

The closely related GTPases RalA and RalB are required for assembly of tight junction gate, but not fence, function. These activities depend on direct binding to the exocyst complex. Whereas RalA–exocyst complexes are required for exocytosis of junction proteins, RalB–exocyst complexes are required for endocytosis of these components.
Abstract
Tight junctions (TJs) are structures indispensable to epithelial cells and are responsible for regulation of paracellular diffusion and maintenance of cellular polarity. Although many interactions between TJ constituents have been identified, questions remain concerning how specific functions of TJs are established and regulated. Here we investigated the roles of Ral GTPases and their common effector exocyst complex in the formation of nascent TJs. Unexpectedly, RNA interference–mediated suppression of RalA or RalB caused opposing changes in TJ development. RalA reduction increased paracellular permeability and decreased incorporation of components into TJs, whereas RalB reduction decreased paracellular permeability and increased incorporation of components into TJs. Activities of both Ral GTPases were mediated through the exocyst. Finally, we show that TJ-mediated separation of apical–basal membrane domains is established prior to equilibration of barrier function and that it is unaffected by Ral knockdown or specific composition of TJs.
INTRODUCTION
Tight junctions (TJs) are key structures responsible for the ability of epithelial cells to serve as barriers between lumenal and interstitial tissue compartments. Located at the apex of lateral membranes, TJs have both barrier and “fence” functions. The barrier function describes a selectively permeable filter that regulates paracellular diffusion of ions and solutes based on charge and size, respectively (Diamond, 1977). Barrier function is tightly regulated by a specific set of TJ proteins, the claudins (Tsukita et al., 2008). TJs also serve as a molecular “fence” in that they separate apical and basolateral membrane domains, thereby preventing diffusion of integral membrane proteins and outer leaflet lipids from one membrane domain to the other (Diamond, 1977). This function is also regulated, and yet early studies suggested that these two TJ functions are independent (Mandel et al., 1993; Takakuwa et al., 2000). Despite significant progress made in identifying protein complexes that specify spatiotemporal aspects of TJ development (St. Johnston and Ahringer, 2010) and in dissecting interactions between them, mechanisms underlying development of barrier function and formation of apical–basal distinction of these structures are only partially understood.
Once sites at which TJs will form are established, cells assemble junctions by transporting TJ components to these locations and modulate abundance of these proteins via exocytosis and endocytosis (Yu and Turner, 2008). Excellent candidates responsible for these regulatory functions are Ral GTPases and the exocyst. RalA and RalB comprise the Ral subfamily of the larger Ras superfamily of GTPases and are activated downstream of Ras by specific guanine nucleotide exchange factors (GEFs) such as RalGDS family members (van Dam and Robinson, 2006). Once activated, RalA and RalB bind effectors, known examples of which include the exocyst complex, filamin, RalBP1, and ZONAB (van Dam and Robinson, 2006). The exocyst, an evolutionarily conserved heterooctameric protein complex, localizes to sites of membrane remodeling and contains two Ral-binding effectors, Sec5 and Exo84 (Moskalenko et al., 2003; Yeaman et al., 2004). The exocyst is known to be required for anisotropic growth of membrane areas such as bud tips of vegetatively growing yeast (TerBush et al., 1996), growth cones of neurons (Vega and Hsu, 2001), and the lateral membranes (Grindstaff et al., 1998) and developing apical domains (Blankenship et al., 2007; Bryant et al., 2010) of epithelial cells. We hypothesize that Ral engagement of the exocyst (Moskalenko et al., 2002) provides context-specific allosteric regulation of the complex. For example, Ral engagement of the exocyst primes it for a role in cell migration by recruiting paxillin (Spiczka and Yeaman, 2008). Ral–exocyst binding also induces recruitment of Par3 in neuronal cells and of Tank-binding kinase 1 in transformed cells during the innate immune response (Chien et al., 2006; Lalli, 2009).
Although some research indicates that binding of different Ral GTPases to the exocyst triggers distinct functions, other studies have shown that both RalA and RalB engage the exocyst to perform related functions. As an example of evidence for distinct functions, constitutively active RalA, but not RalB, accelerated basolateral trafficking of E-cadherin, presumably through the exocyst (Shipitsin and Feig, 2004); this led to the suggestion that RalA interacts with a factor on recycling endosomes necessary for basolateral trafficking but that RalB does not. In another study, RNA interference–mediated reduction of RalB expression but not of RalA expression reduced exocyst-dependent membrane trafficking to the leading edge of migrating fibroblasts (Rosse et al., 2006). In contrast to these results, a growing number of studies have shown that RalA–exocyst and RalB–exocyst complexes collaborate to promote complicated cellular events, such as tumor-cell invasion (Spiczka and Yeaman, 2008) and cytokinesis (Cascone et al., 2008). Coordinated actions of these complexes are likely also required for development of epithelial polarity. Indeed, indiscriminate loss of Ral function interferes with delivery of several basolateral proteins (vesicular stomatitis virus glycoprotein, epidermal growth factor receptor, gp58; Moskalenko et al., 2002). Of note, formation of apical–basal membrane domains and TJ barrier functions is expected to be associated with significant trafficking to and recycling from developing junctions. Given the well-described involvement of Ral and the exocyst in basolateral trafficking, we hypothesize that Ral–exocyst interactions are also required for development of these functions.
Here we show that RalA, RalB, and the exocyst are necessary for normal formation of TJs. Each has a different role in this process, however, as knockdown individually has opposite effects on development of transepithelial resistance (TER) and incorporation of components into TJs but not development of distinct apical–basal membrane domains. Furthermore, Ral GTPase activation is differentially regulated following induction of intercellular adhesion. Finally, we show that the functions of RalA and RalB in TJ formation are mediated through interactions with the exocyst.
RESULTS
RalA and RalB have opposing roles in the development of barrier function but not polarity during TJ assembly
Transducing cells with lentiviruses coding short hairpin RNAs (shRNAs) specific to RalA or RalB (short hairpin RalA [shRalA] and short hairpin RalA [shRalB] cells, respectively) reduced RalA or RalB expression in Madin–Darby canine kidney strain II (MDCK II) cells. In cell lines stably expressing these shRNAs, knockdown efficiency was ∼70% for RalA and >95% for RalB (Figure 1A). A control cell line was also transduced with a Lentivirus containing a nonspecific shRNA into MDCK II cells (short hairpin nonspecific control [shCtrl] cells). Specificities of RalA and RalB depletion were determined by immunoblotting and immunofluorescence labeling of endogenous proteins; both RalA and RalB localized to the plasma membrane in subconfluent MDCK II cells, and this localization was unaffected in shCtrl cells (Figure 1B). In shRalA cells, membrane-associated RalA labeling was completely lost, whereas RalB localization was unaffected. In contrast, membrane-associated RalB labeling was lost in shRalB cells, but RalA localization was not altered (Figure 1B). MDCK II cells exhibited not only plasma membrane labeling, but also a diffuse cytosolic and/or nuclear labeling for both RalA and RalB; however, this labeling was nonspecific, as neither signal was reduced in knockdown cell lines. Hairpin-resistant (hr) variants of RalA and RalB were used to rescue knockdown in respective cell lines (hrRalA and hrRalB, respectively), and expression levels and localizations were determined to be similar to those of endogenous proteins in shCtrl cells (Figure 1).
FIGURE 1:

RalA and RalB knockdown is specific. (A) Knockdown efficiency and rescue, as determined by immunoblotting for RalA, RalB, and β-tubulin (loading control). Cell lysates were generated after selection for cells stably expressing shCtrl, shRalA, or shRalB and a hairpin-resistant (hr) RalA or RalB (harboring silent point mutations in the short hairpin targeting sequence) in respective knockdown cells. (B) Immunofluorescence labeling of endogenous RalA and RalB in subconfluent MDCK II, shCtrl, shRalA, and shRalB cells. Localization of endogenous RalA and RalB at the plasma membrane was unaffected in shCtrl cells, but each protein was specifically lost from this site in shRalA or shRalB cell lines, respectively. Bar, 20 μm.
Because TJ development is promoted by E-cadherin–mediated cell–cell adhesion (Gumbiner and Simons, 1986), we sought to determine whether RalA or RalB becomes activated in response to establishment of cell–cell contacts. A glutathione S-transferase (GST)–Exo84 Ral-binding domain (RBD) fusion protein was used to precipitate RalAGTP at various time points after inducing cell–cell adhesion. Levels of RalAGTP increased steadily following initiation of cell–cell adhesion (Figure 2, A and B); within 2 h of Ca2+ addition, RalAGTP levels were increased threefold to fourfold higher than baseline (Figure 2B).
FIGURE 2:

RalA is activated, and RalB is inactivated, following initiation of E-cadherin–mediated cell–cell adhesion. (A, C) Immunoblots of RalA and RalB show activation and inactivation, respectively, of each GTPase following induction of cell–cell adhesion. Serum-starved MDCK II cells stably expressing myc-tagged RalA or RalB were subjected to a Ca2+ switch using serum-free LCM and HCM. GTP pulldowns were performed at the indicated times, and immunoblotting for RalA or RalB was performed with total and pulldown samples. Closed arrows denote ectopic, myc-tagged Ral, and open arrows denote endogenous Ral. (B, D) Quantification of A and C was performed using a phosphorimager screen and ImageQuant software. Immunoblots and quantitations are representative of three independent experiments.
RalBGTP was precipitated from cells using a GST-Sec5 RBD fusion protein. In contrast to increased RalAGTP levels, we observed a substantial decrease in the amount of RalBGTP following induction of cell–cell adhesion (Figure 2, C and D). The amount of RalBGTP fell to <50% of baseline levels within 0.5 h of cell–cell contact initiation and remained reduced for at least 2 h. That RalA was activated and RalB was inactivated in response to E-cadherin–mediated adhesion suggests that the two GTPases are regulated independently during TJ formation.
A common technique for monitoring the formation of functional TJs is to measure the TER of cell monolayers after inducing synchronous cell–cell adhesion by raising extracellular calcium (Ca2+) levels (Ca2+ switch; Gumbiner and Simons, 1986; Gonzalez-Mariscal et al., 1990). Following a Ca2+ switch, MDCK II cells displayed a characteristic TER overshoot, with TER subsequently returning to baseline levels (Figure 3A; Cereijido et al., 1978; Griepp et al., 1983). In shRalA cells this overshoot was completely suppressed, with TER slowly increasing to steady-state levels. Conversely, in shRalB cells the TER overshoot was greatly exaggerated but returned to steady-state levels comparable to those in control and shRalA cells (Figure 3A). The TER profiles of shRalA and shRalB cells were restored to normal by expression of hrRalA or hrRalB, confirming that these effects on TJ formation were specific to Ral GTPases (Figure 3B).
FIGURE 3:

RalA and RalB knockdown differentially affect barrier function during TJ formation. (A) RalA knockdown caused loss of the TER overshoot, whereas RalB knockdown drastically exaggerated the overshoot. MDCK II, shRalA and shRalB cells were seeded at confluent densities on Transwell filters in low-Ca2+ medium for 3 h before high-Ca2+ medium was added (Ca2+ switch), and the TER was measured every hour for 30 h and at 6-h intervals thereafter. TER measurement every hour caused frequent changes in media temperature, which caused increased TER values and slowed kinetics of TJ formation. Thus TER of subsequent Ca2+-switch experiments was measured starting 6–10 h following addition of HCM. (B) Expression of hrRalA or hrRalB in respective knockdown cell lines rescued knockdown TER phenotypes. A Ca2+ switch was performed with the indicated cell lines, as described in A. TER was measured every hour starting 6 h after HCM addition. (C) Ral knockdown has no effect on the ability of 3H-inulin to pass across the pores of developing TJs. A Ca2+ switch was performed on 12-mm Transwell filters with indicated cell lines, and 3H-inulin was applied to apical chambers of filters. Medium from basal chambers was removed, and the amount of 3H-inulin present was quantified using a scintillation counter. Error bars, SD of the mean of triplicate readings from three independent filters.
To further investigate the integrity of newly forming TJs in Ral-knockdown cells, diffusion of 3H-inulin was used to analyze passage of large, noncharged solutes across the monolayer by way of the pore pathway. 3H-Inulin was added to apical medium of control and knockdown cells 9 h following a Ca2+ switch. Samples were removed from both chambers and quantified to determine diffusion rates of 3H-inulin across developing monolayers. As expected, there was substantial diffusion across blank Transwell filters; however, there were no statistical differences between the amount of 3H-inulin that diffused across developing TJs in shCtrl and either Ral-knockdown cell line (Figure 3C). Taken together, these data suggest that RalA and RalB are uninvolved with generation of the large solute barrier but are specifically involved with formation of the charge selective barrier function of the TJ.
It is unknown whether the TJ develops barrier and “fence” functions simultaneously, although previous work established that these functions are differentially regulated (Mandel et al., 1993; Takakuwa et al., 2000). To determine whether Ral GTPases are important during development of distinct apical and basal domains at the time when maximal differences in electrical barrier function are observed, we directly assayed for presence of the “fence” function 10 h after a Ca2+ switch. BODIPY-FL-C5 sphingomyelin was used to label the outer leaflet of the apical plasma membrane of shCtrl, shRalA, and shRalB cells. Cells were fixed and imaged either immediately (Figure 4A) or following a 1-h incubation on ice (Figure 4B). Surprisingly, the fluorescent lipid was restricted to the apical plasma membrane in all cell types, indicating that apical and basal membrane domains were separated by TJs at this time point. To confirm that the lipid can diffuse along all exterior plasma membranes in absence of TJs, each cell line was maintained in low-calcium medium (LCM) prior to labeling and imaging (Figure 4C). To quantify differences in fluorescent lipid that may have diffused through TJs of shCtrl, shRalA, and shRalB cells, lipid was back-extracted from apical or basolateral membranes of monolayers incubated in absence or presence of extracellular Ca2+. This analysis showed that the amounts of lipid back-extracted from basolateral and apical membranes of cells maintained in LCM were nearly identical in all cell types, indicating a lack of TJs (Figure 4E). Similar amounts of fluorescent lipid were also back-extracted from monolayers of shCtrl, shRalA, and shRalB cell lines in the presence of extracellular Ca2+ (Figure 4D). This fluorescence labeling and quantification-based analysis demonstrates that TJ separation of apical and basal membrane domains is achieved before the period at which TJ electrical barrier function most significantly differs between control and Ral knockdown cells and that neither RalA nor RalB contributes to this function.
FIGURE 4:

Formation of distinct apical–basal membrane domains is established prior to equilibration of TJ barrier function and is independent of both RalA and RalB. (A–C) The “fence” function is present in shCtrl, shRalA, and shRalB cells but not in cells without TJs. (A) A Ca2+ switch was performed on each cell type, and after 10 h, samples were labeled with BODIPY-FL-C5 sphingomyelin and imaged immediately. (B) Samples were processed as in A, but were washed and incubated on ice for 60 min before imaging. (C) Samples were labeled before the switch to HCM and immediately processed for imaging. Bars, 10 μm. (D, E) Samples treated as in B and C, respectively, were incubated for 1 h in buffer containing defatted BSA after labeling. Fluorescent signals in apical and basolateral samples were quantified. Error bars, SD of the mean of triplicate readings of three independent filters.
RalA and RalB distinctly alter TJ composition
To further elucidate the mechanisms underlying the roles of RalA and RalB in development of barrier function, we performed immunofluorescence labeling studies, assessing whether knockdown of each GTPase affected protein incorporation into newly forming TJs. This analysis was performed in cells 10 h after cell–cell adhesion was induced, the time at which differences in barrier function were greatest between knockdown and control cell lines (Figure 2). Compared to TJs of MDCK II and shCtrl cells, those of shRalA cells contained considerably less ZO-1, whereas considerably more ZO-1 was incorporated into TJs of shRalB cells (Figure 5, A and B). Similarly, the levels of claudin4 (Figure 5, C and D), claudin1, and claudin7 (Supplemental Figure S1, A and B) were significantly decreased in TJs of shRalA cells and increased in those of shRalB cells compared with TJs of MDCK II and shCtrl cells. TJ incorporation of occludin was unaffected in shRalB cells but significantly decreased in shRalA cells (Supplemental Figure S1, C and F). Of interest, the fluorescence intensity of claudin2 in shRalB cells did not show the same distribution as other TJ proteins in these cells; it was decreased in TJs to a greater extent than in shRalA cells (Figure 5, E and F). These results are consistent with a model in which RalA is necessary for delivery of components to newly forming TJs, whereas RalB is necessary for endocytosis of these same components, and that certain proteins (e.g., claudin2) have more complex levels of regulation.
FIGURE 5:

Ral knockdown directly alters TJ composition. (A, C, E) Immunofluorescence labeling of ZO-1, claudin4, and claudin2 in TJs is dependent on RalA and RalB. MDCK II, shCtrl, shRalA, and shRalB cells were subjected to a Ca2+ switch, and samples were processed for immunofluorescence 10 h after HCM addition. For each protein examined (ZO-1, claudin4, and claudin2), optical sections were merged into a single stacked image and inverted. Bars, 20 μm. (B, D, F) Fluorescence intensities of TJ labeling of each protein in each cell type were traced and quantified using ImageJ software. Images are representative of three independent experiments. Error bars, SD of five lengths of TJ. *p < 0.05.
We sought to confirm these results by biochemically analyzing and quantifying the solubility of TJ-associated claudins in Ral-knockdown cells. The results of these experiments were consistent with those of immunofluorescence analysis. Specifically, in shRalA cells, the amounts of insoluble claudin1, 2, and 4 were significantly decreased relative to those in MDCK II cells (Supplemental Figure S2, A, E, and I). The amount of insoluble claudin3 was only modestly reduced, however, and that of claudin7 was not affected (Supplemental Figure S2, C and G). RalB knockdown caused the opposite effect of RalA knockdown; all claudins, with the exception of claudin2, became more insoluble when RalB expression was suppressed, suggesting increased amounts of components in the TJs of these cells. Specifically, incorporation of claudin1, 4, and 7 in TJs of shRalB cells was considerably increased (Supplemental Figure S2, A, E, and G) and that of claudin3 was slightly increased (Supplemental Figure S2C). Substantially less claudin2 was insoluble in shRalB cells than in control and shRalA cells (Supplemental Figure S2I). Taken together, these biochemical analyses of TJ component incorporation and immunofluorescence studies indicate that RalA and RalB directly alter TJ composition, likely by exocytosis and endocytosis.
Because the amounts of TJ components incorporated into junctions changed as a result of Ral GTPase knockdown, we postulated that the stability of these proteins might be affected. In the case of shRalA cells, we expected to observe a general decrease in stability of TJ components because overall incorporation into junctions was decreased. Conversely, in cells lacking RalB we anticipated components of TJs to be more stable. To assess the stability of TJ resident proteins, we treated cells with cyclohexamide for various lengths of time before processing for immunoblotting and analysis (Supplemental Figure S3). Although the stability of certain TJ proteins was only slightly affected in shRalA cells (Supplemental Figure S3, C and D), that of others (e.g., claudin1, occludin, and ZO-1) was much lower than in MDCK II and shRalB cells (Supplemental Figure S3, A, E, and G). In contrast, the stability of several other TJ components (claudin1, claudin4, occludin, and ZO-1) was much higher in shRalB cells than in MDCK II and/or shRalA cells (Supplemental Figure S3). These results confirm that RalA and RalB differentially regulate either the delivery of proteins to or their incorporation into TJs during development of epithelial polarity.
RalB regulates TJ assembly via endocytosis
Observed differences in TER profiles could theoretically result from altered trafficking of TJ components to and from plasma membrane sites of cell–cell contacts. Alternatively, lateral diffusion of components within the TJ could be involved, as transient chelation of extracellular Ca2+ is known to rapidly compromise TJ barrier function without promoting internalization of TJ components (Cereijido et al., 1978; Siliciano and Goodenough, 1988). Furthermore, this disruption has been observed to result from cytoskeleton remodeling in response to acute Ca2+ depletion (Ivanov et al., 2004). Given that RalA and RalB are known to be involved with cytoskeleton remodeling in some contexts (Lalli and Hall, 2005), we sought to exclude the possibility that these GTPases were acting in this manner. Within 10 min of acute Ca2+ depletion, TER of control and Ral knockdown cells was drastically reduced, but it returned to baseline levels 3–4 h after physiologic levels of extracellular Ca2+ were restored (Figure 6A). ZO-1 distribution at the TJ was not affected during Ca2+ chelation and restoration, confirming that neither endocytosis nor exocytosis of TJ components was involved in these transient changes in electrical barrier function (Figure 6B). Therefore, despite having contrasting effects on barrier function during TJ development, Ral GTPases do not appear to regulate TJ barrier function via cytoskeleton remodeling. RalA and RalB are thus likely required for the trafficking of components to and from the plasma membrane during TJ formation.
FIGURE 6:

Ral knockdown does not affect reorganization of TJ components within the lateral membrane. (A) The TER of MDCK II, shCtrl, shRalA, and shRalB cells is recovered with comparable kinetics after acute Ca2+ chelation. Polarized MDCK II, shCtrl, shRalA, and shRalB cells at steady state on Transwell filters were treated with EGTA for 10 min, after which the medium was replaced with HCM. TER measurements were recorded immediately before EGTA treatment and at regular intervals after Ca2+ addition. Error bars, SD of the mean of triplicate readings from three independent filters. (B) ZO-1 is not internalized after 10 min of EGTA treatment. Samples were fixed and processed for immunofluorescence localization of ZO-1 after 10 min of EGTA treatment. Inset, magnification of TJ segment. Bars, 20 μm.
Because reductions of RalA and RalB cause opposite effects on TER development (Figure 3) and RalA promotes exocytosis of junction components (Shipitsin and Feig, 2004; Figure 6), we hypothesized that RalB is required for endocytosis of junction proteins. Because biotinylation of TJ proteins is inefficient in MDCK cells, the internalization efficiency of an adherens junction protein, E-cadherin, was analyzed. Cells were biotinylated 9 h following a Ca2+ switch using a cleavable form of biotin, sulfosuccinimyl 2-(biotinamido)ethyl-l,3 di-thioproprionate (NHS-SS-biotin). This form of biotin allowed for determination of the maximum biotinylatable material, background following cleavage, and amount endocytosed (Figure 7A). The amount of E-cadherin endocytosed during a 30-min uptake phase was quantified by immunoblot in triplicate. Although there was no difference between the amount of E-cadherin endocytosed in shCtrl and shRalA cells, shRalB cells internalized significantly less (p < 0.05; Figure 7B).
FIGURE 7:

Endocytosis of E-cadherin is specifically disrupted by loss of RalB but not RalA. (A, B) RalB knockdown results in decreased endocytosis of E-cadherin. Indicated cell lines were subjected to a Ca2+ switch after seeding on 24-mm Transwell filters and were biotinylated 9 h following HCM addition. Three filters of each cell type were not reduced (NR), reduced with no 37°C incubation (T-0), and incubated for 30 min at 37°C (T-30). Avidin pulldowns were completed, and E-cadherin immunoblots were performed with total and pulldown samples. Immunoblots were visualized with a ChemiDoc-It Imaging System and quantified with Visionworks software. (C) Transferrin internalization rate is not affected by Ral knockdown. 125I-Transferrin was bound to cells infected with human transferrin receptor and warmed for indicated times (data points). Both internal and surface 125I-transferrin were quantified and applied to a model in which clearance from the surface into early endosomes is represented by the equation  . The lines represent best-fit curves generated by the model (shCtrl, solid line; shRalA, dashed line; shRalB, dotted line). (D) Inhibition of dynamin during TJ assembly causes a TER profile that phenocopies RalB knockdown. A Ca2+ switch was performed with MDCK II cells treated with DMSO or a dynamin inhibitor (dynasore, 80 μM). Triplicate TER readings of triplicate filters were recorded every hour for 20 h. (E) Dynasore inhibits endocytosis of transferrin. MDCK cells expressing human transferrin receptor were treated with DMSO or 80 μM dynasore. Samples were processed for immunofluorescence after addition of fluorescent human transferrin. Arrows indicate origin of the xy-slice within the z-series. Error bars, SD of triplicate samples. *p < 0.05. Bars, 20 μm.
. The lines represent best-fit curves generated by the model (shCtrl, solid line; shRalA, dashed line; shRalB, dotted line). (D) Inhibition of dynamin during TJ assembly causes a TER profile that phenocopies RalB knockdown. A Ca2+ switch was performed with MDCK II cells treated with DMSO or a dynamin inhibitor (dynasore, 80 μM). Triplicate TER readings of triplicate filters were recorded every hour for 20 h. (E) Dynasore inhibits endocytosis of transferrin. MDCK cells expressing human transferrin receptor were treated with DMSO or 80 μM dynasore. Samples were processed for immunofluorescence after addition of fluorescent human transferrin. Arrows indicate origin of the xy-slice within the z-series. Error bars, SD of triplicate samples. *p < 0.05. Bars, 20 μm.
To investigate whether loss of RalB caused a global disruption in endocytosis, 125I-transferrin internalization was analyzed in control and Ral-knockdown cell lines. 125I-Transferrin was bound to surface receptors on shCtrl, shRalA, and shRalB cells and internalized for indicated times, and the amount of endocytosed material was quantified with a scintillation counter. To determine the endocytic rate, percentage surface transferrin was plotted, and best-fit curves were applied using the equation 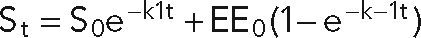 . No significant differences were observed in the rate of transferrin internalization in control or Ral-knockdown cells (Figure 7C).
. No significant differences were observed in the rate of transferrin internalization in control or Ral-knockdown cells (Figure 7C).
To further study the role of RalB in endocytosis during TJ development, we specifically blocked endocytosis with dynasore, a specific inhibitor of dynamin-mediated endocytic pathways. Other studies determined that dynasore does not affect dynamin-independent pathways (Macia et al., 2006). Immunofluorescence analysis was also performed to verify that dynasore treatment effectively inhibited endocytosis. MDCK T cells stably expressing human transferrin receptor were treated with DMSO or dynasore before labeling with fluorescent human transferrin. Confocal xy- and xz-series show that transferrin was internalized in cells treated with DMSO but not dynasore (Figure 7E). A Ca2+ switch was performed with MDCK II cells treated with DMSO or dynasore, beginning with the readdition of high-calcium medium (HCM). Although slightly lower than typically observed, the MDCK II cells treated with DMSO displayed a characteristic TER overshoot and subsequent return to basal levels (Figure 7D). Dynasore treatment, however, caused an extensive exaggeration of the TER overshoot (Figure 7D) that closely resembled the TER profile of shRalB cells (Figure 3). These data, combined with the E-cadherin endocytosis results, indicate that reduced endocytic rates in shRalB cells may be specific for junctional components.
RalA and RalB affect development of TJ barrier function via interactions with the exocyst
We next sought to determine the effects of Ral knockdown on exocyst assembly and stability. Exocyst stability in shRalA or shRalB cells was assessed as a function of Sec6 and Sec8 half-life and was found to be unaffected compared with exocyst stability in control MDCK II cells (Supplemental Figure S4, A and B). To determine whether Ral GTPases regulated assembly of the exocyst holocomplex, the exocyst was immunoprecipitated with Sec8 antibodies following a brief pulse labeling (Supplemental Figure S4C). Ratios of newly synthesized Sec6 and Sec15 to Sec8 were compared in control cells, Ral-knockdown cells, and cells overexpressing constitutively active RalA23V or RalB23V. One-way analysis of variance (ANOVA) with Tukey's post test was performed, which indicated that these ratios (e.g., Sec6:Sec8 and Sec15:Sec8) were not significantly different, regardless of the presence or absence of each GTPase (Supplemental Figure S4, D and E). These observations confirm that Ral GTPases are not strictly required for exocyst assembly and suggest that they are likely involved in regulating other exocyst activities, consistent with previous observations (Spiczka and Yeaman, 2008).
To determine whether the effects of RalA on development of TJ barrier function were mediated via the exocyst, we introduced RalA point mutations that specifically and severely reduce its binding affinity for either Sec5 or Exo84 (Fukai et al., 2003; Jin et al., 2005) in the context of a constitutively active mutant (Q72L). Specifically, Sec5-uncoupled (E38R) and Exo84-uncoupled (A48W) variants were generated. These constructs were used to establish tetracycline-inducible cell lines that expressed mutant proteins at levels comparable to those of endogenous RalA within 2 d of induction (Figure 8A).
FIGURE 8:

Ral engagement of the exocyst is necessary for formation of TJ barrier function. (A) RalA72L, RalA38R, and RalA48W mutants are not expressed in the presence of doxycycline but are expressed at levels comparable to endogenous RalA 2 d after doxycycline removal (induction). Open arrows indicate positions of endogenous RalA, and closed arrows indicate myc-tagged, ectopic RalA mutant proteins. (B–D) A Ca2+ switch was performed with induced and noninduced cells, and TER measurements were recorded every hour for 25 h after HCM addition. (E) Interaction of RalB with Sec5 is required for normal TJ barrier function. shRalB cells were transiently transfected with RalB23V or RalB49E and subjected to a Ca2+ switch. Error bars, SD of the mean of triplicate readings from three independent filters.
Although the RalA38R and RalA48W point mutations selectively interfere with Sec5 and Exo84 binding in vitro (Jin et al., 2005), whether these mutations cause similar binding disruptions when expressed in cells is not known. We found that RalA38R was significantly impaired in its ability to bind Sec5 but not RalBP1 or Exo84 in cells. In contrast, RalA48W was impaired in its binding to Exo84 but not RalBP1 or Sec5 (Supplemental Figure S5A). As expected, RalA72L bound all three effectors similarly (Supplemental Figure S5A). This analysis also revealed that the subcellular localization of RalA mutants depended on Sec5- or Exo84-binding capacity. Immunofluorescence labeling and differential sedimentation approaches demonstrated that RalA72L was present on both intracellular vesicles and the plasma membrane (Supplemental Figure S5, B and C), as reported previously (Shipitsin and Feig, 2004). RalA38R, however, was localized primarily on endomembranes and vesicles (Supplemental Figure S5, B and D), whereas RalA48W was enriched on the plasma membrane (Supplemental Figure S5, B and E). These results suggest that RalA may engage Sec5 and Exo84 at distinct subcellular locations.
We next determined whether the development of normal TJ barrier function requires RalA to engage the exocyst. As expected, control cell lines expressing only endogenous RalA displayed the characteristic TER overshoot (Figure 8, B–D). The TER profile of RalA72L-expressing cells was similar to that of noninduced cells, although the overshoot occurred at a substantially expedited rate (Figure 8B). Induction of either RalA38R or RalA48W, however, caused loss of the TER overshoots, which phenocopied the TER profile observed in shRalA cells (Figures 3A and 8, C and D).
Transient expression of point-mutant RalB variants was used to investigate the requirement of RalB–exocyst interactions in normal TJ formation. Constitutively active (23V) and Sec5-uncoupled (49E) RalB variants were used to rescue the exaggerated TER overshoot observed in shRalB cells. The TER following a Ca2+ switch was rescued to lower levels by expression of the RalB23V variant but not the RalB49E variant (Figure 8E). These data indicate that RalA must engage each of its exocyst-localized effectors, Sec5 and Exo84, in order to properly function in the process of TJ formation.
DISCUSSION
RalA and RalB differentially regulate development of TJ barrier function through recruitment of the exocyst but do not affect formation of distinct apical basal membrane domains. This conclusion is based on several findings: 1) knockdown of RalA or RalB had opposing effects on TER development (Figure 3); 2) TER differences were mirrored by differential incorporation of proteins into TJs (Figure 5 and Supplemental Figure S1); 3) trafficking of junctional components is impaired by RalA and RalB knockdown (Figures 6 and 7); and 4) the exocyst is a common Ral effector in TJ development (Figure 8). Furthermore, Ral knockdown had no effect on transepithelial diffusion of outer leaflet phospholipids (Figure 4). Therefore we suggest that RalA regulates trafficking of components to newly forming TJs, RalB regulates endocytosis of junctional components, and both processes require the exocyst. Together, these opposing activities likely serve to fine tune TJ barrier function in response to specific cellular or environmental demands.
Ral GTPases have opposing roles in development of barrier function but not polarity during TJ assembly
That RalA and RalB differentially affect TJ formation is consistent with evidence showing that Ral GTPases serve opposing or complementary roles in diverse cellular processes. During cytokinesis, for example, RalA is first required for stabilization and elongation of the intracellular bridge, whereas RalB is recruited later to complete abscission (Cascone et al., 2008). Of note, cytokinesis-associated activities of both RalA and RalB require binding to the exocyst (Cascone et al., 2008). In tumor cells, Ral GTPases regulate the balance between proliferation and survival. Specifically, RalA-mediated promotion of cell proliferation is offset by RalB-mediated inhibition of apoptosis (Bodemann and White, 2008). During tumor cell invasion, RalA and RalB mediate complementary but distinct functions, as each must engage the exocyst at different subcellular locations to properly localize the complex to cell protrusions (Spiczka and Yeaman, 2008). Therefore contrasting functions of Ral GTPases during TJ development are consistent with an emerging paradigm concerning the actions of these proteins.
Antagonistic effects of Ral GTPases on TJ barrier function and composition predict that cells independently regulate activities each GTPase during TJ formation. This may be achieved by recruiting each to distinct subcellular localizations or by selective activation or inactivation at those sites. Sites of Ral activation have been demonstrated with Ras and interacting protein chimeric unit (Raichu) probes, which revealed that Ral GEFs activate RalA specifically at the leading edge of lamellipodia in epidermal growth factor–stimulated Cos7 cells (Takaya et al., 2004) and at recycling endosomes in MDCK cells (Takaya et al., 2007). Similar studies revealed that changing cellular demands caused relocalization-specific Ral-GEFs and guanine-activating proteins, resulting in spatiotemporal regulation of Ral activities (Ohba et al., 2003). Our finding that RalA and RalB are inversely regulated following induction of E-cadherin–mediated cell–cell adhesion is consistent with these findings. Rapid activation of RalA would promote exocytosis as cell contacts are initially forming (Figure 2; Shipitsin and Feig, 2004). Concurrent inactivation of RalB, which signals internalization of junction proteins, would synergize with RalA activation to promote efficient junction assembly.
During development of epithelial polarity, TJs assemble to maintain distinct membrane domains and provide a size- and charge-selective barrier for paracellular diffusion of solutes and ions. Although much research concerning epithelial TJ barrier function has been performed, less attention has been given to the relationship between barrier function and the establishment of apical–basal polarity. However, it is known that barrier function and maintenance of apical–basal polarity are differentially regulated. For example, barrier function is abolished by ATP depletion and actin disruption, and yet apical–basal polarity remains intact (Mandel et al., 1993; Takakuwa et al., 2000). Of interest, one study revealed that resident apical and basolateral proteins remained polarized after complete loss of TJs in a mouse epithelial cell line, whereas biotin diffused down the lateral membrane (Umeda et al., 2006). Nonetheless, it is unknown whether these functions were established concurrently, whether specific TJ composition affected maintenance of apical–basal polarity, and to what extent interactions with the submembrane cytoskeleton contributed to the steady-state polarity of the transmembrane protein markers examined.
Our investigation of RalA and RalB involvement in assembly of TJs indicated that these GTPases serve opposing roles in this process. Knockdown of RalA resulted in loss of the TER overshoot, whereas RalB knockdown caused an exaggerated overshoot (Figure 3A), which represents a deregulation of the small claudin pores that establish the TJ charge-selective barrier. To determine whether the effects of Ral knockdown on TER resulted from disruption of both size- and charge-selective barrier functions, we measured diffusion of 3H-inulin (Figure 3C). That there was no effect of Ral knockdown on paracellular diffusion of 3H-inulin indicates that the TJ size-selective barrier was functional and suggests that TJs were physically present.
Given these dramatic differences in effects of RalA and RalB knockdown on the charge-selective but not size-selective barrier of the TJ (Figure 3), we sought to determine whether these GTPases were also involved in formation of separate apical–basal membrane domains. Surprisingly, reduction of neither GTPase affected diffusion of outer leaflet lipids between apical and lateral membrane domains, and separation between these domains was established prior to equilibration of the electrical solute barrier (Figure 4). This observation initially appears to contradict data showing that RalA knockdown reduced incorporation of TJ components (Figure 5). However, it is possible that fewer components are required to retain discrete apical–basal membrane domains than are necessary for normal TJ barrier formation. Thus, although TJ strands are likely to be less complex at early time points following initiation of cell–cell adhesion than they are in mature TJs, there may still be a critical mass of components present within the plasma membrane to generate a functional apical–basal division to prevent diffusion of outer leaflet lipids between membrane domains. Conversely, perhaps the requirements of forming distinct apical and basal domains are not as complex as those for barrier function. Lateral oligomeric interactions of TJ proteins that are required for “fence” function might be established immediately upon insertion into the plasma membrane and thus be independent of specific TJ composition. Therefore distinct apical–basal membrane domains could be developed before the specific protein composition of the TJ is formed, thus preceding equilibration of barrier function.
The exaggerated differences in TER resulting from Ral knockdown suggest that components are differentially incorporated into TJs, including claudins. Our immunofluorescence and biochemical studies verified this hypothesis, as some components were reduced in RalA-knockdown cells and increased in RalB-knockdown cells (Figure 5 and Supplemental Figure 2). The pattern, quantity, and various electrostatic characteristics of claudins in TJs of an epithelial monolayer determine the permeability electrical barrier (Van Itallie and Anderson, 2006; Angelow et al., 2008). The role of RalA in exocytic trafficking has been well documented (Moskalenko et al., 2002; Shipitsin and Feig, 2004; Chen et al., 2007), and we propose that RalA regulates, in part, delivery of components to sites of TJ assembly. Furthermore, the increased amount of components in TJs of shRalB cells is consistent with a role of RalB in endocytosis of these proteins.
Ral GTPases mediate TJ assembly via endocytosis and exocytosis
Knockdown of RalA and RalB had opposing consequences on incorporation of multiple components into assembling TJs (Figure 5). We propose that these effects are due to perturbed trafficking of TJ components to and from developing TJs as a consequence of Ral knockdown. According to our model, RalA is required for exocytic trafficking during TJ formation, whereas RalB is required later, to mediate endocytosis of certain proteins to regulate TJ composition in order to meet specific cellular demands. Several lines of evidence support this model. RalA and RalB do not affect reorganization of TJ components within the junction nor are they involved with cytoskeleton-mediated remodeling during TJ assembly (Figure 6). Internalization of E-cadherin is significantly reduced during TJ formation in cells lacking RalB, and inhibition of dynamin-mediated endocytosis phenocopies the TER profile of RalB-knockdown cells during TJ assembly (Figure 7, B and D). That RalA knockdown caused decreased stability of TJ components and RalB knockdown caused increased stability of these components further suggests a defect in trafficking (Supplemental Figure S3). In shRalA cells, TJ cargo that is not delivered to developing TJs is likely degraded, as there are no intracellular accumulations of these proteins. Conversely, the increased stability of TJ proteins in shRalB cells suggests that they are not internalized and recycled from TJs. Furthermore, the exocyst has also been implicated in endocytosis, as it has been localized to endocytic vesicles in Drosophila oocytes (Sommer et al., 2005). Thus RalA and RalB may engage the exocyst to regulate exocytic and endocytic trafficking of TJ components.
Interactions between Ral and the exocyst are necessary for normal TJ formation
Use of Ral point mutants has facilitated study of specific Ral–Sec5, and Ral–Exo84 functions; for example, RalA–Sec5 binding is required in murine embryonic fibroblasts during cell spreading and lipid-raft exocytosis, and RalB–Sec5 binding is required during host–defense signaling, specifically for recruitment of Tank-binding kinase 1 to the exocyst (Chien et al., 2006; Balasubramanian et al., 2010). RalA–Exo84 interaction was shown to suppress early stages of squamous cell carcinoma progression by regulating plasma membrane delivery of E-cadherin (Sowalsky et al., 2010), and RalB–Exo84 binding is required for starvation-induced autophagocytosis (Bodemann et al., 2011). Disruption of RalA-Sec5, RalA–Exo84 or RalB-exocyst binding disrupted TJ biogenesis (Figures 3A and 8, B-E). Although RalA must to bind either Sec5 or Exo84 to support normal TJ assembly, the two interactions do not necessarily serve the same function. Indeed, we observed, by both immunofluorescence and differential sedimentation, that the two RalA mutants localize to different subcellular sites (Supplemental Figure S5). The shift of RalA38R to a primarily intracellular localization and of RalA47E to a primarily plasma membrane localization provides novel insights into roles of each interaction during basolateral trafficking. RalA may bind Sec5 at the trans-Golgi network/recycling endosome to facilitate trafficking to the plasma membrane, where RalA–Exo84 subsequently mediates vesicle tethering to sites of fusion. Disruption of either interaction would impair exocytosis.
In summary, our findings expand knowledge of the processes in which Ral GTPases are involved to include TJ biogenesis. In addition, they further elucidate the differences in regulation of TJ barrier and formation of apical–basal polarity. Ral GTPases are antagonistic regulators of TJ electrical barrier formation and composition and recruit the exocyst to function in distinct processes to mediate TJ assembly.
MATERIALS AND METHODS
Cell culture
MDCK strain II cells were maintained in low-glucose DMEM (LG-DMEM) with 1.8 mM Ca2+ containing 1 g/l sodium bicarbonate and supplemented with 10% fetal bovine serum (FBS; Cell Generation, Fort Collins, CO), penicillin, streptomycin, and gentamicin (PSG; HCM) and grown at 37°C with 5% CO2. shRalA and shRalB cells were generated by stable integration of shRNAs targeting canine RalA or RalB (RalA: sense, 5′-CGAGCTAATGTTGACAAGGTA-3′; RalB: sense, 5′-GAGTTTGTAGAAGACTATGAA-3′), respectively. shRNAs were delivered via transduction with recombinant lentiviral vectors that were pseudotyped with vesicular stomatitis virus G protein. Cells were selected and maintained in medium containing 5 μg/ml puromycin. Negative controls were generated by transducing MDCK II cells with lentiviral vectors encoding a nontargeting shRNA (sense, 5′-CCAGACCTTCAAGGAATCCAT-3′) and selecting them in puromycin. RalA rescue cell lines were generated by introducing three wobble-base point mutations into the first three codons of the target RalA hairpin sequence of simian RalA cDNA. This cDNA was ligated into pQCXIN and delivered into shRalA cells via retroviral transduction. Transduced cells were selected using 400 μg/ml G418 and assayed by immunofluorescence and immunoblotting for hairpin resistant RalA. The same process was performed to generate and screen RalB rescue cells. Inducible RalA mutant cell lines were generated using the MDCK T23 cell line, which expresses the tetracycline-repressible transactivator (Barth et al., 1997). The 72L, 38R, and 47E point mutations were introduced into a RalA cDNA that was a generous gift from Larry Feig (Tufts University School of Medicine, Boston, MA), using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA). These cDNAs were introduced into MDCK T23 cells as described previously (Jou and Nelson, 1998). Transient transfections were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the suggested protocol. Tissue culture plates, 10 cm, were transfected with DNA or vehicle only 24 h prior to seeding in triplicate 12-mm, 0.4-μm Transwell polycarbonate filters following the protocol described for measurement of transepithelial resistance.
Antibodies and reagents
Mouse monoclonal antibodies (mAbs) against Sec6 (9H5) and Sec8 (2E12, 5C3, 10C2) were described previously (Hsu et al., 1996; Kee et al., 1997; Yeaman, 2003). mAbs against claudin4, ZO-1, and occludin, as well as rabbit polyclonal antibodies against claudin1, claudin3, and claudin7, were obtained commercially (Zymed Laboratories, San Francisco, CA). RalA mAb (BD Transduction Laboratories, San Jose, CA), claudin2 mAb (Invitrogen), and myc mAb (Millipore, Billerica, MA) were also obtained commercially. A RalB mAb was a generous gift from Michael White (University of Texas Southwestern Medical Center, Dallas, TX). A rabbit polyclonal antibody against E-cadherin was a generous gift from W. James Nelson (Stanford University, Stanford, CA; Marrs et al., 1993). Fluorescein isothiocyanate (FITC)–goat anti–mouse and Texas red–donkey anti–rabbit immunoglobulin G (IgG) were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Horseradish peroxidase (HRP)–conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from Promega (Madison, WI). 125I-Labeled goat anti-mouse IgG, 125I-labeled goat anti–rabbit IgG, and 35S-methionine were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA).
Measurement of transepithelial resistance
A well-described Ca2+-switch method was used with cultures of MDCK II, shCtrl, shRalA, shRalB, RalA rescue, and RalB rescue cells (Gumbiner and Simons, 1986; Gonzalez-Mariscal et al., 1990). Each cell line was seeded on triplicate 0.4-μm Transwell polycarbonate filters at confluent density (8 × 105 cells/12-mm filter) in LG-DMEM with low Ca2+ (LCM; 5 μM Ca2+), supplemented with 1 g/l sodium bicarbonate, 5% dialyzed FBS (Cell Generation), and PSG. Cells were cultured at 37°C for 3 h to allow for attachment to the filter. TER measurements were recorded both before and after replacing LCM with HCM. Two filters were left free of cells but otherwise treated identically, so that the intrinsic resistance of the membrane could be determined. For each filter, three independent readings were taken at each time point, using a Millicell (Millipore) electrical resistance device. Final values were obtained by subtracting the averaged blank value from the averaged sample reading and multiplying by the surface area of the filter. The final results are expressed as ohms · cm2. Readings of samples in Figure 3A were recorded every hour after initial attachment in LCM. This technique resulted in frequent changes in media temperature, which caused higher TER values and decreased kinetics of TJ formation. In subsequent experiments, TER was not recorded until 6–10 h after HCM addition.
Immunofluorescence labeling
Samples were fixed on ice with 4% paraformaldehyde for 20 min and quenched with Ringer's saline (154 mM NaCl, 1.8 mM Ca2+, 7.2 mM KCl, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4) containing 50 mM NH4Cl. Samples were then permeabilized with CSK buffer (1% Triton X-100, 10 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 50 mM NaCl, 300 mM sucrose, 3 mM MgCl2) for 10 min or with Ringer's saline containing 0.1% SDS for 5 min; both buffers were supplemented with protease inhibitors (1 mM Pefabloc [Sigma-Aldrich, St. Louis, MO] and 10 μg/ml each of aprotinin, antipain, leupeptin, and pepstatin A). After samples were blocked with 0.2% fish-skin gelatin in Ringer's saline (blocking buffer) for 1 h, primary antibodies were diluted in blocking buffer and applied for 1 h at room temperature. After five washes with blocking buffer, FITC- or Texas red–conjugated secondary antibodies and 4′,6-diamidino-2-phenylindole were applied for 30 min at room temperature. Samples were washed five times with blocking buffer, and filters and coverslips were mounted onto slides using Elvanol (Dupont, Wilmington, DE)–paraphenylene diamine. Images were obtained using either a Zeiss (Thornwood, NY) 510 scanning confocal microscope (63× objective) equipped with a krypton/argon laser (FITC excitation using 488-nm laser line, Texas red excitation using 543-laser line) or with a Leica (Wetzlar, Germany) DMI 6000B microscope equipped with a BD CARV II (BD Biosciences, San Diego, CA) spinning disk confocal imager. Stacks of confocal images were collected from several different fields using a 63× objective and a Photometrics (Tucson, AZ) QuantEM 512SC EM-CCD high-speed camera. For studies examining immunofluorescence intensity at TJs, confluent monolayers of cells were subjected to a Ca2+ switch and processed as described at 10 h following Ca2+ addition. Acquired z-series optical sections were merged, and the immunofluorescence intensity along the TJ was traced and quantified using the wand tool with ImageJ software (National Institutes of Health, Bethesda, MD). One-way ANOVA with Tukey's posttest statistical analyses was performed for each protein and cell type, and differences are considered significant when p < 0.05.
Inulin diffusion
A Ca2+ switch was performed on MDCK II, shCtrl, shRalA, and shRalB cells seeded on triplicate 12-mm, 0.4-μm Transwell polycarbonate filters. At 9 h following Ca2+ readdition, 1 × 106 dpm of 3H-inulin was added in 0.5 ml of warm media to the apical chambers of all cell types, including bank filters. Filters were placed at 37°C, except for removal of 100 μl from basal chambers immediately after inulin addition and every 30 min thereafter, for a total of 4 h. After removal of basal samples, 100 μl of nonlabeled fresh media was applied to basal chambers at each time point. The basal samples were placed in 3 ml of scintillation fluid, and each was counted with a Beckman LS 6500 multiplier purpose scintillation counter (Beckman Coulter, Brea, CA) for 0.3 min in triplicate.
Biotin assay for endocytosis
Totals of 3 × 106 shCtrl, shRalA, and shRalB cells were seeded on triplicate 24-mm, 0.4-μm–pore size Transwell polycarbonate filters, and a Ca2+ switch was performed as described. At 9 h following Ca2+ readdition, filters were placed on ice and washed twice with Ringer's saline. Apical and basal chambers were biotinylated with 0.5 mg/ml NHS-SS-biotin (Thermo Scientific, Waltham, MA). All filters were washed three times with DMEM/0.2% BSA, and three filters of each cell type were lysed immediately after the last wash. Three filters of each cell type were placed into warm media and incubated at 37°C for 30 min, whereas the other three remained on ice. Endocytosis was stopped by placing warm filters on ice and washing with ice-cold Ringer's saline. Filters were reduced and quenched following a method described previously (Graeve et al., 1989). Filters were lysed with radioimmunoprecipitation assay buffer (RIPA; 50 mM Tris-HCl, pH 7.5, 1% NP40, 0.25% sodium deoxycholate, 150 mM NaCl, and 1 mM EDTA) containing protease inhibitors. Following centrifugation for 10 min at 14,000 rpm, 10% of the total extract was removed for analysis, whereas the remainder was incubated with streptavidin agarose resin (Thermo Scientific) overnight at 4°C. Beads were pelleted by gentle centrifugation and washed as described for immunoprecipitation. Immunoprecipitates were eluted from beads with SDS–PAGE sample buffer and processed for SDS–PAGE as described. Immunoblots were performed using antibodies specific to E-cadherin, and band intensities were quantified, as described.
Inhibition of endocytosis
MDCK II cells stably expressing the human transferrin receptor were seeded at confluent density on 12-mm Transwell polycarbonate filters, and a Ca2 -switch was performed as described. HCM readdition was supplemented with DMSO or 80 μM dynasore (Sigma-Aldrich). TER of triplicate filters was recorded in triplicate beginning 8 h after HCM readdition. For immunofluorescence, samples were treated with DMSO or 80 μM dynasore for 2 h before addition of fluorescent human transferrin for 25 min. Samples were fixed for immunofluorescence as described but not permeabilized.
Transferrin internalization
Human transferrin receptor was expressed in shCtrl, shRalA, and shRalB cells by transduction with a replication-deficient adenoviral vector. Cells were transduced 1 d after seeding at confluent density on 12-mm Transwell filters in HCM. Three days after transduction, cells were incubated in serum-free DMEM at 37°C for 30 min. Filters were then placed on ice and washed with PBS2+ (phosphate-buffered saline [PBS] with 0.9 mM CaCl2 and 0.5 mM MgCl2) before labeling with 125I-transferrin on ice for 45 min. All filters were washed five times with cold PBS2+. Triplicate filters of each cell type were then warmed to 37°C for indicated times prior to immediate transfer to cold PBS2+. All filters were then washed twice, alternating between DMEM, pH 3.5, and PBS2+. Media and PBS2+ from these washes were saved in 5-ml glass tubes. Following the final wash, filters were excised from collars and transferred to 5-ml glass tubes. Radioactivity associated with filters and media samples were quantified using a Titertek Plus Series Automatic Gamma Counter (Huntsville, AL). The model used, in which clearance from the cell surface into early endosomes assumes a first-order rate constant, is represented by the equation 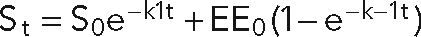 , where St is the percentage of total transferrin at the cell surface at time t, S0 is at time zero, EE0 is the percentage of total transferrin in early endosomes at time zero, k1 is the internalization rate, k–1 is the rate of transfer from early endosomes to the cell surface. t is time in minutes, and e is the natural logarithm base. The internalization rate of transferrin was determined using a model described previously (Sheff et al., 1999).
, where St is the percentage of total transferrin at the cell surface at time t, S0 is at time zero, EE0 is the percentage of total transferrin in early endosomes at time zero, k1 is the internalization rate, k–1 is the rate of transfer from early endosomes to the cell surface. t is time in minutes, and e is the natural logarithm base. The internalization rate of transferrin was determined using a model described previously (Sheff et al., 1999).
Ral GTP pulldown
Cells expressing myc-tagged RalA or RalB were seeded at confluent density (14 × 106 cells) in LCM lacking serum on six 10-cm tissue culture plates. They were subjected to the described Ca2+-switch protocol using HCM without serum. Plates were cultured in HCM at 37°C for the indicated length of time before being placed on ice and washed twice with Ringer's saline. Each plate was lysed with 1 ml of GTP lysis buffer (1% NP40, 50 mM Tris, pH 7.5, 200 mM NaCl, 10 mM MgCl2, and 10 mM dithiothreitol) containing protease inhibitors (1 mM Pefabloc and 10 μg/ml each of aprotinin, antipain, leupeptin, and pepstatin A). Lysates were cleared at 20,000 × g, and 10% of the supernatant from each sample was set aside for assessment of total protein levels; the remaining supernatants were transferred to glutathione–agarose beads (Pierce; 25-μl bead volume/sample) prebound to GST-Exo84 PH RBD (RalA wild-type samples) or GST-Sec5 RBD (RalB wild-type samples). Samples were rotated for 45 min at 4°C and then washed twice with GTP lysis buffer and prepared for SDS–PAGE as described previously (Pasdar and Nelson, 1988).
Fluorescence labeling of the lipid membrane
BODIPY-FL-C5 sphingomyelin (Molecular Probes, Invitrogen) and defatted BSA were used to prepare sphingomyelin/BSA complexes (5 nmol/ml) in P buffer (10 mM HEPES, pH 7.4, 1 mM sodium pyruvate, 10 mM glucose, 3 mM CaCl2, and 145 mM NaCl). A Ca2+ switch was performed on shCtrl, shRalA, and shRalB cells seeded at confluent density on 12-mm Transwell filters, as described previously. Five filters of each cell type were placed on ice 10 h after the switch to HCM, and apical and basal chambers were washed twice with cold P buffer. For labeling, 0.5 ml of 5 μM sphingomyelin/BSA complex was added to apical chambers, and 1 ml of P buffer was added to basal chambers. Filters were incubated on ice for 10 min and then washed extensively with P buffer. For each cell type, one filter was immediately processed for confocal microscopy, as described previously, and one additional filter was incubated in P buffer on ice for 1 h and then processed for confocal microscopy. The remaining three filters of each cell type were incubated on ice for 1 h in P buffer containing 1 mg/ml defatted BSA (0.5 ml apical, 1 ml basal). The solutions from apical and basal compartments were collected at the end of this incubation, and fluorescence intensities were quantified using a Wallac Victor2 1420 Multi Label Counter (PerkinElmer).
Gel electrophoresis and immunoblotting
Protein samples were incubated in SDS–PAGE sample buffer for 10 min at 65°C before separation on 10, 12.5, or 15% SDS polyacrylamide gels. Proteins were electrophoretically transferred onto Immobilon polyvinylidene fluoride membrane (Millipore) and blocked with BLOTTO (5% nonfat dry milk, 0.5% normal goat serum, and 0.1% sodium azide in Tris-buffered saline [TBS]) overnight at 4°C. Immunoblots were incubated with primary antibodies overnight at 4°C and then washed five times with TST (TBS containing 0.1% Tween-20), 10 min each. Finally, immunoblots were incubated with either 125I-labeled goat anti-mouse or goat anti-rabbit (Amersham, GE Healthcare, Piscataway, NJ) or HRP-labeled goat anti-mouse or goat anti-rabbit antibodies (GE Healthcare) for 45 min at room temperature. Immunoblots were washed as described, then twice with TBS. The immunoblots were exposed to either phosphorimager screens (Molecular Dynamics, Sunnyvale, CA) and signal was quantified using a phosphorimager (Typhoon, Molecular Dynamics) and ImageQuant software, version 5.0 (GE Healthcare), or incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and visualized with a ChemiDoc-It Imaging System and quantified with Visionworks software (UVP, Upland, CA).
Detergent solubility assay
MDCK II, shRalA, and shRalB cells were seeded on 0.4-μm Transwell polycarbonate filters at confluent densities (3 × 106 cells/24-mm filter) and subjected to a Ca2+. Samples were lysed in CSK containing 0.5% Triton-X 100 plus protease inhibitors (as described earlier) 0, 10, or 65 h after the switch to HCM. After a 10-min incubation on ice, lysates were centrifuged at 20,000 × g for 10 min. The supernatant was removed, and the pellet was resuspended in CSK containing 1% SDS and processed by SDS–PAGE, as described.
Protein stability assay
Confluent plates of cells were left nontreated or were treated with 4 μg/ml cyclohexamide for 3, 6, or 9 h at 37°C. Following treatment, samples were placed on ice and washed twice with Ringer's buffer. Samples were then lysed for 20 min on ice with CSK plus protease inhibitors. Lysates were centrifuged for 10 min at 20,000 × g, and supernatants were analyzed by SDS–PAGE.
Ca2+ depletion assay
TER of confluent cell monolayers on Transwell filters was measured before washing twice with Hank's buffered salt solution (HBSS; 5.33 mM KCl, 0.441 mM KH2PO4, 4.17 mM NaHCO3, 137.93 mM NaCl, 0.338 mM Na2HPO4, 5.56 mM dextrose, 0.0266 mM phenol red). Samples were incubated in HBSS plus 2.5 mM ethylene glycol tetraacetic acid (EGTA) for 10 min at 37°C, and the TER of all filters was measured. All filters were then washed twice with HBSS before HCM was added. Cells were cultured at 37°C for the times indicated in Figure 3, at which points TER was measured again. One filter of each cell type was processed for immunofluorescence (as described) immediately following the calcium chelation, to determine the subcellular localization of occludin.
Immunoprecipitation/pulse labeling
Confluent monolayers of MDCK II, shRalA, and shRalB cells on Transwell polycarbonate filters were washed twice with LG-DMEM without cystine or methionine and then incubated in the same medium for 1 h at 37°C. Samples were then labeled with 170 μCi of 35S-methionine per Transwell filter for 1 h at 37°C. Samples were washed three times on ice with Ringer's buffer and lysed with RIPA. Lysates were precleared with Pansorbin (Calbiochem, San Diego, CA), and 10% of the starting extract was removed for analysis, whereas the remainder was incubated with anti-Sec8 primary antibodies (mAbs 2E12, 5C3, 10C2) prebound to protein A–Sepharose (GE Healthcare) for 2 h at 4°C. Beads were pelleted by gentle centrifugation and washed under stringent conditions; twice with HS- buffer (0.1% SDS, 0.5% deoxycholate, 0.5% Triton X-100, 20 mM Tris-HCl, pH 7.5, 120 mM NaCl, 25 mM KCl, 5 mM EDTA, and 5 mM EGTA), twice with HS- buffer containing 1 M NaCl, and once with low-salt washing buffer (10 mM Tris-HCl, pH 7.5, and 2 mM EDTA). Immunoprecipitates were eluted from beads with SDS–PAGE sample buffer and processed for SDS–PAGE as described. Intensities of the Sec8, Sec6, and Sec15 bands were quantified using ImageQuant software, and the ratios of Sec8:Sec6 and Sec8:Sec15 were compared. Sec6 and Sec15 were identified by molecular weight, and their identities were verified by immunoprecipitation with specific antibodies to each protein.
Supplementary Material
Acknowledgments
We thank Nicholas Andersen and Iwona Driscoll for help with generating inducible RalA mutant cell lines. We also thank Amit Choudhury and Alexander Sandra for helpful discussions concerning membrane-labeling experiments and Krystle Peterson for discussions regarding lentiviral transduction and cloning techniques. This work was supported by a grant from the National Institutes of Health (GM067002) and a predoctoral fellowship from the American Heart Association (11PRE5670009).
Abbreviations used:
- GEF
guanine nucleotide exchange factor
- HCM
high-calcium medium
- LCM
low-calcium medium
- RBD
Ral-binding domain
- shCtrl
short hairpin nonspecific control
- shRalA
short hairpin RalA
- shRalB
short hairpin RalB
- TER
transepithelial resistance
- TJ
tight junction
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-07-0657) on October 19, 2011.
REFERENCES
- Angelow S, Ahlstrom R, Yu AS. Biology of claudins. Am J Physiol Renal Physiol. 2008;295:F867–F876. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian N, Meier JA, Scott DW, Norambuena A, White MA, Schwartz MA. RalA-exocyst complex regulates integrin-dependent membrane raft exocytosis and growth signaling. Curr Biol. 2010;20:75–79. doi: 10.1016/j.cub.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AI, Pollack AL, Altschuler Y, Mostov KE, Nelson WJ. NH2-terminal deletion of beta-catenin results in stable colocalization of mutant beta-catenin with adenomatous polyposis coli protein and altered MDCK cell adhesion. J Cell Biol. 1997;136:693–706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JT, Fuller MT, Zallen JA. The Drosophila homolog of the Exo84 exocyst subunit promotes apical epithelial identity. J Cell Sci. 2007;120:3099–3110. doi: 10.1242/jcs.004770. [DOI] [PubMed] [Google Scholar]
- Bodemann BO, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–267. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–140. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, White M, Camonis J. Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J. 2008;27:2375–2387. doi: 10.1038/emboj.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereijido M, Robbins ES, Dolan WJ, Rotunno CA, Sabatini DD. Polarized monolayers formed by epithelial cells on a permeable and translucent support. J Cell Biol. 1978;77:853–880. doi: 10.1083/jcb.77.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell. 2007;13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Chien Y, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Diamond JM. Twenty-first Bowditch Lecture. The epithelial junction: bridge, gate, and fence. Physiologist. 1977;20:10–18. [PubMed] [Google Scholar]
- Fukai S, Matern HT, Jagath JR, Scheller RH, Brunger AT. Structural basis of the interaction between RalA and Sec5, a subunit of the sec6/8 complex. EMBO J. 2003;22:3267–3278. doi: 10.1093/emboj/cdg329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Contreras RG, Bolivar JJ, Ponce A, Chavez De Ramirez B, Cereijido M. Role of calcium in tight junction formation between epithelial cells. Am J Physiol. 1990;259:C978–C986. doi: 10.1152/ajpcell.1990.259.6.C978. [DOI] [PubMed] [Google Scholar]
- Graeve L, Drickamer K, Rodriguez-Boulan E. Polarized endocytosis by Madin-Darby canine kidney cells transfected with functional chicken liver glycoprotein receptor. J Cell Biol. 1989;109:2809–2816. doi: 10.1083/jcb.109.6.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griepp EB, Dolan WJ, Robbins ES, Sabatini DD. Participation of plasma membrane proteins in the formation of tight junctions by cultured epithelial cells. J Cell Biol. 1983;96:693–702. doi: 10.1083/jcb.96.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindstaff KK, Yeaman C, Anandasabapathy N, Hsu SC, Rodriguez-Boulan E, Scheller RH, Nelson WJ. Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell. 1998;93:731–740. doi: 10.1016/s0092-8674(00)81435-x. [DOI] [PubMed] [Google Scholar]
- Gumbiner B, Simons K. A functional assay for proteins involved in establishing an epithelial occluding barrier: identification of a uvomorulin-like polypeptide. J Cell Biol. 1986;102:457–468. doi: 10.1083/jcb.102.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, Ting AE, Hazuka CD, Davanger S, Kenny JW, Kee Y, Scheller RH. The mammalian brain rsec6/8 complex. Neuron. 1996;17:1209–1219. doi: 10.1016/s0896-6273(00)80251-2. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol Biol Cell. 2004;15:2639–2651. doi: 10.1091/mbc.E04-02-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Junutula JR, Matern HT, Ervin KE, Scheller RH, Brunger AT. Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J. 2005;24:2064–2074. doi: 10.1038/sj.emboj.7600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou TS, Nelson WJ. Effects of regulated expression of mutant RhoA and Rac1 small GTPases on the development of epithelial (MDCK) cell polarity. J Cell Biol. 1998;142:85–100. doi: 10.1083/jcb.142.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, Yoo JS, Hazuka CD, Peterson KE, Hsu SC, Scheller RH. Subunit structure of the mammalian exocyst complex. Proc Natl Acad Sci USA. 1997;94:14438–14443. doi: 10.1073/pnas.94.26.14438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli G. RalA and the exocyst complex influence neuronal polarity through PAR-3 and aPKC. J Cell Sci. 2009;122:1499–1506. doi: 10.1242/jcs.044339. [DOI] [PubMed] [Google Scholar]
- Lalli G, Hall A. Ral GTPases regulate neurite branching through GAP-43 and the exocyst complex. J Cell Biol. 2005;171:857–869. doi: 10.1083/jcb.200507061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Mandel LJ, Bacallao R, Zampighi G. Uncoupling of the molecular “fence” and paracellular “gate” functions in epithelial tight junctions. Nature. 1993;361:552–555. doi: 10.1038/361552a0. [DOI] [PubMed] [Google Scholar]
- Marrs JA, Napolitano EW, Murphy-Erdosh C, Mays RW, Reichardt LF, Nelson WJ. Distinguishing roles of the membrane-cytoskeleton and cadherin mediated cell-cell adhesion in generating different Na+,K(+)-ATPase distributions in polarized epithelia. J Cell Biol. 1993;123:149–164. doi: 10.1083/jcb.123.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, White MA. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278:51743–51748. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- Ohba Y, Kurokawa K, Matsuda M. Mechanism of the spatio-temporal regulation of Ras and Rap1. EMBO J. 2003;22:859–869. doi: 10.1093/emboj/cdg087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdar M, Nelson WJ. Kinetics of desmosome assembly in Madin-Darby canine kidney epithelial cells: temporal and spatial regulation of desmoplakin organization and stabilization upon cell-cell contact. II. Morphological analysis. J Cell Biol. 1988;106:687–695. doi: 10.1083/jcb.106.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Hatzoglou A, Parrini MC, White MA, Chavrier P, Camonis J. RalB mobilizes the exocyst to drive cell migration. Mol Cell Biol. 2006;26:727–734. doi: 10.1128/MCB.26.2.727-734.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipitsin M, Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol. 2004;24:5746–5756. doi: 10.1128/MCB.24.13.5746-5756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Goodenough DA. Localization of the tight junction protein, ZO-1, is modulated by extracellular calcium and cell-cell contact in Madin-Darby canine kidney epithelial cells. J Cell Biol. 1988;107:2389–2399. doi: 10.1083/jcb.107.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B, Oprins A, Rabouille C, Munro S. The exocyst component Sec5 is present on endocytic vesicles in the oocyte of Drosophila melanogaster. J Cell Biol. 2005;169:953–963. doi: 10.1083/jcb.200411053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowalsky AG, Alt-Holland A, Shamis Y, Garlick JA, Feig LA. RalA suppresses early stages of Ras-induced squamous cell carcinoma progression. Oncogene. 2010;29:45–55. doi: 10.1038/onc.2009.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiczka KS, Yeaman C. Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J Cell Sci. 2008;121:2880–2891. doi: 10.1242/jcs.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Johnston D, Ahringer J. Cell polarity in eggs and epithelia: parallels and diversity. Cell. 2010;141:757–774. doi: 10.1016/j.cell.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Takakuwa R, Kokai Y, Kojima T, Akatsuka T, Tobioka H, Sawada N, Mori M. Uncoupling of gate and fence functions of MDCK cells by the actin-depolymerizing reagent mycalolide B. Exp Cell Res. 2000;257:238–244. doi: 10.1006/excr.2000.4887. [DOI] [PubMed] [Google Scholar]
- Takaya A, et al. R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol Biol Cell. 2007;18:1850–1860. doi: 10.1091/mbc.E06-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya A, Ohba Y, Kurokawa K, Matsuda M. RalA activation at nascent lamellipodia of epidermal growth factor-stimulated Cos7 cells and migrating Madin-Darby canine kidney cells. Mol Biol Cell. 2004;15:2549–2557. doi: 10.1091/mbc.E03-11-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TerBush DR, Maurice T, Roth D, Novick P. The exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 1996;15:6483–6494. [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Yamazaki Y, Katsuno T, Tamura A. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene. 2008;27:6930–6938. doi: 10.1038/onc.2008.344. [DOI] [PubMed] [Google Scholar]
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741–754. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- van Dam EM, Robinson PJ. Ral: mediator of membrane trafficking. Int J Biochem Cell Biol. 2006;38:1841–1847. doi: 10.1016/j.biocel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- Vega IE, Hsu SC. The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J Neurosci. 2001;21:3839–3848. doi: 10.1523/JNEUROSCI.21-11-03839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman C. Ultracentrifugation-based approaches to study regulation of Sec6/8 (exocyst) complex function during development of epithelial cell polarity. Methods. 2003;30:198–206. doi: 10.1016/s1046-2023(03)00026-4. [DOI] [PubMed] [Google Scholar]
- Yeaman C, Grindstaff KK, Nelson WJ. Mechanism of recruiting Sec6/8 (exocyst) complex to the apical junctional complex during polarization of epithelial cells. J Cell Sci. 2004;117:559–570. doi: 10.1242/jcs.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Turner JR. Stimulus-induced reorganization of tight junction structure: the role of membrane traffic. Biochim Biophys Acta. 2008;1778:709–716. doi: 10.1016/j.bbamem.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.