

Dominant Cx26 mutations that cause keratitis-ichthyosis-deafness syndrome (KIDS) show increased hemichannel activity. Transgenic expression of these mutations recapitulates human skin disease in mice. Excess hemichannel activity persists in diseased epidermis from the transgenic mice. Thus hemichannel activity may be a novel therapeutic target in the treatment of KIDS.
Abstract
Mutations in the GJB2 gene (Cx26) cause deafness in humans. Most are loss-of-function mutations and cause nonsyndromic deafness. Some mutations produce a gain of function and cause syndromic deafness associated with skin disorders, such as keratitis-ichthyosis-deafness syndrome (KIDS). Cx26-G45E is a lethal mutation linked to KIDS that forms constitutively active connexin hemichannels. The pathomechanism(s) by which mutant Cx26 hemichannels perturb normal epidermal cornification are poorly understood. We created an animal model for KIDS by generating an inducible transgenic mouse expressing Cx26-G45E in keratinocytes. Cx26-G45E mice displayed reduced viability, hyperkeratosis, scaling, skin folds, and hair loss. Histopathology included hyperplasia, acanthosis, papillomatosis, increased cell size, and osteal plugging. These abnormalities correlated with human KIDS pathology and were associated with increased hemichannel currents in transgenic keratinocytes. These results confirm the pathogenic nature of the G45E mutation and provide a new model for studying the role of aberrant connexin hemichannels in epidermal differentiation and inherited connexin disorders.
INTRODUCTION
Connexins (Cx) were first identified as the subunit proteins of the intercellular membrane channels that cluster in the cell communication structures known as gap junctions (Revel and Karnovsky, 1967; Goodenough, 1974). Connexin proteins have been studied for 50 years in the context of these intercellular channels and have been well characterized with regard to their role in mediating the direct exchange of ions, second messengers, metabolites, and short interfering RNAs between adjacent coupled cells (Robertson, 1963; Kanno and Loewenstein, 1964; Lawrence et al., 1978; Bruzzone et al., 1996; Harris, 2001; Wei et al., 2004; Valiunas et al., 2005). More recently, connexins have been shown to also be capable of forming functional hemichannels in nonjunctional plasma membranes that can enable the exchange of molecules between the cytosol and the extracellular space, creating the possibility of additionally influencing neighboring cells through paracrine signaling mechanisms (Bennett et al., 2003; Goodenough and Paul, 2003; Saez et al., 2003; Evans et al., 2006). The role played by connexin hemichannels in normal physiology is poorly understood (Scemes et al., 2009; Scemes, 2011), but there is accumulating evidence showing that their activity can be deleterious under certain pathological conditions, including inherited disorders caused by connexin mutations (Lee and White, 2009; Schalper et al., 2009; Saez et al., 2010; Levit et al., 2011).
Several human hereditary diseases, including hearing loss and skin disorders, have been linked to mutations in connexin genes (White and Paul, 1999; Richard, 2005; Lee and White, 2009). For example, mutations in GJB2 (Cx26) can cause both nonsyndromic hearing loss and syndromic hearing loss associated with a variety of skin disorders, including palmoplantar keratoderma, Vohwinkel syndrome, Bart–Pumphrey syndrome, and keratitis–ichthyosis–deafness syndrome (KIDS; Richard et al., 1998, 2002, 2004; Maestrini et al., 1999; van Steensel et al., 2002). Functional characterization of the disease-associated GJB2 mutations has alluded to different pathomechanisms underlying nonsyndromic sensorineural hearing loss (SNHL) and syndromic hearing loss associated with skin disorders. Cx26 mutants causing nonsyndromic SNHL without skin manifestations have been shown to cause either a partial or total loss of gap junction function (White, 2000; Mese et al., 2007; Lee and White, 2009). In contrast, mutations leading to syndromic SNHL and skin disorders were shown 1) to have impaired intracellular trafficking, leading to the accumulation of mutant connexin proteins within cells; 2) to inhibit transport and/or function of other coexpressed wild-type connexins (trans-dominant inhibition); and 3) to generate constitutively active connexin hemichannels. Thus mutant connexin proteins causing skin disease seem to acquire novel properties, resulting in a gain-of-function effect (Rouan et al., 2001; Bakirtzis et al., 2003; Bruzzone et al., 2003; Mese et al., 2004, 2008; Montgomery et al., 2004; Beltramello et al., 2005; Gerido et al., 2007; Lee et al., 2009). In addition, a dramatic increase in Cx26 expression has been observed in human epidermis following pathological or experimentally induced changes in keratinocyte proliferation and differentiation, such as in response to retinoic acid treatment, in psoriatic skin lesions, in palmoplantar keratoderma, and in experimentally wounded skin (Masgrau-Peya et al., 1997; Rivas et al., 1997; Labarthe et al., 1998; Lucke et al., 1999; Rouan et al., 2001). Taken together, these studies establish a strong correlation between increased Cx26 expression and increased keratinocyte proliferation and differentiation. In turn, the increased Cx26 expression would also amplify any pathological gain of function induced by Cx26 mutations associated with skin disease, forming a positively reinforcing loop of mutation-driven disease.
Adult skin is composed of the mesodermally derived dermis and the ectodermally derived epidermis. The epidermis mainly contains keratinocytes forming four layers: the basal layer, the spinous layer, the granular layer, and the cornified layer. Cells in the basal layer continuously proliferate and terminally differentiate as they move upward into the spinous, granular, and cornified layers prior to being ultimately shed (Blanpain and Fuchs, 2006, 2009). Prior studies reported differential expression of several connexins in keratinocytes of the basal, spinous, and granular layers (Caputo and Peluchetti, 1977; Risek et al., 1992; Goliger and Paul, 1994). At least nine connexin genes, including Cx26, are expressed during keratinocyte differentiation, and they show complex overlapping spatial and temporal expression patterns (Salomon et al., 1994; Di et al., 2001; Richard, 2005; Mese et al., 2007). For the epidermis to function properly, basal keratinocytes must maintain a delicate balance between proliferation and differentiation mechanisms (Blanpain and Fuchs, 2009). Any perturbation of this balance can lead to skin abnormalities, like the hyperkeratotic skin disorders caused by dominant gain-of-function mutations affecting Cx26 (Richard, 2000).
The most severe genetic disorder due to GJB2 (Cx26) mutations is the keratitis-ichthyosis-deafness syndrome. KIDS is an ectodermal dysplasia characterized by photophobia and vascularizing keratitis, SNHL or deafness, erythrokeratoderma, nail and hair abnormalities, and an increased prevalence of squamous cell carcinoma (Richard et al., 2002; van Steensel et al., 2002; van Steensel, 2004; Mazereeuw-Hautier et al., 2007). KIDS patients also have recurrent cutaneous infections that can lead to lethal septicemia in pediatric patients, particularly those carrying the Cx26-G45E mutation (Janecke et al., 2005; Griffith et al., 2006; Jonard et al., 2008; Sbidian et al., 2010; Koppelhus et al., 2011). The phenotypic spectrum of KIDS is broad and may also include features of a follicular occlusion triad encompassing dissecting folliculitis, hidradenitis suppurativa, and cystic acne (Montgomery et al., 2004; Maintz et al., 2005), mucositis (Brown et al., 2003; Lazic et al., 2008), or KIDS with proliferative pilar cysts (Nyquist et al., 2007). Current treatment of KIDS is symptomatic and aims to alleviate symptoms, and includes topical or systemic antibiotics/antifungals, keratolytics, and moisturizers (Richard, 2005; Abdollahi et al., 2007; Braun-Falco, 2009).
At least 10 different Cx26 mutations have been linked to KIDS in humans, and in vitro analysis demonstrated that the majority of these mutations lead to constitutively active hemichannels that significantly compromise cell viability and intracellular calcium homeostasis (Montgomery et al., 2004; Stong et al., 2006; Gerido et al., 2007; Lee et al., 2009; Sanchez et al., 2010; Terrinoni et al., 2010). Although the in vitro evidence generated thus far strongly suggests that hemichannel dysfunction significantly compromises cell homeostasis, the role of mutant Cx26 hemichannels in vivo during epidermal differentiation is still poorly understood. To better characterize the mechanisms leading to KIDS in affected individuals and to evaluate the role of aberrant hemichannels in an animal model of this disorder, we generated an inducible transgenic mouse that expresses the Cx26-G45E KIDS mutation in basal keratinocytes.
RESULTS
Inducible expression of Cx26-G45E in epithelial cells leads to increased dye uptake
It was previously demonstrated that constitutive Cx26-G45E expression caused cell death linked to increased hemichannel activity in both Xenopus oocytes and mammalian cells (Stong et al., 2006; Gerido et al., 2007). To develop a doxycycline-inducible expression system, we subcloned wild-type Cx26 or Cx26-G45E into pTRE2, including an internal ribosome entry site (IRES) after the coding sequence, followed by the enhanced green fluorescent protein (EGFP), and cotransfected it into HeLa cells with the pTet-On plasmid. Exposure of untransfected HeLa cells (Figure 1A) to doxycycline did not induce EGFP (Figure 1B) or Cx26-G45E (Figure 1C) protein expression. In contrast, cells cotransfected with pTRE2-Cx26-G45E-IRES-EGFP and pTet-On (Figure 1D) produced a robust EGFP signal (Figure 1E) and expressed high levels of Cx26-G45E (Figure 1F) after 24 h of incubation in doxycycline. To determine whether the expressed Cx26-G45E exhibited increased hemichannel activity, cells transfected with either wild-type Cx26 or Cx26-G45E were incubated with neurobiotin tracer in the extracellular medium and washed, fixed, and stained with rhodamine-conjugated avidin. Wild-type Cx26-expressing cells (Figure 1G) showed a strong EGFP signal (Figure 1H) but limited or no dye uptake (Figure 1I). Cells expressing Cx26-G45E (Figure 1J) displayed an equivalent EGFP signal (Figure 1K) in conjunction with a greatly increased accumulation of cytoplasmic neurobiotin (Figure 1L). These data show that doxycycline-inducible expression of Cx26-G45E resulted in increased dye uptake in transfected HeLa cells.
FIGURE 1:

Inducible expression of Cx26-G45E in HeLa cells increases dye uptake. Untransfected HeLa cells (A, blue DAPI stain) failed to express either EGFP (B, green) or Cx26 (C, red) when treated with doxycycline. Cells cotransfected with pTRE2-Cx26-G45E-IRES-EGFP and pTet-On (D) showed a strong EGFP signal (E) and expressed high levels of Cx26-G45E (F) 24 h after doxycycline addition. To test for increased hemichannel activity, cells expressing either wild-type Cx26 or Cx26-G45E were incubated with neurobiotin tracer. Even though wild-type Cx26 expressing cells (G) showed a strong EGFP signal (H), they had very limited or no dye uptake from the extracellular medium (I, red). Cells expressing Cx26-G45E (J) displayed a similar EGFP signal as wild-type Cx26 expressing cells (K) but showed a dramatic increase in the accumulation of neurobiotin (L), consistent with an elevation of hemichannel activity due to the Cx26-G45E mutation. Scale bar, 5 μm.
Expression of Cx26-G45E increases whole-cell membrane current
If the increased dye uptake seen in Cx26-G45E–expressing cells was mediated by the opening of connexin hemichannels, then one would predict that whole-cell membrane currents would also be greatly increased and that unitary channel conductance could be resolved. To test this hypothesis, we transfected N2A cells with either wild-type Cx26 or Cx26-G45E and measured membrane currents and channel conductance by whole-cell patch clamp electrophysiology (Figure 2). Untransfected N2A cells displayed minimal membrane currents when stepped to membrane potentials between −90 and +90 mV (Figure 2A). Membrane currents recorded from N2A cells transfected with wild-type Cx26 (Figure 2B) were not appreciably different from untransfected cells. However, Cx26-G45E–expressing N2A cells exhibited much larger whole-cell membrane currents at both hyperpolarizing and depolarizing potentials (Figure 2C). Plotting the current density as a function of membrane potential (Figure 2D) showed that at all voltages tested, Cx26-G45E currents were at least twofold to threefold larger (p < 0.01, analysis of variance [ANOVA]) than those recorded in either untransfected or wild-type Cx26 transfected cells. In some cells, the activity of single Cx26-G45E hemichannels could be resolved (Figure 2E). In this example, hemichannels had a unitary conductance value of ∼320 pS at a membrane potential of −30 mV measured in 120 mM K+ aspartate−. This value is in good agreement with recently reported human Cx26 hemichannel data (Sanchez et al., 2010). Thus expression of Cx26-G45E resulted in a significantly increased membrane current in transfected N2A cells mediated by connexin hemichannels.
FIGURE 2:

Cx26-G45E expression induces hemichannel currents. Untransfected N2A cells (A) and N2A cells transfected with wild-type Cx26 (B) had similar membrane currents when stepped to membrane potentials between −90 and +90 mV. N2A cells expressing Cx26-G45E induced much larger whole-cell membrane currents at both hyperpolarizing and depolarizing potentials (C). A plot of current density vs. voltage (D) showed that Cx26-G45E currents were significantly larger than those recorded in either untransfected or wild-type Cx26 expressing cells at all tested voltages (p < 0.01, ANOVA). In addition to whole-cell membrane currents, single-channel activity was observed in Cx26-G45E–transfected cells with a unitary conductance value of ∼320 pS (E), consistent with the size expected for a Cx26 hemichannel.
Generation of inducible transgenic mice expressing Cx26-G45E in the epidermis
Using in vitro assays (Figures 1 and 2), we identified significant changes in the functional behavior of the Cx26-G45E mutation (Gerido et al., 2007; Sanchez et al., 2010). To test whether the aberrant hemichannel properties of Cx26-G45E generate epidermal pathology in vivo in an animal model, we generated transgenic mice with an inducible expression of Cx26-G45E in basal keratinocytes of the epidermis under the control of a tetracycline-responsive promoter (TRE-minCMV). The transgene contained the Cx26-G45E coding sequence, followed by an IRES and EGFP, producing a bicistronic mRNA encoding G45E and EGFP as independent proteins (Figure 3A). This TRE-G45E construct was injected into zygotic pronuclei, and positive founder mice were interbred with a second transgenic strain expressing the reverse tetracycline transactivator (rtTA) under the control of the keratin14 (K14) promoter (Nguyen et al., 2006) to produce doubly transgenic animals. Following induction with doxycycline, K14-rtTA transgenic mice only produced rtTA transcripts in total epidermal RNA samples, whereas K14-rtTA plus TRE-G45E double-transgenic animals made both rtTA and G45E transcripts when assayed by reverse transcription (RT)-PCR (Figure 3, B–E). Transgenic animals were then backcrossed to the SKH1 hairless mouse strain (Benavides et al., 2009) to facilitate direct observation of the epidermis. As the multiple crosses required to generate these mice resulted in a mixed genetic background, K14-rtTA plus TRE-G45E double-transgenic animals were always compared with either TRE-G45E or K14-rtTA single-transgenic littermates as controls in all experiments. Examination of frozen sections of head epidermis by fluorescence microscopy showed an absence of EGFP and Cx26 in single K14-rtTA transgenic animals (Figure 4A). In contrast, K14-rtTA plus TRE-G45E double-transgenic mice showed a large induction of EGFP in keratinocytes and a punctate pattern of Cx26-G45E staining (Figure 4B). In vivo fluorescence detection of EGFP expression in whole animals after 2 wk of doxycycline induction showed that transgene activity was induced throughout the epidermis (Figure 4, C and D). These data document that we generated a new transgenic model of KIDS in which we can detect the inducible expression of a transgenic bicistronic transcript and its protein products in the epidermis.
FIGURE 3:

Generation of doxycycline-inducible Cx26-G45E transgenic mice. In the diagram of the construct used for generation of doxycycline-inducible Cx26-G45E transgenic mice (A), the TRE-minCMV promoter drives transcription of an mRNA encoding Cx26-G45E followed by an IRES and the EGFP. Cx26-G45E animals were interbred with mice expressing the reverse tetracycline transactivator (rtTA) controlled by the keratin14 (K14) promoter to produce double-transgenic animals, which were genotyped by PCR of tail DNA (B, D) and analyzed for gene expression by RT-PCR of skin RNA (C, E). After doxycycline induction, K14-rtTA single-transgenic mice (animals 1 and 2) produced only rtTA transcripts in total epidermal RNA samples, whereas K14-rtTA plus TRE-G45E double-transgenic animals made both rtTA and G45E transcripts (animals 3 and 4).
FIGURE 4:

Transgenic expression of Cx26-G45E in mouse epidermis. Frozen sections of head epidermis from the single K14-rtTA transgenic animals (A) showed no EGFP (green) or Cx26 expression (red). K14-rtTA plus TRE-G45E double-transgenic mice showed a large induction of EGFP in keratinocytes and a punctate pattern of Cx26-G45E staining (B). Whole-animal imaging of double transgenic mice (C, right) after 2 wk of doxycycline induction demonstrated skin abnormalities, including erythrokeratoderma and epidermal scaling that was not seen in single transgenic control animals (C, left). Skin abnormalities correlated with activation of the TRE-G45E transgene, which was monitored by EGFP fluorescence in living animals (D, right). Scale bar, A and B, 15 μm.
Cx26-G45E transgenic mice develop epidermal abnormalities
In patients with KIDS the epidermal differentiation process is severely disturbed, as evidenced by hyperkeratosis of the skin, erythrokeratoderma, and osteal plugging (Richard et al., 2002; van Steensel et al., 2002). Patients with the lethal Cx26-G45E mutation also suffer bacterial infections and succumb to septicemia within the first year of life (Janecke et al., 2005; Griffith et al., 2006; Jonard et al., 2008; Sbidian et al., 2010). To determine whether the inducible Cx26-G45E transgenic mice developed epidermal pathology consistent with KIDS, we first analyzed neonatal mice that had been induced with doxycycline from conception. When the TRE-G45E transgene was induced in utero and expression was maintained postnatally, K14-rtTA plus TRE-G45E double-transgenic mice had a mortality rate exceeding 50% by weaning and surviving animals were generally in poor health (data not shown). In contrast, K14-rtTA plus TRE-G45E double-transgenic animals raised without doxycycline exhibited normal postnatal development, reproductive capability, and lifespan. When the TRE-G45E transgene was first induced with doxycycline in adults (5 wk of age), K14-rtTA plus TRE-G45E double-transgenic mice developed skin abnormalities within 7–14 d of induction. These changes manifested with erythrokeratoderma and epidermal scaling (Figure 4C). When mice induced as adults were maintained for longer periods, the skin pathology progressively worsened. After 10 wk of transgene expression, severe skin changes were evident, with an extensively thickened epidermis leading to scaling and deepened skin folds with a rough, coarse-grained appearance (Figure 5). The skin phenotype and greatly reduced survival of induced K14-rtTA plus TRE-G45E double-transgenic mice is consistent with the disease seen in human KIDS patients with the G45E mutation.
FIGURE 5:
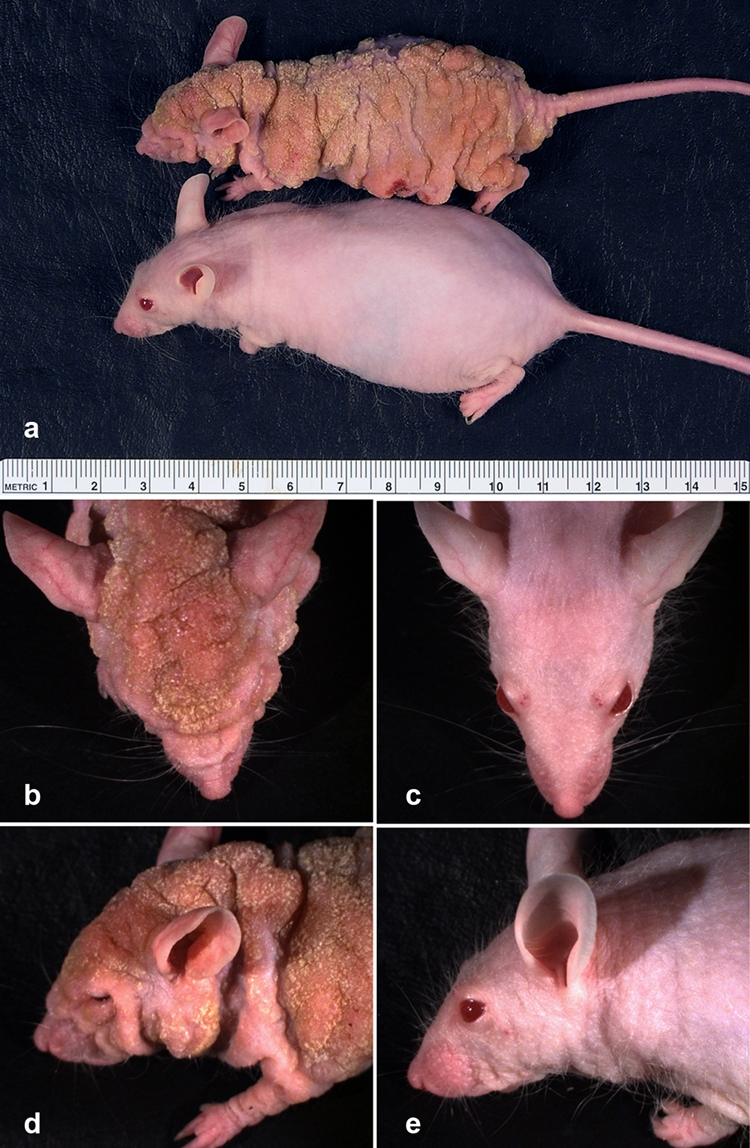
Cx26-G45E transgenic mice develop skin abnormalities. Animals were maintained on doxycycline for 10 wk after initial induction at 5 wk of age. K14-rtTA plus TRE-G45E double-transgenic mice showed severely altered skin compared with single-transgenic control littermates (A). Higher-power views of the head revealed an extensively thickened epidermis with deepened skin folds and a coarse-grained appearance in the double-transgenic mouse (B, D) compared with the normal adult skin of the control mouse (C, E). The major units of the ruler in A are centimeters.
Histological examination of adult head skin from control (Figure 6A) or K14-rtTA plus TRE-G45E double-transgenic mice induced for 4 wk (Figure 6B) showed a noticeably thickened epidermis (acanthosis) and greater undulation of the epidermis into the dermis (papillomatosis) in Cx26-G45E animals compared with control littermates. The stratum corneum was thickened and compact but orthokeratotic. The hair follicle epithelium was also acanthotic and hyperkeratotic, leading to horny cysts and follicular plugging. Keratinocytes appeared large, and there was also an increased number of mitotic epidermal cells in Cx26-G45E animals compared with the control littermates, suggesting possible hyperproliferation. The subepidermal dermis showed elongated and dilated capillaries and a mild lymphocytic infiltration. Similar histological features were seen in skin samples taken from trunk, tail, or paw (data not shown). After 10 wk of transgene induction, the epidermis displayed a massive hyperkeratosis with frequent keratotic plugging and extensive hair follicle and sebaceous gland atrophy (Figure 6C). These histological findings are consistent with the epidermal pathology seen in human KIDS patients, especially those manifesting the lethal form of the disease (Sbidian et al., 2010; Koppelhus et al., 2011). Thus K14-rtTA plus TRE-G45E double-transgenic mice offer a promising model for studies of epidermal pathology that arise from Cx26 mutations linked to the lethal form of KIDS.
FIGURE 6:

Histological analysis of Cx26-G45E transgenic skin. Adult head skin samples from control animals (A) displayed normal epidermal and dermal morphology after 4 wk of doxycycline application. In contrast, K14-rtTA plus TRE-G45E double-transgenic mice (B) induced for 4 wk demonstrated massive skin hyperplasia with greatly increased thickening of the epidermis (acanthosis), papillomatosis (undulation of the epidermis into the dermis), and a thickened, compact stratum corneum (hyperkeratosis). Animals maintained on doxycycline for 10 wk had pronounced hyperkeratosis with frequent keratotic plugging and sebaceous gland atrophy (C). A and B were photographed with a 20× objective (scale bar, 50 μm), and C was photographed with a 4× objective (scale bar, 100 μm).
Increased apoptosis and mitosis in Cx26-G45E transgenic mice
Epidermal thickness is maintained by a balance between cell proliferation in the basal layer and cell shedding in the cornified layer. To gain insight into how this balance is perturbed in the skin of K14-rtTA plus TRE-G45E double-transgenic mice, we examined apoptosis and cell proliferation by staining frozen sections from control and Cx26-G45E animals with antibodies to either cleaved caspase-3 or phospho-histone-3. Staining for the apoptotic marker cleaved caspase-3 was absent in both the dermal and epidermal layers of adult control head skin (Figure 7A). In sections of head skin from K14-rtTA plus TRE-G45E double-transgenic mice that had been induced with doxycycline for 3 wk, a substantial increase in cleaved caspase-3 staining was observed, particularly in the dermis, in the thickened hair follicle epithelium, and possibly within gland structures (Figure 7B). Adult control head skin did show sparse staining of the mitotic marker phospho-histone-3 primarily in the epidermal layer, with additional staining in follicular and glandular structures of the dermis (Figure 7C). In K14-rtTA plus TRE-G45E double-transgenic mice that had been induced for 3 wk, phospho-histone-3 staining was markedly increased in the thickened epidermal layer (Figure 7D). These data suggest that both cell division and cell death signaling pathways are modulated in the diseased skin of Cx26-G45E animals, although in different locations, with increased apoptosis occurring primarily in the dermis and cell proliferation being increased in the epidermis.
FIGURE 7:

Altered apoptosis and mitosis in Cx26-G45E transgenic skin. Cell death and proliferation were determined by immunofluorescence staining using antibodies against either cleaved caspase-3 or phospho-histone-3, respectively. Frozen sections of head skin from adult control animals (A, blue DAPI stain) showed no staining for the apoptotic marker cleaved caspase-3 (red) in either the dermal or epidermal layers. There was a substantial increase in cleaved caspase-3 staining, especially in the dermis of the double-transgenic animals (B, green EGFP) after 3 wk of induction by doxycycline. Inset, stained cells in thickened hair follicle epithelium. For the proliferation-specific marker phospho-histone-3, adult control head skin (C) had sparse staining (red) primarily in the epidermal layer, as well as in follicular and glandular structures of the dermis. In Cx26-G45E transgenic mice induced with doxycycline for 3 wk (D), an increase in phospho-histone-3 staining was observed in the thickened epidermal layer (inset). Scale bar, 50 μm.
Transgenic Cx26-G45E keratinocytes display increased hemichannel activity
Transgenic expression of Cx26-G45E in the epidermis recapitulated the histopathology seen in human KIDS patients. If the aberrant hemichannel activity shown by this mutation in vitro contributed to epidermal pathology observed in vivo, then primary keratinocytes isolated from transgenic skin should also display increased hemichannel activity when compared with control keratinocytes. To test this hypothesis, we isolated keratinocytes from control or K14-rtTA plus TRE-G45E double-transgenic mice and measured whole-cell membrane currents and single-channel conductance by patch clamp electrophysiology. Neonatal K14-rtTA plus TRE-G45E double-transgenic mice (P2) that had been induced with doxycycline in utero were easily distinguished from their control littermates by their reduced size, reddish appearance (Figure 8A), and uniform epidermal expression of EGFP (Figure 8B). Isolated Cx26-G45E keratinocytes retained EGFP expression (Figure 8C), facilitating their identification for whole-cell patch clamp analysis. Keratinocytes isolated from control mice displayed modest membrane currents when stepped to membrane potentials between −90 and +90 mV (Figure 8D). In contrast, Cx26-G45E keratinocytes exhibited significantly increased whole-cell membrane currents at both hyperpolarizing and depolarizing membrane potentials (Figure 8E). Plotting the current density as a function of membrane potential (Figure 8F) showed that K14-rtTA plus TRE-G45E double-transgenic currents were greater than fivefold larger (p < 0.01, Student's t test) than those recorded in control keratinocytes. In some primary K14-rtTA plus TRE-G45E double-transgenic cells, the activity of single Cx26-G45E hemichannels could be resolved (Figure 8G). In the example shown, the hemichannels had a unitary conductance value of ∼315 pS at a membrane potential of −30 mV measured in 120 mM K+ aspartate−, a value in excellent agreement with the Cx26-G45E hemichannel conductance recorded in transfected N2A cells (Figure 2E). No single channels of this size were observed in control keratinocytes. Thus the increased hemichannel activity seen for Cx26-G45E in exogenous expression systems persists in primary isolated keratinocytes from K14-rtTA plus TRE-G45E double-transgenic mice.
FIGURE 8:

Increased hemichannel activity in transgenic Cx26-G45E keratinocytes. Whole-cell membrane currents and hemichannel activities of the primary keratinocytes isolated from neonatal control and K14-rtTA plus TRE-G45E double-transgenic mice induced with doxycycline in utero were measured by patch clamp electrophysiology. Neonatal double-transgenic mice were smaller than the control littermates and had a red, shiny appearance (A), which was correlated with transgene activation as seen by whole-animal EGFP fluorescence (B). Keratinocytes isolated from K14-rtTA plus TRE-G45E double-transgenic mice retained EGFP expression (C, scale bar, 5 μm). Keratinocytes isolated from control animals displayed modest whole-cell membrane currents (D) when stepped to membrane potentials between −90 and +90 mV. In contrast, Cx26-G45E keratinocytes exhibited significantly increased membrane currents (E) compared with controls. Plotting the current density as a function of membrane potential (F) quantified greater than fivefold increase (p < 0.01, Student's t test) in membrane currents recorded from K14-rtTA plus TRE-G45E double-transgenic keratinocytes. When the single-channel activity of the Cx26-G45E hemichannels was detected, the unitary conductance was observed to be ∼315 (G), comparable to that of Cx26-G45E hemichannels recorded from transfected N2A cells (Figure 2).
Cx26-G45E keratinocytes have a significantly increased cell size
In human patients carrying the lethal Cx26-G45E mutation, histological examination of skin biopsies suggested that keratinocytes in the epidermis were swollen (Sbidian et al., 2010). To determine whether our transgenic mice replicated this aspect of KIDS pathology, we measured keratinocyte circumference in histological sections and freshly isolated keratinocyte cell membrane capacitance (a measure of cell surface area) by patch clamp. Skin sections from control (Figure 9A) or K14-rtTA plus TRE-G45E double-transgenic (Figure 9B) animals that were stained with rhodamine-conjugated wheat germ agglutinin showed clearly demarcated keratinocyte cell borders. Cell perimeters from control (Figure 9, C and D) and Cx26-G45E (Figure 9, E and F) mice were traced across the entire epidermal layer using cellSens imaging software and normalized to the average perimeter value for control keratinocytes. K14-rtTA plus TRE-G45E double-transgenic mice had a 28% increase in cell perimeter (p < 0.01, Student's t test) compared with littermate controls (Figure 9G). This finding was confirmed by capacitance measurements made from postnatal day 2 (P2) Cx26-G45E mice that had been induced with doxycycline in utero and 10-wk-old Cx26-G45E mice (P70) that had been induced with doxycycline for 5 wk (Figure 9H). There was no significant difference between the membrane capacitance of control keratinocytes measured at P2 and P70 (p = 0.34). In contrast, membrane capacitance was increased 51% (p < 0.01) in P2 and 74% (p < 0.01) in P70 K14-rtTA plus TRE-G45E double-transgenic keratinocytes. These data show that when measured by two independent methods, keratinocytes from Cx26-G45E transgenic mice were significantly larger than their littermate controls and that cell size increased with disease progression.
FIGURE 9:

Transgenic Cx26-G45E keratinocytes are larger. Keratinocyte size was compared between double-transgenic mice and the control animals by two independent methods. Cell perimeters in sections of control (A) and K14-rtTA plus TRE-G45E double-transgenic skin (B) were labeled with rhodamine-conjugated wheat germ agglutinin. Cell perimeters were measured in control (C, D) and Cx26-G45E sections (E, F) using the tracing function of cellSens imaging software. Quantification of these data showed that double-transgenic animals had a 28% increase in keratinocyte perimeter compared with control mice (G; p < 0.01, Student's t test). Measurement of membrane capacitance from isolated primary keratinocytes by whole-cell patch clamp (H) showed 51% (p < 0.01) and 74% (p < 0.01) increases in cell capacitance in K14-rtTA plus TRE-G45E double-transgenic keratinocytes compared with control cells on postnatal days 2 and 70, respectively. Scale bar, 15 μm.
DISCUSSION
We generated an animal model replicating the skin pathology of the lethal form of KIDS by inducible transgenic expression of the human Cx26-G45E mutation in mouse keratinocytes. Cx26-G45E mice displayed most major features of the human disease, including greatly reduced viability, hyperkeratosis, acanthosis, papillomatosis, and extensive ichthyosiform scaling. Transgenic Cx26-G45E keratinocytes also showed significantly increased whole-cell capacitance compared with controls, features consistent with histological observations of increased cell size in skin biopsies from human KIDS patients. These skin changes were correlated with a significant increase in whole-cell membrane hemichannel currents detected in freshly isolated primary keratinocytes that were biophysically indistinguishable from those measured following inducible expression of Cx26-G45E in tissue culture cells. These results confirm the pathogenic nature of the Cx26-G45E mutation and provide a new model for future studies of the effects of aberrant connexin hemichannels in human connexin disorders of epithelial differentiation and cornification.
There are two other transgenic models of skin disease caused by human Cx26 mutations that can be compared with the Cx26-G45E mice. Expression of Cx26-D66H in suprabasal keratinocytes under the control of the keratin 10 promoter mimicked many features of Vohwinkel syndrome (VS) seen in human patients carrying this mutation (Bakirtzis et al., 2003). Instead of localizing at intercellular gap junctions, the transgenic Cx26-D66H accumulated within the keratinocyte cytoplasm, indicating abnormal trafficking of mutant Cx26. Consistent with in vitro data demonstrating a trans-dominant effect of mutant Cx26 on other coexpressed wild-type connexins (Rouan et al., 2001), Cx30 was also trapped into the cytoplasm of keratinocytes in Cx26-D66H transgenic mice. Early postnatal animals showed epidermal scaling with zonal hyperkeratosis of the tail and digits producing constriction bands, a hallmark feature of VS. Microscopic analysis revealed areas of hyperkeratosis and a thickened stratum corneum associated with an increase in keratinocyte apoptosis, as well as increased proliferation in the lesional areas. These results were consistent with the skin pathology seen in human patients (Maestrini et al., 1999) and suggested that the D66H mutation caused premature keratinocyte death in the upper epidermis, with compensatory hyperproliferation in the basal layer, leading to a thickened stratum corneum. Although Cx26-G45E mice also showed increased epidermal cell proliferation, our assay showed only increased apoptosis in the dermis, localizing mainly to hair follicles and possibly sweat glands, which may be related to the atrophy of these structures in transgenic skin and human patients.
In a recently developed model of KIDS, a conditional mouse mutant was generated to express the Cx26-S17F mutation under control of the endogenous murine Cx26 promoter after deletion of the floxed wild-type Cx26 coding region (Schutz et al., 2011). Breeding this line with a phosphoglycerate kinase Cre-expressing strain produced a ubiquitous knock-in of the human Cx26-S17F allele in the native mouse Cx26 locus. Homozygous mutant animals were not viable, which could be predicted by the complete lack of any channel function by Cx26-S17F in vitro (Richard et al., 2002; Lee et al., 2009) and the fact that homozygous knockout Cx26 mice are embryonic lethal (Gabriel et al., 1998). Surviving heterozygous mice displayed hyperplasia of the tail and foot epidermis and annular tail restrictions, a milder epidermal phenotype than shown by the Cx26-G45E animals, which is consistent with a milder epidermal disease in humans carrying Cx26-S17F (Richard et al., 2002; Mazereeuw-Hautier et al., 2007). Unlike the majority of other KIDS mutations, Cx26-S17F fails to form hemichannels in vitro (Lee et al., 2009), and this may partially explain the milder phenotype in humans and transgenic mice. The authors also reported that the total number of heterozygous Cx26-S17F mice born was much lower than that given by the expected Mendelian ratio. This was surprising, as human Cx26-S17F patients have not been reported to have the infant mortality associated with Cx26-G45E (Janecke et al., 2005; Jonard et al., 2008; Sbidian et al., 2010). The increase in mouse mortality may have been due to differences in the expression pattern of Cx26 in human and murine epidermis. In human skin, Cx26 is found in a patchy distribution in basal keratinocytes but is more highly expressed in palmer and plantar epidermis, eccrine sweat glands, hair follicles, and all epidermal layers after injury or in hyperproliferative disease (Richard, 2001). In mice, Cx26 is present in the upper suprabasal/granular layers of the epidermis in tail skin (Schutz et al., 2011). Further comparison between these animal models may be helpful in characterizing the extensive phenotypic overlap among different skin disease syndromes associated with connexin mutations (van Steensel et al., 2004).
The vast majority of Cx26 mutations linked to KIDS have been shown to display increased hemichannel activity (Montgomery et al., 2004; Stong et al., 2006; Gerido et al., 2007; Lee et al., 2009; Sanchez et al., 2010; Terrinoni et al., 2010). Biophysical studies of Cx26-A40V and Cx26-G45E hemichannels found that both showed impaired regulation by extracellular Ca2+, increasing the likelihood of aberrant hemichannel opening. Cx26-G45E hemichannels specifically exhibited a substantial increase in permeability to Ca2+ ions compared with wild-type Cx26, providing a route for excessive entry of Ca2+ into keratinocytes in epidermal tissue (Sanchez et al., 2010). Extracellular calcium plays important roles in normal epidermal differentiation, regulating cell proliferation, terminal differentiation, and cell-to-cell adhesion, as well as being implicated in the pathogenesis of some types of skin disease (Hennings et al., 1980; Fairley, 1991; Bikle et al., 2004; Tu et al., 2004). Changes in the extracellular Ca2+ concentration are believed to help drive a developmental switch from keratinocyte proliferation to terminal differentiation by providing an extracellular reservoir of Ca2+ ions to influence intracellular calcium-dependent signaling processes. In the case of the Cx26-G45E mutation, the combined effects of increased hemichannel activity and increased hemichannel Ca2+ permeability would be expected to wreak havoc on the delicate balance between keratinocyte proliferation and differentiation mechanisms. The availability of a mouse model that faithfully recapitulates Cx26-G45E–induced skin disease will facilitate future studies to further explore the relationship between increased hemichannel activity and epidermal homeostasis, with the ultimate goal of developing new therapies to treat this lethal human disorder.
MATERIALS AND METHODS
Molecular cloning
pIRES2-EGFP (Clontech Laboratories, Mountain View, CA) was digested with SacII and NotI to liberate the IRES2-EGFP cassette, which was then subcloned into the SacII-NotI sites of pTRE2 (Clontech Laboratories). The Cx26-G45E cDNA was then subcloned into BamHI site of the pTRE2-IRES2-EGFP construct in order to generate the plasmid pTRE2-Cx26-G45E-IRES2-EGFP. This construct was used for cotransfection of intercellular communication–deficient neuro-2A (N2A) or HeLa cells together with the pTet-On vector (Clontech Laboratories) for immunocytochemical, dye uptake, and patch clamp studies. The construct was also digested with AatII and DrdI to obtain a ∼4-kb linear transgene free of plasmid sequences (abbreviated TRE-G45E) for zygotic pronuclear injection to generate transgenic mice.
Cell transfection
N2A or HeLa cells were transiently cotransfected either with a 1:5 mixture of pTet-On vector (1 μg) and pTRE2-Cx26-G45E-IRES2-EGFP (5 μg) or with a 1:5 mixture of pTet-On vector and pTRE2-Cx26-IRES2-EGFP using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. Briefly, 1 d before transfection, cells were plated so that they would be 70–95% confluent on the day of transfection. Lipofectamine 2000 reagent, plasmids, and OPTI-MEM medium (Invitrogen) were brought to room temperature. DNA (total of 6 μg) and Lipofectamine 2000 (6 μl) were individually diluted in OPTI-MEM. Following 20 min of incubation at 37°C, the DNA and Lipofectamine mixture was added onto the cells drop by drop, and cells were incubated at 37°C for 18–24 h. The following day, the medium was changed to regular medium supplemented with a final concentration of 1 μg/μl doxycycline (Sigma-Aldrich, St. Louis, MO) and 2 mM CaCl2, and cells were incubated for an additional 24 h to allow the induction of gene expression. Cells were then used for immunofluorescence staining, patch clamp electrophysiology, or dye uptake assays.
Immunocytochemistry
Transiently transfected HeLa cells were fixed with 1% paraformaldehyde in phosphate-buffered saline (PBS), blocked, and permeabilized with 5% bovine serum albumin (BSA) in PBS with 0.1% Triton X-100. Cells were stained with a polyclonal rabbit anti-Cx26 (Zymed, San Francisco, CA), washed with 0.1% Triton X-100/PBS, incubated with a Cy3-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA), washed with 0.1% Triton X-100/PBS, and mounted on slides using Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Skin samples comprising the epidermis and dermis were dissected and fixed in a 1% formaldehyde solution in PBS for 1 h at room temperature. Fixed tissues were rinsed with PBS, immersed in Optimal Cutting Temperature compound (OCT; Ted Pella, Redding, CA), and frozen. Sections of thickness 8–10 μm were cut on a cryotome, dried onto glass slides, and stained with polyclonal rabbit antibodies against Cx26 (Zymed), phospho-histone-3 (Cell Signaling Technology, Danvers, MA), or cleaved caspase-3 (Cell Signaling Technology), washed with 0.1% Triton X-100/PBS, incubated with Cy3-conjugated anti-rabbit secondary antibodies, washed with 0.1% Triton X-100/PBS, and mounted on slides using Vectashield with DAPI. Stained slides were viewed on a BX51 microscope and photographed with a DP72 digital camera (Olympus, Lake Success, NY).
Neurobiotin uptake assay
Neurobiotin (Vector Laboratories) was dissolved at 0.1 mg/ml in PBS without calcium. Cells grown on coverslips were first washed with calcium-free medium for 20 min, followed by incubation in neurobiotin solution for 20 min at 37°C. Cells were then washed three times with PBS supplemented with 2 mM CaCl2 for 10 min at 37°C and fixed with 1% paraformaldehyde in PBS for 30 min at room temperature. After fixation, cells were washed three times with PBS for 5 min, permeabilized with 0.1% Triton X-100/PBS for 20 min, and blocked with 3% BSA/0.1% Triton X-100/PBS for 1 h at room temperature. Cells were then incubated with 1:1000 dilution of tetramethylrhodamine isothiocyanate–conjugated avidin in 3% BSA/0.1% TritonX-100/PBS (Vector Laboratories) for 1 h at room temperature in the dark, washed three times for 10 min with 0.1% Triton X-100/PBS, and mounted on slides using Vectashield with DAPI. Images were acquired at the same exposure time for all conditions with a DP72 digital camera on a BX51 microscope (Olympus).
Patch clamp electrophysiology
Experiments were carried out on transiently transfected N2A cells or primary isolated mouse keratinocytes at room temperature. Cells transfected with wild-type Cx26 or the G45E mutation were grown in medium supplemented with 2 mM CaCl2 until the measurements were performed. Cells on glass coverslips were transferred to the experimental chamber with a bath solution containing (in mM) 137.7 NaCl, 5.4 KCl, 2.3 NaOH, 1 MgCl2, 2 CsCl2, 4 BaCl2 10 glucose, and 5 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), where CaCl2 was omitted from the bath solution. Patch pipettes were pulled from glass capillaries with a horizontal puller (Sutter Instruments, Novato, CA) and filled with a pipette solution of 120 mmol/l K aspartate, 5 mM HEPES, 10 mM ethylene glycol tetraacetic acid, and 3 mM NaATP (pH 7.2). Single cells were selected and used to measure the membrane capacitance, as well as the membrane currents (Im) invoked while the membrane potential (Vm) was stepped from −90 to +90 mV in 20-mV increments.
Generation of transgenic mice
All animal work was done with approval of the Stony Brook University Institutional Animal Care and Use Committee. TRE-G45E transgenic mice were generated by zygotic pronuclear injection followed by embryo transfer into pseudopregnant female animals. Positive founder mice were interbred with a second transgenic strain (Nguyen et al., 2006) expressing the reverse tetracycline transactivator under the control of the keratin14 (abbreviated K14-rtTA) promoter to produce doubly transgenic animals. Transgenic mice were identified by amplification of tail genomic DNA using the PCR Extender System (5 PRIME, Gaithersburg, MD). The TRE-G45E transgene was detected using the primer pair 5′-CATTGTCATGTACGACGGCTTCTC-3′ and 5′-GGTACCTTCTGGGCATCCTT-3′. The K14-rtTA transgene was detected using the primer pair 5′-CACGATACACCTGACTAGCTGGGTG-3′ and 5′-CATCACCCACAGGCTAGCGCCAACT-3′. PCR products were run on 2% agarose gels, visualized with ethidium bromide, and photographed. Transgenic animals were backcrossed to the SKH1 hairless mouse strain (Charles River, Wilmington, MA) to facilitate visual observation of the epidermis. For in vivo fluorescence detection of EGFP expression, mice were anesthetized with isoflurane and imaged using a Maestro small-animal imaging system (CRi, Woburn, MA).
RT-PCR analysis
Total mRNA was prepared from mouse skin samples using the RNeasy Kit (Qiagen, Germantown, MD) following the manufacturer's protocol. RNA samples were treated with DNase I for 30 min, extracted with phenol/chloroform, ethanol precipitated, and resuspended in diethylpyrocarbonate-treated water. cDNAs were synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) following the manufacturer's protocol using random hexamer primers. cDNAs were used as templates in PCRs to amplify the TRE-G45E transgenic transcript using the primer pair 5′-GACTGTCTTCACAGTGTTCATGATT-3′ and 5′-TGTATCTTATACACGTGGCTTTTGG-3′ or the K14-rtTA transgenic transcript using the primer pair 5′-ACTGGACAAGAGCAAAGTCATAAAC-3′ and 5′-CTCTTTCCTCTTTTGCTACTTGATG-3′. PCR products were run on a 2% agarose gel, visualized with ethidium bromide, and photographed.
Histology
Skin samples comprising the epidermis and dermis were dissected and fixed in a 4% formaldehyde solution in PBS for 16–24 h at room temperature. Fixed tissues were rinsed with PBS, dehydrated through an ethanol series, and embedded in paraffin. Sections of thickness 2–3 μm were cut on a diamond knife, deparaffinized, and stained with hematoxylin–eosin. Some sections were deparaffinized, rehydrated, blocked with 3% BSA/0.1% Triton X-100/PBS for 1 h at room temperature, and stained with rhodamine-conjugated wheat germ agglutinin in 3% BSA/0.1% Triton X-100/PBS (Vector Laboratories) for 1 h at room temperature in the dark, washed three times for 10 min with 0.1% Triton X-100/PBS, and mounted on slides using Vectashield with DAPI. Sections were viewed on a BX51 microscope and photographed with a DP72 digital camera. Cell perimeters were traced and measured using cellSens digital imaging software (Olympus).
Acknowledgments
We thank Wenhao Xu of the Gene Targeting and Transgenic Facility at the University of Virginia (Charlottesville, VA) for assistance in the generation of the Cx26-G45E mice. This work was supported by National Institutes of Health Grants R01 AR59505 (T.W.W.) and R01 GM088181 (V.V.).
Abbreviations used:
- Cx
connexin
- EGFP
enhanced green fluorescent protein
- IRES
internal ribosome entry site
- KIDS
keratitis-ichthyosis-deafness syndrome
- rtTA
reverse tetracycline transactivator
- TRE
tetracycline response element
- VS
Vohwinkel syndrome
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-09-0778) on October 26, 2011.
REFERENCES
- Abdollahi A, Hallaji Z, Esmaili N, Valikhani M, Barzegari M, Akhyani M, Toosi S, Miresmaili A. KID syndrome. Dermatol Online J. 2007;13:11. [PubMed] [Google Scholar]
- Bakirtzis G, et al. Targeted epidermal expression of mutant connexin 26(D66H) mimics true Vohwinkel syndrome and provides a model for the pathogenesis of dominant connexin disorders. Hum Mol Genet. 2003;12:1737–1744. doi: 10.1093/hmg/ddg183. [DOI] [PubMed] [Google Scholar]
- Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol. 2005;7:63–69. doi: 10.1038/ncb1205. [DOI] [PubMed] [Google Scholar]
- Benavides F, Oberyszyn TM, VanBuskirk AM, Reeve VE, Kusewitt DF. The hairless mouse in skin research. J Dermatol Sci. 2009;53:10–18. doi: 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MV, Contreras JE, Bukauskas FF, Saez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26:610–617. doi: 10.1016/j.tins.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikle DD, Oda Y, Xie Z. Calcium and 1,25(OH)2D: interacting drivers of epidermal differentiation. J Steroid Biochem Mol Biol. 2004;89–90:355–360. doi: 10.1016/j.jsbmb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339–373. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun-Falco M. Hereditary palmoplantar keratodermas. J Dtsch Dermatol Ges. 2009;7:971–984. doi: 10.1111/j.1610-0387.2009.07058.x. [DOI] [PubMed] [Google Scholar]
- Brown CW, et al. A novel GJB2 (connexin 26) mutation, F142L, in a patient with unusual mucocutaneous findings and deafness. J Invest Dermatol. 2003;121:1221–1223. doi: 10.1046/j.1523-1747.2003.12550_4.x. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D'Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Caputo R, Peluchetti D. The junctions of normal human epidermis. A freeze-fracture study. J Ultrastruct Res. 1977;61:44–61. doi: 10.1016/s0022-5320(77)90005-3. [DOI] [PubMed] [Google Scholar]
- Di WL, Rugg EL, Leigh IM, Kelsell DP. Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. J Invest Dermatol. 2001;117:958–964. doi: 10.1046/j.0022-202x.2001.01468.x. [DOI] [PubMed] [Google Scholar]
- Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairley JA. Calcium metabolism and the pathogenesis of dermatologic disease. Semin Dermatol. 1991;10:225–231. [PubMed] [Google Scholar]
- Gabriel HD, Jung D, Butzler C, Temme A, Traub O, Winterhager E, Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J Cell Biol. 1998;140:1453–1461. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerido DA, DeRosa AM, Richard G, White TW. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol. 2007;293:C337–C345. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- Goliger JA, Paul DL. Expression of gap junction proteins Cx26, Cx31.1, Cx37, and Cx43 in developing and mature rat epidermis. Dev Dyn. 1994;200:1–13. doi: 10.1002/aja.1002000102. [DOI] [PubMed] [Google Scholar]
- Goodenough DA. Bulk isolation of mouse hepatocyte gap junctionsCharacterization of the principal protein, connexin. J Cell Biol. 1974;61:557–563. doi: 10.1083/jcb.61.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- Griffith AJ, et al. Cochleosaccular dysplasia associated with a connexin 26 mutation in keratitis-ichthyosis-deafness syndrome. Laryngoscope. 2006;116:1404–1408. doi: 10.1097/01.mlg.0000224549.75161.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980;19:245–254. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Hennies HC, Gunther B, Gansl G, Smolle J, Messmer EM, Utermann G, Rittinger O. GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am J Med Genet A. 2005;133:128–131. doi: 10.1002/ajmg.a.30515. [DOI] [PubMed] [Google Scholar]
- Jonard L, et al. A familial case of keratitis-ichthyosis-deafness (KID) syndrome with the GJB2 mutation G45E. Eur J Med Genet. 2008;51:35–43. doi: 10.1016/j.ejmg.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Kanno Y, Loewenstein WR. Low-resistance coupling between gland cells. some observations on intercellular contact membranes and intercellular space. Nature. 1964;201:194–195. doi: 10.1038/201194a0. [DOI] [PubMed] [Google Scholar]
- Koppelhus U, Tranebjaerg L, Esberg G, Ramsing M, Lodahl M, Rendtorff ND, Olesen HV, Sommerlund M. A novel mutation in the connexin 26 gene (GJB2) in a child with clinical and histological features of keratitis-ichthyosis-deafness (KID) syndrome. Clin Exp Dermatol. 2011;36:142–148. doi: 10.1111/j.1365-2230.2010.03936.x. [DOI] [PubMed] [Google Scholar]
- Labarthe MP, Bosco D, Saurat JH, Meda P, Salomon D. Upregulation of connexin 26 between keratinocytes of psoriatic lesions. J Invest Dermatol. 1998;111:72–76. doi: 10.1046/j.1523-1747.1998.00248.x. [DOI] [PubMed] [Google Scholar]
- Lawrence TS, Beers WH, Gilula NB. Transmission of hormonal stimulation by cell-to-cell communication. Nature. 1978;272:501–506. doi: 10.1038/272501a0. [DOI] [PubMed] [Google Scholar]
- Lazic T, Horii KA, Richard G, Wasserman DI, Antaya RJ. A report of GJB2 (N14K) Connexin 26 mutation in two patients—a new subtype of KID syndrome? Pediatr Dermatol. 2008;25:535–540. doi: 10.1111/j.1525-1470.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- Lee JR, Derosa AM, White TW. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J Invest Dermatol. 2009;129:870–878. doi: 10.1038/jid.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert Rev Mol Med. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- Levit NA, Mese G, Basaly MG, White TW. Pathological hemichannels associated with human Cx26 mutations causing keratitis-ichthyosis-deafness syndrome. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbamem.2011.09.003. doi:10.1016/j.bbamem.2011.09.003 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke T, Choudhry R, Thom R, Selmer IS, Burden AD, Hodgins MB. Upregulation of connexin 26 is a feature of keratinocyte differentiation in hyperproliferative epidermis, vaginal epithelium, and buccal epithelium. J Invest Dermatol. 1999;112:354–361. doi: 10.1046/j.1523-1747.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- Maintz L, Betz RC, Allam JP, Wenzel J, Jaksche A, Friedrichs N, Bieber T, Novak N. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur J Dermatol. 2005;15:347–352. [PubMed] [Google Scholar]
- Masgrau-Peya E, Salomon D, Saurat JH, Meda P. In vivo modulation of connexins 43 and 26 of human epidermis by topical retinoic acid treatment. J Histochem Cytochem. 1997;45:1207–1215. doi: 10.1177/002215549704500904. [DOI] [PubMed] [Google Scholar]
- Mazereeuw-Hautier J, et al. Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol. 2007;156:1015–1019. doi: 10.1111/j.1365-2133.2007.07806.x. [DOI] [PubMed] [Google Scholar]
- Mese G, Londin E, Mui R, Brink PR, White TW. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet. 2004;115:191–199. doi: 10.1007/s00439-004-1142-6. [DOI] [PubMed] [Google Scholar]
- Mese G, Richard G, White TW. Gap junctions: basic structure and function. J Invest Dermatol. 2007;127:2516–2524. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- Mese G, Valiunas V, Brink PR, White TW. Connexin26 deafness associated mutations show altered permeability to large cationic molecules. Am J Physiol Cell Physiol. 2008;295:C966–C974. doi: 10.1152/ajpcell.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JR, White TW, Martin BL, Turner ML, Holland SM. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol. 2004;51:377–382. doi: 10.1016/j.jaad.2003.12.042. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Rendl M, Fuchs E. Tcf3 governs stem cell features and represses cell fate determination in skin. Cell. 2006;127:171–183. doi: 10.1016/j.cell.2006.07.036. [DOI] [PubMed] [Google Scholar]
- Nyquist GG, et al. Malignant proliferating pilar tumors arising in KID syndrome: a report of two patients. Am J Med Genet A. 2007;143:734–741. doi: 10.1002/ajmg.a.31635. [DOI] [PubMed] [Google Scholar]
- Revel JP, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard G. Connexins: a connection with the skin. Exp Dermatol. 2000;9:77–96. doi: 10.1034/j.1600-0625.2000.009002077.x. [DOI] [PubMed] [Google Scholar]
- Richard G. Connexin disorders of the skin. Adv Dermatol. 2001;17:243–277. [PubMed] [Google Scholar]
- Richard G. Connexin disorders of the skin. Clin Dermatol. 2005;23:23–32. doi: 10.1016/j.clindermatol.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2. J Invest Dermatol. 2004;123:856–863. doi: 10.1111/j.0022-202X.2004.23470.x. [DOI] [PubMed] [Google Scholar]
- Richard G, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- Risek B, Klier FG, Gilula NB. Multiple gap junction genes are utilized during rat skin and hair development. Development. 1992;116:639–651. doi: 10.1242/dev.116.3.639. [DOI] [PubMed] [Google Scholar]
- Rivas MV, Jarvis ED, Morisaki S, Carbonaro H, Gottlieb AB, Krueger JG. Identification of aberrantly regulated genes in diseased skin using the cDNA differential display technique. J Invest Dermatol. 1997;108:188–194. doi: 10.1111/1523-1747.ep12334217. [DOI] [PubMed] [Google Scholar]
- Robertson JD. The occurrence of a subunit pattern in the unit membranes of club endings in Mauthner cell synapses in goldfish brains. J Cell Biol. 1963;19:201–221. doi: 10.1083/jcb.19.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. trans-Dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114:2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- Saez JC, Contreras JE, Bukauskas FF, Retamal MA, Bennett MV. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol Scand. 2003;179:9–22. doi: 10.1046/j.1365-201X.2003.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MV. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–2389. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- Salomon D, Masgrau E, Vischer S, Ullrich S, Dupont E, Sappino P, Saurat JH, Meda P. Topography of mammalian connexins in human skin. J Invest Dermatol. 1994;103:240–247. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbidian E, Feldmann D, Bengoa J, Fraitag S, Abadie V, de Prost Y, Bodemer C, Hadj-Rabia S. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: pre-natal diagnosis in a familial lethal form. Clin Genet. 2010;77:587–592. doi: 10.1111/j.1399-0004.2009.01339.x. [DOI] [PubMed] [Google Scholar]
- Scemes E. Nature of plasmalemmal functional “hemichannels.”. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbamem.2011.06.005. doi:10.1016 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Spray DC, Meda P. Connexins, pannexins, innexins: novel roles of “hemi-channels.”. Pflugers Arch. 2009;457:1207–1226. doi: 10.1007/s00424-008-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalper KA, Orellana JA, Berthoud VM, Saez JC. Dysfunctions of the diffusional membrane pathways mediated by hemichannels in inherited and acquired human diseases. Curr Vasc Pharmacol. 2009;7:486–505. doi: 10.2174/157016109789043937. [DOI] [PubMed] [Google Scholar]
- Schutz M, Auth T, Gehrt A, Bosen F, Korber I, Strenzke N, Moser T, Willecke K. The connexin26 S17F mouse mutant represents a model for the human hereditary keratitis-ichthyosis-deafness syndrome. Hum Mol Genet. 2011;20:28–39. doi: 10.1093/hmg/ddq429. [DOI] [PubMed] [Google Scholar]
- Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116:2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- Terrinoni A, Codispoti A, Serra V, Didona B, Bruno E, Nistico R, Giustizieri M, Alessandrini M, Campione E, Melino G. Connexin 26 (GJB2) mutations, causing KID syndrome, are associated with cell death due to calcium gating deregulation. Biochem Biophys Res Commun. 2010;394:909–914. doi: 10.1016/j.bbrc.2010.03.073. [DOI] [PubMed] [Google Scholar]
- Tu CL, Oda Y, Komuves L, Bikle DD. The role of the calcium-sensing receptor in epidermal differentiation. Cell Calcium. 2004;35:265–273. doi: 10.1016/j.ceca.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Valiunas V, et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J Physiol. 2005;568:459–468. doi: 10.1113/jphysiol.2005.090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004;131:12–19. doi: 10.1002/ajmg.c.30030. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Steijlen PM, Bladergroen RS, Hoefsloot EH, van Ravenswaaij-Arts CM, van Geel M. A phenotype resembling the Clouston syndrome with deafness is associated with a novel missense GJB2 mutation. J Invest Dermatol. 2004;123:291–293. doi: 10.1111/j.0022-202X.2004.23204.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol. 2002;118:724–727. doi: 10.1046/j.1523-1747.2002.01735.x. [DOI] [PubMed] [Google Scholar]
- Wei CJ, Xu X, Lo CW. Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- White TW. Functional analysis of human Cx26 mutations associated with deafness. Brain Res Brain Res Rev. 2000;32:181–183. doi: 10.1016/s0165-0173(99)00079-x. [DOI] [PubMed] [Google Scholar]
- White TW, Paul DL. Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]