurons were not immunoreactive for tau. This somatodendritic relocalization of tau upon overexpression was previously described (30, 31). In the cortex, the synaptic-like pattern of tau immunostaining in the neuropil was again evident (Figs. 2C, 3C). Although the majority of neuronal cell bodies were negative for RD4, accumulation of tau was detectable in some neuronal cell bodies and extended into dendrites, especially in the more lateral cortical regions (Fig. 2C, inset). In addition, there was a granular dot-like pattern of staining in the cingulate gyrus (CG) and supracallosal region that resembled abnormal neuritic processes (Fig. 3C, asterisks and inset). This staining was more prevalent in deeper layers of the CG.
In the mouse line expressing lower levels of E391-truncated 4R-tau (E391-3608), the normal hilar/mossy fiber axonal staining seen in NTG and E391-3610 mice was prominent using the 4R-tau specific antibody; however, the inner dentate molecular layer was negative (Fig. 2B). As detected in E391-3610 mice, a somatodendritic re-distribution was apparent but the pattern of this distribution was strikingly different, especially in the hippocampus. Dentate granule neurons were mostly negative, except for the outer most superficial granule cells (SGCs), located at the border of the granule cell layer facing the molecular layer (Fig. 2B, G). These SGCs stained intensely for tau in both the cell bodies and dendrites projecting to the dentate molecular layer and were especially evident in the dorsal blade compared to the ventral blade of the dentate gyrus. CA pyramidal neurons were mostly negative, except for scattered neurons in the CA1, primarily in the medial region, in which several soma and dendritic processes projecting through the stratum radiatum stained intensely for tau (Fig. 2B, I). We also observed robust tau immunoreactivity in neuropil and neuronal cell bodies in the amygdala, a result not seen in E391-3610 mice or NTG mice (Fig. 4A, B). Cortical tau immunoreactivity was similar to that observed in E391-3610 mice, except that more neuronal cell bodies and dendrites were positively stained (Figs. 2B, 3B). In addition, neuronal cell bodies and processes were positively stained in the entorhinal cortex (EC), a result that was not apparent in E391-3610 or NTG mice (Fig. 4E, F). We also observed the abnormal granular dot-like staining in the CG and supracallosal region (Figs. 3B, 5B–D, asterisks). These presumptive neuritic processes were also apparent in the EC as well as medial temporal regions such as the amygdala. Pyramidal neurons in the cortex often appeared irregular, with thickened, atypical apical dendrites (Fig. 5A, F; arrowheads). In addition, the 4R-tau specific antibody revealed occasional atypical dystrophic neurites that were non-symmetrical and often abnormally oriented to the cortical surface (Fig. 5B, D–F; arrows), similar to tau-positive dystrophic neurites observed in AD. These dystrophic neurites were most prevalent in the CG but also appeared in the EC.
Figure 4.
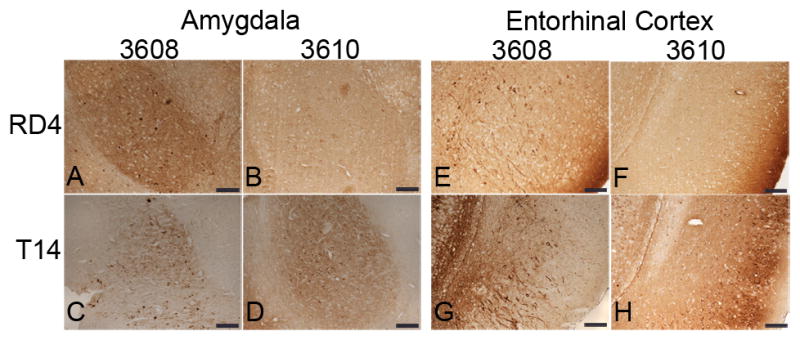
Somatodendritic distribution of 4R-tau in the amygdala and entorhinal cortex (EC) of E391 transgenic mice. (A–H) Coronal paraffin sections were stained with the 4R-tau specific antibody RD4, which recognizes both mouse and human 4R-tau (A, B, E, F) or with the human specific tau antibody T14 (C, D, G, H). Somatodendritic distribution of tau is detectable with both antibodies in the amygdala and EC in E391-3608 mice (lower expressers). Only the human specific tau antibody demonstrates somatodendritic localization in the amygdala and EC in E391-3610 mice (higher expressers). Scale bars: 100 μm
Figure 5.
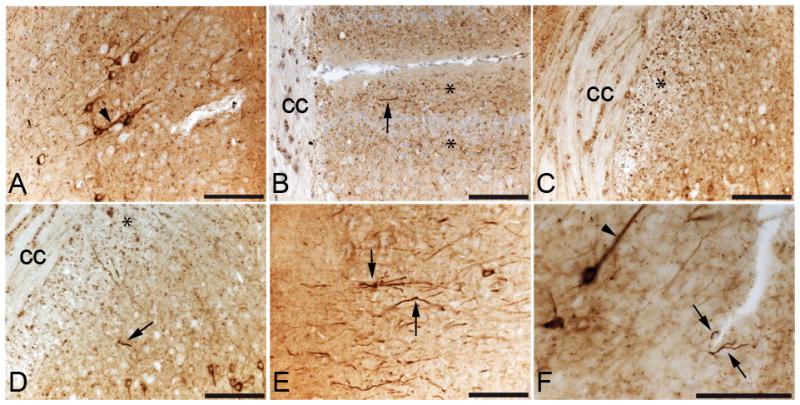
Tau pathology in E391-3608 mice. (A–F) Coronal paraffin sections stained with the 4R-tau specific antibody RD4 (A–E) or Tau2, an antibody that recognizes a pathological conformation of tau (F). Irregular apical dendrite (arrowhead) in the cingulate gyrus (CG) (A) Abnormal granular neuritic processes (*) and dystrophic neurites (arrow) in the CG (B) and in the supracallosal region (C, D); dystrophic neurites (arrows) (E) in the entorhinal cortex; (F) irregular apical dendrites (arrowhead), abnormal granular dot-like neuritic processes and dystrophic neurites (arrow) in the CG detected with Tau2 (F). cc, corpus callosum. Scale bars: 100 μm
In the transgenic E391-3608 mouse line, we observed mild staining of glial cells, similar to that observed in NTG mice. Tau immunoreactivity in glial cells in NTG and transgenic animals appeared to be specific for mouse tau rather than human tau because the mouse tau-specific antibody T49 displayed robust staining in glial cells in all mice (NTG, 3608 and 3610), whereas the human-specific tau antibody T14 failed to detect this glial immunoreactivity (data not shown). Additionally, glial immunoreactivity of mouse-specific tau appeared to be age-dependent, i.e. young adult animals (postnatal day 24) did not show immunoreactivity for T49 in glial cells, whereas aged animals (1 and 2 years) displayed robust T49 immunoreactivity in glial cells (data not shown). Tau immunoreactivity in glial cells under normal conditions was somewhat surprising because this has not been previously documented. However, we did not pursue this finding because it was specific for mouse tau also in NTG mice and not specific to the tau transgene under study.
The E391 Transgene Drives tau Pathology
Our 2 transgenic mouse lines express human E391 truncated 4R-tau on a mouse tau background. Thus the tau immunostaining described above using the 4R-tau specific antibody (RD4) could be due to expression of human and/or mouse tau. To determine the contribution of the human E391 truncated 4R-tau, we immunostained with T14, a tau antibody that is specific for human tau. As expected, T14 did not detect tau in NTG mice (Figs. 2D, 3D). In transgenic mice, the pattern of immunostaining observed using the human-specific antibody was similar to that obtained with the RD4 antibody, although less robust (Figs. 2E, F, K–N, 3E, F). Normal axonal distribution was apparent in the mossy fibers and hilus, although the synaptic-like pattern of tau immunostaining in the cortex, stratum radiatum and stratum oriens (staining of neuropil) was less evident with the human specific antibody, particularly in the lower expressing mouse line (E391-3608). In contrast, the somatodendritic redistribution of tau was more prevalent (more cell body and dendritic staining) using the human-specific antibody, particularly in the higher expressing mouse line (E391-3610). Interestingly, the human tau-specific antibody positively stained neurons in the amygdala and EC in both mouse lines, whereas only the lower expressing mouse line (E391-3608) demonstrated tau immunoreactivity in these regions using the 4R-tau specific antibody (Fig. 4). The human tau-specific antibody also detected the granular dot-like staining of presumptive neuritic processes in the CG in both mouse lines, although at low levels. Only the lower expressing mouse line displayed this abnormal granular staining in the EC and amygdala. In addition, the human specific tau antibody detected the irregularly shaped dystrophic neurites in the lower expressing mouse line, similar to what was observed using the 4R-tau specific antibody. Thus, the truncated human tau expressed in these mice exhibited some normal axonal distribution as well as somatodendritic redistribution and later stages of tau pathology, including atypical neurites.
E391 Mice Exhibit Conformational Changes in tau Associated with tau Pathology
The antibodies Alz50, MC1, and Tau2 recognize a pathological conformation of tau that is accompanied by the assembly of tau aggregates similar to those seen in AD. The conformation of tau recognized by these antibodies is an earlier stage of tau pathology, i.e. preceding tangle formation. As expected, NTG mice were not immunoreactive for Alz50, MC1 and Tau2 immunostaining (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A279). Mice expressing higher levels of human E391-truncated 4R-tau (E391-3610) positively stained with these antibodies; the distribution was similar to that observed with the 4R-tau-specific antibody RD4, and the human tau-specific antibody T14, with a few exceptions. Immunostaining with Alz50 demonstrated the normal axonal distribution of tau in the hippocampus, but not the somatodendritic redistribution observed with the RD4 or T14 antibodies (Fig. 6B, D, Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A279). Furthermore, neither cortical neuropil nor neurons (including in CG and EC) were immunoreactive for Alz50; however, abnormal neuritic processes were immunoreactive (Fig. 7B, asterisks). In contrast, immunostaining using the MC1 and Tau2 antibodies did demonstrate a somatodendritic distribution in cortical and hippocampal neurons (Figs. 6, 7, Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A279). None of these conformation-specific tau antibodies positively stained neuronal soma in the amygdala (Fig. 7H, J, L) or EC (data not shown), whereas all 3 stained abnormal neuritic processes in the CG (Fig. 7B, D, F, asterisks) and EC.
Figure 6.
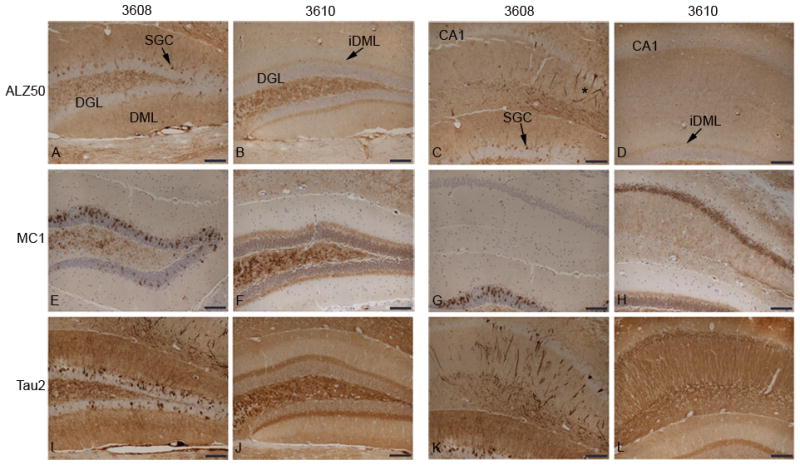
Conformational changes of tau in the hippocampus in E391 transgenic mice. (A–L) Coronal paraffin sections were stained with ALZ50 (A–D), MC1 (E–H) and Tau2 (I–L). E391-3608 mice (lower expressers) exhibit widespread somatodendritic distribution of conformational tau when immunostained with Alz50 (A, C), and Tau2 (I, K). Somatodendritic distribution of conformational tau detected by MC1 (E, G) is much more limited and does not extend into the CA1 apical dendrites or cell bodies. E391-3610 mice (higher expressers) exhibit somatodendritic distribution of conformational tau when immunostained with MC1 (F, H) and Tau2 (J, L), but only an axonal distribution of conformational tau when stained with Alz50 (B, D). DGL, dentate granule layer; DML, dentate molecular layer; iDML, inner dentate molecular layer; SGC, superficial granule cells; *CA1 apical dendrites. Scale bars: 100 μm.
Figure 7.
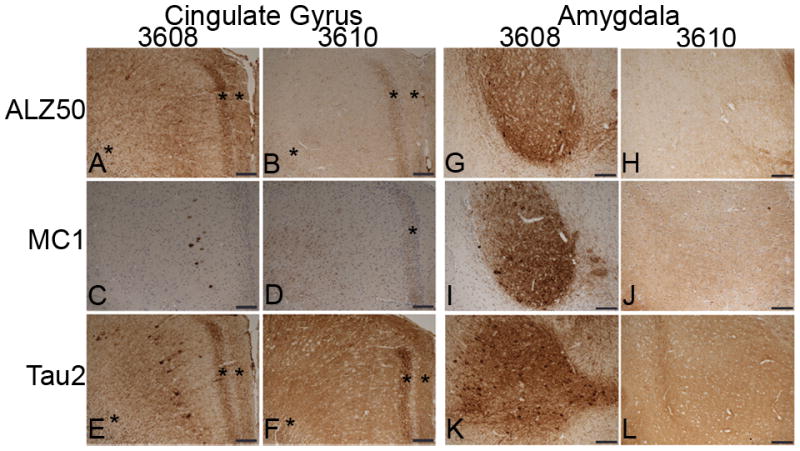
Conformational changes of tau in the cingulate gyrus (CG) and amygdala in E391 transgenic mice. (A–L) Coronal paraffin sections were stained with ALZ50 (A, B, G. H), MC1 (C, D, I, J) and Tau2 (E, F, K, L). Somatodendritic distribution of conformational tau is apparent in E391-3608 mice (lower expressers) in the CG and amygdala. Little to no somatodendritic localization of conformational tau is detectable in the CG with the Alz50 or MC1 antibody in E391-3610 mice (higher expressers), although mild somatodendritic localization is detected in the CG with Tau2. The amygdala is not immunoreactive for conformation-specific antibodies in E391-3610 mice. *Abnormal granular dot-like neuritic processes in CG and supracallosal regions. Scale bars: 100 μm
Mice expressing lower levels of E391-truncated 4R-tau (E391-3608) also stained positively with the conformation-specific tau antibodies. Immunostaining with Alz50 and Tau2 were identical to that observed using the RD4 and T14 antibodies, with both neuropil and somatodendritic distribution in the hippocampus, CG, EC, and amygdala (Figs. 6, 7, Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A279), as well as atypical neurites. Neuronal accumulation of tau appeared as dense staining surrounding the nucleus and extended into dendrites, and in some cases, Tau2 immunoreactivity completely filled the soma (Fig. 5F). The MC1 antibody demonstrated redistribution to neuronal cell bodies, but the dendritic staining was absent. Immunoreactive cell bodies were limited to the cortex (including the CG and EC), amygdala and SGCs of the dentate gyrus (Figs. 6, 7, Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A279). The neuronal cell bodies and apical dendrites that stained positively with other tau antibodies in the medial region of CA1 pyramidal cells were negative for MC1 (Fig. 6G). Furthermore, while neuronal cell bodies were positive in the cortex, staining of neuropil was not apparent with the MC1 antibody, although axonal distribution was robust in hippocampal mossy fibers and hilus. As with Alz50 and Tau2, atypical neurites (granular dot-like neuritic processes and dystrophic neurites) in the EC and amygdala were detected by MC1, although much less apparent in the CG.
Hyperphosphorylated tau Accumulates in E391 Mice
Antibody AT8 detects dual phosphorylation sites (phospho-Ser 202 and phospho-Thr 205) characteristic of tau in AD. This pair-wise phosphorylation of tau indicates advanced stage aggregation. As expected, NTG mice were negative for AT8 immunostaining (Fig. 8A–C). Mice expressing higher levels of E391-truncated 4R-tau (E391-3610) showed strong AT8 immunopositivity. We again observed the somatodendritic redistribution detected using the other tau antibodies (RD4, T14, conformation-specific antibodies), and the neuronal accumulation of tau was abundant throughout the cortex and hippocampus (Figs. 8, 9). Interestingly, AT8 stained neurons in the EC and amygdala of these mice (Fig. 9D, F), similar to what was observed using the human tau-specific antibody T14 (Fig. 4). The normal axonal distribution (mossy fibers, hilus, inner dentate molecular layer, neuropil staining of cortex) detected with the other tau antibodies was less apparent with AT8, with the exception of neuropil staining in the EC, amygdala and the molecular layer of the dentate gyrus. Finally, AT8-immunoreactive granular dot-like neuritic processes were detected in the EC, but not the CG.
Figure 8.
E391 truncated tau accumulates in the hippocampus and cortex as AT8-positive deposits. (A–C) Non-transgenic (NTG) mice do not show immunoreactivity for AT8. (D–F) E391-3608 mice (lower expressers) exhibit somatodendritic distribution of phospho- Ser 202/Thr 205 tau throughout the cortex, in hippocampal SGCs of the dentate gyrus, and in CA1 apical dendrites and cell bodies. (G–I) E391-3610 mice (higher expressers) exhibit robust somatodendritic distribution of phospho- Ser 202/Thr 205 tau throughout the cortex, in hippocampal CA3 pyramidal neurons, CA1 apical dendrites and cell bodies and dentate granule cells. Little to no axonal distribution is apparent in either mouse line. DGL, dentate granule layer; SGC, superficial granule cells; *CA1 apical dendrites. Scale bars: A, D, G, 500 μm; B, C, E, F, H, I, 100 μm.
Figure 9.

E391 truncated tau accumulates in the cingulate gyrus (CG), entorhinal cortex and amygdala as AT8-positive deposits. Neuronal cell bodies and dendrites are immunoreactive for AT8 in the CG (A) entorhinal cortex (C, D) and amygdala (E, F) of E391 transgenic mice. Little to no axonal distribution is apparent in either mouse line. Scale bars: 100 μm
Immunostaining for AT8 was also apparent in the E391-3608 line. The somatodendritic redistribution was evident but was not as robust as in E391-3610 mice (Figs. 8, 9). The pattern of this neuronal distribution was similar to that detected with other tau antibodies, including SGCs, pyramidal neurons and apical dendrites in the medial region of CA1, cortical neurons, (including CG and EC) and in the amygdala. As in E391-3610 mice, little axonal distribution was detected using this phospho-specific antibody. Finally, atypical neurites (granular dot-like neuritic processes and dystrophic neurites) were AT8-immunoreactive in the EC and amygdala. The 2 other phospho-tau antibodies related to tau pathology, AT180 (phospho-Thr 231) and CP13 (phospho-Ser 202) were immunoreactive in both transgenic mouse lines and exhibited a somatodendritic distribution (Figure, Supplemental Digital Content 2, http://links.lww.com/NEN/A280), further indicating that tau is hyperphosphorylated in E391 mice.
In summary, truncated E391 tau exhibits a pathological phenotype (conformational change or hyperphosphorylation) in both transgenic mouse lines, although the distribution and the pathological state differ between the lines (Table). In general, pathological tau in the line expressing lower levels of E391-truncated 4R-tau (E391-3608) was conformationally altered and hyperphosphorylated mainly in the SGCs, CG neuronal soma and neuropil, amygdala and EC. Although pathological tau in CA1 neuronal soma and apical dendrites was rare and sporadic, the neurons that were positive were fairly robustly stained. In contrast, pathological tau in the E391-3610 line was conformationally altered and hyperphosphorylated in the inner dentate molecular layer, CA1 neuronal soma and apical dendrites, and CG neuropil. In dentate granular cells, CG neuronal soma, amygdala and EC, pathological tau was mainly hyperphosphorylated and generally did not adopt the conformationally altered phenotype.
Table.
Pathological tau Immunoreactivity in E391 Mice
| Mouse Lines: | ALZ50 | MC1 | Tau2 | AT8 | ||||
|---|---|---|---|---|---|---|---|---|
| 3608 | 3610 | 3608 | 3610 | 3608 | 3610 | 3608 | 3610 | |
| Brain Region | ||||||||
| Superficial granule cells | + | 0 | ++ | 0 | ++ | 0 | ++ | 0 |
| Dentate granule cells | 0 | 0 | 0 | 0 | 0 | + | 0 | ++ |
| Inner dentate molecular layer | 0 | + | 0 | ++ | 0 | ++ | 0 | + |
| CA1 neuronal soma | +/− | 0 | 0 | ++ | +/− | ++ | +/− | ++ |
| CA1 apical dendrites | +/− | 0 | 0 | 0 | +/− | ++ | +/− | ++ |
| CG neuronal soma | + | 0 | + | 0 | + | +/− | + | ++ |
| CG neuropil | ++ | + | 0 | +/− | ++ | ++ | + | + |
| Amygdala | ++ | 0 | ++ | 0 | ++ | 0 | ++ | + |
| EC neuronal soma | ++ | 0 | ++ | 0 | ++ | 0 | ++ | ++ |
None (0), Rare (+/−), Moderate (+), Abundant (++).
CG, cingulate gyrus; EC, entorhinal cortex.
Lack of Tangle Formation in E391 Mice
Detection of tau in the insoluble fraction, somatodendritic redistribution, tau hyperphosphorylation and conformational changes in tau all indicate “pre-tangle” tau pathology. To determine if these mice develop late-stage tau pathology in the form of frank tangle formation, Gallyas silver stain was used to identify fibrillar tau within neurons. We were unable to detect tau by Gallyas silver stain in either transgenic mouse line expressing human E391-truncated 4R-tau (Figure, Supplemental Digital Content 3, http://links.lww.com/NEN/A281). In contrast, we were able to detect numerous Gallyas-positive silver deposits in human AD cases and in transgenic mice harboring the P301S tau mutation (PS19), as previously reported (32). Likewise, we readily detected thioflavin S-positive deposits in AD brain, but not in the transgenic mice (data not shown). These results indicate that tau does not undergo NFT formation in our E391 truncated tau transgenic mouse model.
DISCUSSION
Truncated tau terminating at glutamic acid 391 (E391) greatly potentiates tau polymerization in vitro and is known to exist in NFTs and abnormal neurites in the brains of AD patients (8, 9). To elucidate the role of this C-terminal truncated tau species in the development of tau pathology we generated a mouse model that expresses human 4R-tau truncated at E391. Many pathological changes in the tau protein occur in AD and other tauopathies, including somatodendritic redistribution, aggregation, adoption of a pathological conformation, hyperphosphorylation, and tangle formation (16, 33–35). In this study, we examined the brains of aged E391-truncated 4R-tau transgenic mice for these pathological changes in tau.
We generated 2 mouse lines that express human 4R-tau truncated at E391 at different levels, with higher and lower expression, i.e. E391-3610 and E391-3608, respectively. Both mouse lines exhibit relatively low levels of transgene expression compared to other mouse models of tauopathy. Previous transgenic models overexpressing full-length or truncated wild type human tau have required high levels of tau overexpression (ranging from 4- to 15-fold) to achieve pathological changes in tau and neurotoxicity (20, 21, 36–38). In our study, despite relatively low levels of transgene expression, we observed E391 truncated tau driving pre-tangle pathological changes in tau, including accumulation of tau in the insoluble fraction, somatodendritic redistribution, formation of pathological conformations, and dual phosphorylation of tau at sites associated with AD pathology. These findings are consistent with tau truncation promoting tau aggregation, as has been suggested in previous in vitro studies (11, 39).
Both transgenic mouse lines also exhibited atypical neurites characterized by granular dot-like staining in the CG and EC, which could be detected with a variety of tau antibodies. In E391-3608 mice these presumptive abnormal neuritic processes were also apparent in medial temporal regions such as the amygdala. In addition to this abnormal granular dot-like staining pattern, E391-3608 mice also displayed atypical neurites resembling dystrophic neurites in the CG, EC and amygdala. These dystrophic neurites were irregularly shaped with abnormal orientation to the cortical surface, similar to tau positive dystrophic neurites observed in AD. In addition, pyramidal neurons in the cortex often appeared irregular, with thickened, atypical apical dendrites. These results indicate that expression of human E391-truncated 4R-tau is sufficient to generate much of the tau pathology characteristic of AD and other tauopathies. However, despite these many pathological changes in tau, neither mouse line developed frank tangles, suggesting that whereas low level expression of E391-truncated 4R-tau is sufficient to generate pre-tangle stage tau pathology, additional factors such as full-length tau or longer lifespan are required for frank tangle formation.
Several studies describe transgenic rat models expressing truncated human tau (37, 38). Overexpression (4–5 fold) of a double truncated (N and C-terminal) 3R or 4R human tau species resulted in NFTs in the brainstem and cortex, respectively. Additionally, accumulation of insoluble tau composed of both human truncated tau and endogenous rat tau was observed. Rats expressing the 4R truncated tau also developed progressive muscle weakness, eventually leading to wasting and death. These studies suggest a role for truncated tau in the development of neurofibrillary pathology. Our mouse model expresses tau cleaved only at the C-terminus, perhaps explaining the lack of tangle formation. Both N and C-terminally truncated tau show an enhanced propensity for aggregation and a doubly truncated protein may be more pathogenic. Indeed, a truncated tau fragment lacking both the N and C-terminus is highly aggregation-prone and a mouse model expressing this fragment exhibits extensive tau pathology (40). In addition, our mouse model expresses tau at relatively low levels. It may be that extensive overexpression is required for NFT formation to develop in many mouse models of tau pathology. Still, our model expressing more biologically significant levels of C-terminally truncated tau develops pre-tangle pathological changes that could potentially develop into NFTs if the lifespan of the animal were longer.
Of interest were the varying degrees of tau pathology in the 2 mouse lines, despite the expression of the same transgene. One might expect that the mouse line expressing higher levels of E391-truncated 4R-tau would exhibit more tau pathology, but that was not the case. We detected granular dot-like tau immunoreactivity, suggestive of abnormal neuritic processes, in the EC, CG, and amygdala in the lower expressing E301-3608 mouse line. The EC is the earliest affected brain region in AD, while the CG and amygdala are generally affected at later stages (41). Thus, the presence of abnormal neuritic processes in all these regions of E391-3608 mice suggests a more advanced stage of tau pathology. In addition to granular dot-like neuritic processes, E391-3608 mice also exhibited atypical neurites in the form of tau-positive irregularly shaped and abnormally oriented dystrophic neurites in the CG, EC and amygdala. These dystrophic neurites, which are similar to tau positive dystrophic neurites observed in AD, were never discernible in E391-3610 mice. At present, the significance of greater pathology associated with lower truncated tau expression is unknown. It is possible that lower levels of truncated tau protein may be more toxic because the lower intracellular concentration of tau remains in an unaggregated state longer and may recruit other tau species into toxic conformations. Alternatively, the varying degrees of tau pathology in the 2 transgenic lines may simply be the result of differences in the integration site of the transgene, rather than differences in expression levels. Although differences in integration site can be responsible for the varying degrees of expression level and pathology, because both mouse lines developed tau pathology, the overall phenotype is due to the presence of the E391 transgene and not a non-specific effect of the integration site.
Immunostaining with a panel of tau antibodies that detect both normal and pathological tau revealed differences in the distribution of tau species between the lines that might play a role in their differing tau pathologies. One of the more interesting differences was the greater tau immunoreactivity in neuronal cell bodies and dendrites in the EC and amygdala in E391-3608 mice vs. E391-3610 mice. Mis-sorting of tau to the somatodendritic compartment may occur when tau levels are increased to a level that overwhelms the normal axonal sorting machinery (31, 42). Despite the lower overall expression of truncated tau in E391-3608 mice, it is possible that localized expression in some regions (the EC and amygdala) may be higher than in E391-3610 mice. Moreover, tau developed a pathological conformation (as detected by Alz50, MC1 and Tau2) in the EC and amygdala in E391-3608, but not E391-3610 mice. A conformational change definedby an increased immunoreactivity against these antibodies isconsidered to be one of the earliest pathological alterationsof tau in AD (43–45). The increased somatodendritic mis-localization of tau and the development of a pathological conformation of tau in these regions could interrupt normal axonal transport. In fact, Garcia-Sierra et al demonstrated the appearance of E391 truncated tau in extracellular NFTs in the EC of AD subjects (detected with mAb423) and postulated a susceptibility of the perforant pathway, the axons providing major input from the EC to the hippocampus (12). In a separate study these authors report that while both E391 truncated tau and Alz-50 immunoreactive tau exist in NFTs in AD brain, they do not colocalize and appear to occur at different stages during tangle development (46). While this may be the case for NFTs in AD, our data suggest this is not the case in the pre-tangle stage of tau pathology in our transgenic mice, given that E391 tau appears to adopt the Alz50 conformation.
Another interesting observation was the detection of tau (with both non-pathological and pathological tau antibodies) in the SGCs of the dentate gyrus in E391-3608 mice. Tau immunoreactivity in these SGCs was more evident in the dorsal blade than in the ventral blade of the dentate gyrus. SGCs typically are larger cells with more complex dendritic trees than the dentate granule cells residing in the deeper layers (47, 48). They are considered to be older cells, often born prenatally or in the early postnatal period, which have migrated to reside in the superficial portion of the dentate gyrus (49–51). They do not undergo adult neurogenesis that has been described in granule cells residing in the subgranular zone of the dentate gyrus. In addition, the dorsal blade develops relatively early compared to the ventral blade (52). Thus, it is postulated that these SGCs of the dorsal blade are more mature, less plastic, and more susceptible to toxic insult (53). The vulnerability of this subset of neurons to AD pathology has been suggested in an AD mouse model overexpressing human amyloid precursor protein (PDAPP mice), in which the authors describe substantial dendritic pathology (decreased dendritic length) that specifically occurs in these neurons well before the accumulation of amyloid pathology (53). Thus, expression of tau, especially conformationally altered and hyperphosphorylated tau, in these neurons is of great interest.
A third major difference in tau distribution between the 2 mouse lines concerned the hippocampal CA1 pyramidal neurons. Tau expression in this region in E391-3610 mice was robust and homogeneous within the soma and apical dendrites. This immunostaining was detectable with a number of tau antibodies, including RD4 and T14, as well as pathology-specific antibodies AT8, Tau2 and MC1, though MC1 staining appeared primarily in the soma. The somatodendritic redistribution to this region has been described previously and has been attributed to tau overexpression (31). In contrast, tau immunoreactivity in the CA1 region of E391-3608 mice was restricted to scattered cell bodies and apical dendrites, mainly in the medial CA1 pyramidal layer, and the dendrites appeared irregular and discontinuous. This staining was consistent with all of the tau antibodies used in this study (RD4, T14, AT8, Alz50, Tau2), with the exception of MC1, which did not immunostain this region. CA1 apical dendrites receive input via the CA3 Schaffer collaterals and the presence of pathological tau in this region hints at possible alterations of this pathway in our E391 truncated 4R-tau mouse lines. The discontinuous and irregular nature of the apical dendrites in the low expressing mouse line could indicate more severe alterations.
In summary, we describe 2 transgenic mouse models for tauopathy in which approximately endogenous levels of transgenically expressed E391 truncated human tau are sufficient to initiate early stages of tau pathology. These pathological changes occur in the absence of FTDP-17 mutations or other insults on a background of wild type mouse tau. Previous mouse models have shown that human full-length wild type tau is relatively non-toxic as compared to mutant tau (2). In our E391 mice, truncated but otherwise wild type human tau is sufficient to drive pathological changes. These findings suggest that changes in tau proteolysis could give rise to early tauopathy phenotypes.
Supplementary Material
Acknowledgments
This work was supported by National Institute on Aging Grant AG17586 to GDS, Department of Veterans Affairs Merit Review Grant to BCK, and Veterans Affairs, NINDS 5P50NS062684-02 and NIA 2P50AG005136-27 to JBL.
We thank the University of Washington Nathan Shock Center on aging research for supporting mouse transgenics. We thank Elaine Loomis, Leo Anderson and Lynne Greenup for outstanding technical assistance. We thank Virginia Lee and Peter Davies for tau antibodies.
References
- 1.Buee L, Bussiere T, Buee-Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 2.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 3.Mandelkow E, von Bergen M, Biernat J, et al. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 5.Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14:453–7. doi: 10.3233/jad-2008-14415. [DOI] [PubMed] [Google Scholar]
- 6.Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Sierra F, Mondragon-Rodriguez S, Basurto-Islas G. Truncation of tau protein and its pathological significance in Alzheimer’s disease. J Alzheimers Dis. 2008;14:401–9. doi: 10.3233/jad-2008-14407. [DOI] [PubMed] [Google Scholar]
- 8.Mena R, Edwards PC, Harrington CR, et al. Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 1996;91:633–41. doi: 10.1007/s004010050477. [DOI] [PubMed] [Google Scholar]
- 9.Novak M, Jakes R, Edwards PC, et al. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7. 51. Proc Natl Acad Sci U S A. 1991;88:5837–41. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, et al. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470–83. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraha A, Ghoshal N, Gamblin TC, et al. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci. 2000;113(Pt 21):3737–45. doi: 10.1242/jcs.113.21.3737. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Sierra F, Wischik CM, Harrington CR, et al. Accumulation of C-terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer’s disease. J Chem Neuroanat. 2001;22:65–77. doi: 10.1016/s0891-0618(01)00096-5. [DOI] [PubMed] [Google Scholar]
- 13.de Silva R, Lashley T, Gibb G, et al. Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol. 2003;29:288–302. doi: 10.1046/j.1365-2990.2003.00463.x. [DOI] [PubMed] [Google Scholar]
- 14.Kosik KS, Orecchio LD, Binder L, et al. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–25. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 15.Ishihara T, Higuchi M, Zhang B, et al. Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J Neurosci. 2001;21:6026–35. doi: 10.1523/JNEUROSCI.21-16-06026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmel G, Mager EM, Binder LI, et al. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–95. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 17.Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer’s disease. J Neurosci Res. 1999;55:713–23. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 18.Jicha GA, Bowser R, Kazam IG, et al. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–32. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 19.Uchihara T, Duyckaerts C, Seilhean D, et al. Exclusive induction of tau2 epitope in microglia/macrophages in inflammatory lesions-tautwopathy distinct from degenerative tauopathies. Acta Neuropathol. 2005;109:159–64. doi: 10.1007/s00401-004-0922-3. [DOI] [PubMed] [Google Scholar]
- 20.Probst A, Gotz J, Wiederhold KH, et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–81. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 21.Ishihara T, Hong M, Zhang B, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–62. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 22.Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18:1825–38. doi: 10.1093/hmg/ddp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janke C, Beck M, Stahl T, et al. Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Brain Res Mol Brain Res. 1999;68:119–28. doi: 10.1016/s0169-328x(99)00079-0. [DOI] [PubMed] [Google Scholar]
- 24.Kampers T, Pangalos M, Geerts H, et al. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer’s disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- 25.Takuma H, Arawaka S, Mori H. Isoforms changes of tau protein during development in various species. Brain Res Dev Brain Res. 2003;142:121–7. doi: 10.1016/s0165-3806(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 26.Bullmann T, de Silva R, Holzer M, et al. Expression of embryonic tau protein isoforms persist during adult neurogenesis in the hippocampus. Hippocampus. 2007;17:98–102. doi: 10.1002/hipo.20255. [DOI] [PubMed] [Google Scholar]
- 27.McMillan P, Korvatska E, Poorkaj P, et al. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–8. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trojanowski JQ, Schuck T, Schmidt ML, et al. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–15. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- 30.Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–90. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 31.Eckermann K, Mocanu MM, Khlistunova I, et al. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007;282:31755–65. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- 32.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Avila J, Perez M, Lim F, et al. Tau in neurodegenerative diseases: tau phosphorylation and assembly. Neurotox Res. 2004;6:477–82. doi: 10.1007/BF03033284. [DOI] [PubMed] [Google Scholar]
- 34.Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–43. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 36.Spittaels K, Van den Haute C, Van Dorpe J, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–65. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filipcik P, Zilka N, Bugos O, et al. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Zilka N, Filipcik P, Koson P, et al. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–8. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 39.von Bergen M, Barghorn S, Biernat J, et al. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005;1739:158–66. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Mocanu MM, Nissen A, Eckermann K, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–48. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 42.Konzack S, Thies E, Marx A, et al. Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci. 2007;27:9916–27. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uboga NV, Price JL. Formation of diffuse and fibrillar tangles in aging and early Alzheimer’s disease. Neurobiol Aging. 2000;21:1–10. doi: 10.1016/s0197-4580(00)00091-9. [DOI] [PubMed] [Google Scholar]
- 44.Weaver CL, Espinoza M, Kress Y, et al. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging. 2000;21:719–27. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 45.Wolozin BL, Pruchnicki A, Dickson DW, et al. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648–50. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Sierra F, Ghoshal N, Quinn B, et al. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- 47.Claiborne BJ, Amaral DG, Cowan WM. Quantitative, three-dimensional analysis of granule cell dendrites in the rat dentate gyrus. J Comp Neurol. 1990;302:206–19. doi: 10.1002/cne.903020203. [DOI] [PubMed] [Google Scholar]
- 48.Green EJ, Juraska JM. The dendritic morphology of hippocampal dentate granule cells varies with their position in the granule cell layer: a quantitative Golgi study. Exp Brain Res. 1985;59:582–6. doi: 10.1007/BF00261350. [DOI] [PubMed] [Google Scholar]
- 49.Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990;301:365–81. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- 50.Bayer SA, Yackel JW, Puri PS. Neurons in the rat dentate gyrus granular layer substantially increase during juvenile and adult life. Science. 1982;216:890–2. doi: 10.1126/science.7079742. [DOI] [PubMed] [Google Scholar]
- 51.Schlessinger AR, Cowan WM, Gottlieb DI. An autoradiographic study of the time of origin and the pattern of granule cell migration in the dentate gyrus of the rat. J Comp Neurol. 1975;159:149–75. doi: 10.1002/cne.901590202. [DOI] [PubMed] [Google Scholar]
- 52.Wang S, Scott BW, Wojtowicz JM. Heterogenous properties of dentate granule neurons in the adult rat. J Neurobiol. 2000;42:248–57. [PubMed] [Google Scholar]
- 53.Wu CC, Chawla F, Games D, et al. Selective vulnerability of dentate granule cells prior to amyloid deposition in PDAPP mice: digital morphometric analyses. Proc Natl Acad Sci U S A. 2004;101:7141–6. doi: 10.1073/pnas.0402147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.