

Highlights
► Coronaviruses maintain a diverse phylogeny by distinct molecular mechanisms. ► BatCoV phylogenetics and receptor use may suggest a direct emergence of SARS-CoV. ► SARS Spike–receptor binding is the minimum determinant of cross-species transmission. ► Mutation and recombination-driven changes to Spike drive CoV host range expansion. ► Novel structural modeling tools allow assessment of receptor-binding determinants.
Abstract
Most new emerging viruses are derived from strains circulating in zoonotic reservoirs. Coronaviruses, which had an established potential for cross-species transmission within domesticated animals, suddenly became relevant with the unexpected emergence of the highly pathogenic human SARS-CoV strain from zoonotic reservoirs in 2002. SARS-CoV infected approximately 8000 people worldwide before public health measures halted the epidemic. Supported by robust time-ordered sequence variation, structural biology, well-characterized patient pools, and biological data, the emergence of SARS-CoV represents one of the best-studied natural models of viral disease emergence from zoonotic sources. This review article summarizes previous and more recent advances into the molecular and structural characteristics, with particular emphasis on host–receptor interactions, that drove this remarkable virus disease outbreak in human populations.
Introduction
Coronaviruses have an established potential for cross-species transmission that became broadly recognized with the emergence of a novel human coronavirus, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), in 2002. SARS was first identified as an atypical pneumonia in isolated patients in Guangdong Province, China. The disease reached epidemic proportions following key super spreader events that were associated with a novel respiratory virus introduction into a globalized community. SARS-CoV caused about 8000 infections and 800 deaths worldwide by July 2003, by which time aggressive public health intervention strategies contained the epidemic absent any effective therapeutics [1]. The decimating lethality of SARS-CoV emergence was borne largely by the elderly, in whom mortality rates approached 50% or more. A subsequent explosion of coronavirus research identified SARS-CoV in several small carnivores (palm civets and raccoon dogs) of the Chinese wet markets and SARS-like CoV in the predicted reservoir host, horseshoe bats (genus Rhinolophus). The vastly expanded CoV phylogeny includes two novel human coronaviruses (NL63 and HKU1) and ultimately tripled the number of full-length genome sequences available in GenBank. SARS-CoV was shown to use a novel host receptor, Angiotensin Converting Enzyme 2 (ACE2), for docking and entry and the viral attachment protein, Spike, was extensively characterized both as a determinant of host specificity and as a therapeutic target. The more recent studies of coronaviruses have progressed to increased surveillance and characterization of numerous new coronaviruses circulating in bats, bids, and other species, integrated bioinformatics and microbiological studies, and extensive evaluations of potential therapeutics [2].
Coronavirus phylogeny and mechanisms of genome diversity
Following the SARS-CoV outbreak a surge in global coronavirus genome sequencing efforts vastly expanded our insight into the CoV phylogeny and resulted in the definition of several subclassifications (Figure 1 ). The greatest contribution of new strains was derived from the newly discovered bat coronavirus (BtCoV), which may be the source of most, if not all, mammalian CoVs identified to date [3, 4, 5, 6, 7, 8, 9]. The high diversity of coronaviruses is attributable to three viral traits [10]. The first characteristic is the potentially high mutation rates associated with RNA replication, generally estimated as 10−3 to 10−5. Surprisingly, the estimated mutation rate for SARS-CoV and other coronaviruses approached 2 × 10−6 [11, 12, 13]. In contrast to other RNA viruses, recent data suggest that coronaviruses encode an RNA proof-reading activity associated with the 3′–5′ exonuclease activity encoded within nsp14 [14]. It is not clear whether RNA proof-reading fidelity is altered in changing environmental settings or during virus replication under stress related conditions, but such possibilities may allow for rapid virus evolution in changing ecologic conditions [14]. Second, recombination frequencies within the coronavirus family have been calculated to be as high as 25% during mixed infection, likely the result of discontinuous RNA transcription and the presence of full length and subgenomic negative strand RNAs that allow for frequent strand switching and recombination between viral genomes and subgenomic replication complexes [15, 16]. The role of discontinuous transcription in recombination is supported by the higher rate of recombination toward the 3′ ends of viral genomes and by targeted RNA recombination techniques designed to genetically manipulate the 3′ end of the genome [17]. Although poorly studied, conservation of transcription regulatory sequence (TRS) sites across viral species may implicate these sequences as foci or hot spots of recombination [17]. Thirdly, as the largest of the RNA viruses at ∼27–31 kb, coronaviruses have both increased opportunity for change and room for modification, clearly evidenced by the presence of numerous unique open reading frames and protein functions encoded toward the 3′ end of the genome [10]. These genomic characteristics allow for rapid adaptation to novel hosts, ecological niches, tissue tropism, and even generation of novel coronavirus species, as seen in the generation of FIPV type II strains from double recombination events between FIPV type I and CCoV [10].
Figure 1.
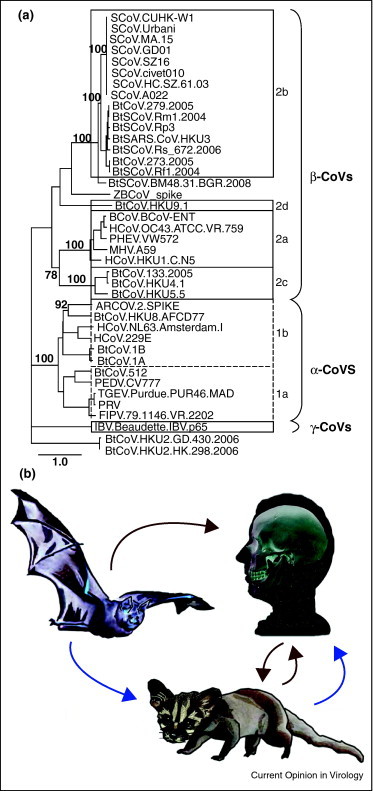
Spike phylogeny of representative CoVs and models of SARS-CoV emergence. (a) The Spike peptide sequence of 40 representative CoVs demonstrates that CoVs make up three distinct groups named alpha, beta, and gamma. These names replaced the former group 1, 2, and 3 designation, respectively. Classical subgroup clusters are marked as 2a–2d for the beta CoVs and 1a and 1b for the alpha CoVs. The tree was generated via Maximum Likelihood using the PhyML package. Major branch labels represent bootstraps that were greater than 70. SCoV: SARS-CoV; BtSCoV: bat SARS-like CoV; BtCoV, ZBCoV, and ARCoV: bat CoVs; HCoV: human; FCoV and FIPV: feline CoVs; BCoV: bovine; IBV: avian; PHEV, TGEV, PRV, PEDV: porcine CoVs; and MHV: murine hepatitis virus. (b) Competing models of SARS-CoV emergence. Early data suggested that SARS-CoV initially jumped from the zoonotic reservoir, bats, to palm civets, followed by a second jump from civets to humans (blue arrow). More recent phylogenetic and receptor analysis studies suggest a direct emergence from bats to humans, with subsequent cross-transmission between humans and civets (red arrow).
Multiple incidents of cross-species transmission
Coronaviruses have a strong history of host shifting as evidenced by phylogenetic incongruences in the family tree [18]. In addition to SARS-CoV, two human coronaviruses, HCoV-OC43 and HCoV-229E, are now also recognized as having likely emerged from animal reservoirs. HCoV-OC43 and bovine coronavirus (BCoV), both betacoronaviruses, have very high sequence similarity suggesting a recent and common origin (Figure 1). Molecular clock analysis of the Spike glycoprotein of both species estimates that HCoV-OC43 originated from a BCoV ancestor around 1890 [19]. Similarly, HCoV-229E likely emerged from a bat alphacoronavirus approximately 200 years ago [20]. In an example of reverse zoonosis, porcine epidemic diarrhea virus emerged suddenly in the early 1980s, most likely originating from HCoV-229E [20]. Additionally, a coronavirus isolated in 1988 from a child with acute diarrhea, HECV-4408, was shown to be closely related to bovine coronavirus (BCoV), indicating the continued introduction of zoonotic coronaviruses into human populations [21]. The origins of HCoV-NL63 and HCoV-HKU1, the most recently discovered human coronaviruses, remain under study. The most recent example of zoonotic emergence of a human coronavirus is the example of SARS-CoV, which had at least two independent emergence events from zoonotic reservoirs, recognized in 2002 and 2003 [22]. The most recent phylogenetic data estimate the emergence of SARS-CoV some seven years earlier, consistent with the identification of low sero-positive cases from archived serum samples in 2001 in China [23].
SARS-related CoVs in bats
Following its emergence in 2003, SARS was quickly identified as a zoonotic virus, and the identification of the wet markets as a potential source may have assisted epidemiological control of the disease [24]. While palm civets, raccoon dogs, and horseshoe bats (Rhinolophus genus) have all been identified as hosts of SARS-like CoVs, it is suggested that only the horseshoe bats are likely reservoir hosts. Bats are widely distributed, highly diverse, and extremely mobile mammals with an established role as hosts of emergent RNA viruses. Coronaviruses occupy an exceptionally wide distribution in bats; recent surveillance studies have extended our recognition of this range to Africa, Europe, South America, and North America [4, 9, 25, 26, 27]. The genetic variation encoded within many recently discovered coronaviruses hosted by bats is far greater than the diversity noted between many human coronaviruses, despite a proportionally small sampling of the ∼1200 bat species, leading some researchers to speculate that all mammalian coronaviruses are derived from bat reservoir strains [4, 28]. The extensive sequence diversity provides considerable opportunity for the emergence of new animal and human coronaviruses, which would be sufficiently antigenically distinct as to not be influenced by preexisting exposure and memory immune responses to established human CoVs. For example, little antigenic cross reactivity exists between the S glycoproteins of more distantly related group 2b bat coronaviruses and the SARS-CoV [29]. From a historic context, the next emergent event is likely dependent only on ecological and epidemiological situations and time, as the viral potential is well established [30, 31].
Repeated efforts have been made in recent years to identify the zoonotic reservoir and path of emergence for SARS-CoV, both by sampling zoonotic populations and by attempting to clarify SARS-CoV receptor usage in alternate hosts. A recent study attempting to address the paucity of bat SARS-related coronavirus sequences gathered and analyzed SARS-related coronaviruses in Rhinolophus bats (SARSr-Rh-BatCoV) (Rp3) genomes from horseshoe bats in China [32•]. Interestingly, several bats sampled were coinfected with HKU2, an alphacoronavirus, providing direct evidence that individual bats can host divergent coronaviruses, even across groups. Further, tagging and clinical assessment of infected bats over a four-year period showed only minor weight loss associated with Rp3 infection, with viral clearance occurring between two weeks and four months. Analysis of the ten novel genomes gathered in this study combined with previously published sequences demonstrated evidence of frequent recombination between the strains. They also note a 26-bp deletion in ORF8 near, but not identical to, the 29-bp deletion seen in human SARS-CoV epidemic strains, suggesting ORF8 may undergo frequent deletions [32•].
ACE2 is the receptor for SARS-CoV, but following the identification of several SARS-like CoVs (SL-CoVs) in horseshoe bats (genus Rhinophus) the ACE2 molecule of R. pearsonii proved incapable of serving a receptor for SARS-CoV [3, 33]. These and other initial studies suggested that the ancestral SARS-CoV strain in bats used an alternate receptor and that the emergence of SARS-CoV was dependent upon either acquisition of an ACE2 binding region or initial utilization of an alternative human receptor [33]. However, while human ACE2 is genetically conserved, the bat ACE2 sequences are highly heterogeneous, with 78–84% amino acid identity between families [34, 35]. Despite this heterogeneity, the residues that interface with the SARS Spike–receptor binding domain (RBD) are more conserved [36]. A recent study determined that a minimum three substitutions in the ACE2 of R. pearsonii (RpACE2) allowed this protein to serve as a receptor for SARS [37]. Looking more broadly at the ACE2 molecules from seven bat species, the ACE2 proteins from Myotis daubentoni and Rhinolophus sinicus are capable of supporting Spike-mediated pseudovirus and SARS-CoV infection, though less efficiently than human ACE2 [34]. Assessment of receptor usage by early phase and civet isolate Spike proteins might better inform our understanding of emergence pathways, determining if SL-CoV jumped directly from bat to human hosts or whether civet or other intermediate hosts were required as early intermediates before human adaptation.
Although original data suggested a bat to civet to human origin, evidence supporting direct bat to human transmission of SL-CoV emerged from recent phylogenetic studies, in addition to the receptor studies mentioned above (Figure 1b). Initially, a reanalysis of published genome sequences developed phylogenies using outgroups that were non-SARS-CoV sequences, designed to test the monophyly of the SARS-CoV sequences [38]. Under this assessment, bat isolates are ancestral host to all SARS-CoVs, while civet and raccoon dog sequences (small carnivores), as well as pig isolates cluster within the human SARS-CoV sequences. The small carnivore CoVs are consistently shown to be terminal branches with human CoVs intermediate, with later transmission of CoV between carnivores and humans responsible for isolated cases such as GD03, a late phase human isolate that phylogenetically clusters with civet rather than human sequences [38]. This phylogeny therefore supports a direct bat to human transmission, with subsequent and bidirectional transmissions between civet cats, raccoon dogs, and humans.
A more recent study analyzed CoV sequences gathered from 24 R. sinicus bats in geographically distant regions of China, characterizing two distinct genotypes, Rs672 and Rs806 [39•]. Interestingly, one sequence (Rs672) and the previously published Rp3 are shown in a monophyly more closely related to human-SCoV than to bat SARS-like CoV strains, based on the strong similarity of Rs672 ORF1a/b region to human SARS sequences. This study also provided further evidence of recombination between Bat-SL-CoV, with a recombination breakpoint identified immediately after the start codon of Spike, identical to the recombination position in the Rp3 genome [39•]. The combination of highly diverse BtCoV species and divergent ACE2 molecules among bat hosts suggests direct bat to human transmission may be feasible. Thus, the field is left with two potentially competing models for the origins of the SARS-CoV epidemic: first, transmission from bats to an intermediate amplifying reservoir in small carnivores, with subsequent transmission to humans or second, direct bat to human transmission followed by cross-transmission between humans and civets and raccoon dogs (Figure 1b). In both models civets and raccoon dogs serve as key amplifying hosts for virus persistence and reintroduction into human populations.
Genesis of an epidemic
A chronological set of SARS-CoV sequence changes spanning the SARS outbreak provided an unparalleled opportunity to identify the genetic basis for zoonotic virus cross-species transmission and human adaptation during an expanding epidemic. Molecular changes noted at with the end of the early phase and expansion into the middle phase of the epidemic include A3047V, A3072V in the replicase and D778Y and perhaps E1163K in the Spike gene. Transition from the middle to late phase of the epidemic included A2552V in ORF1a, E1389D in ORF1b, D77G and T244I in the S gene, respectively (Figure 2 ) [40]. It has been hypothesized that these alterations were key to an expanding epidemic, yet empirical data to support these claims and functional significance of these alterations remain unavailable. For example, it is not clear whether the ORF8 29 bp deletion is central for human adaptation as suggested, or a genetic hitchhiker amplified and maintained following a selective sweep mediated by other beneficial mutations located elsewhere in the genome [3, 40]. In addition to these changes, the SARS-CoV Spike glycoprotein was under strong positive selection, with 23 substitutions evolving during the expanding phases of the epidemic [41]. Experimental evidence suggests both adaptation to ACE2 and antibody selection contributed to Spike changes [40, 42].
Figure 2.
Sequence changes over the SARS-CoV epidemic. Shown here are the most significant changes important for transition of SARS from civet to early, middle and late phases of epidemic strains. Mutations indicative of lineages that were not likely to have contributed to the expansion to other phases have been removed. All other positions in the genome are identical.
Coronavirus cross-species transmission: role of Spike–receptor interactions in viral entry
Coronavirus–receptor interactions are key determinants regulating host range, cross-species transmission, and tissue tropism. The various coronaviruses demonstrate broad receptor and coreceptor usage, from proteases such as aminopeptidase N for transmissible gastroenteritis virus (TGEV), canine-CoV, feline infectious peritonitis virus (FIPV), and HCoV-229E, to cell adhesion molecules such as CEACAM1a for MHV, to sugars as coreceptors for some alpha, beta, and gammacoronaviruses [36, 43, 44]. This diverse receptor usage directly impacts host range and tissue tropism as demonstrated by the closely related PRCoV and TGEV. PRCoV lacks the sugar-binding region of TGEV, and consequently is limited to a respiratory rather than enteric tropism [45]. The recently crystallized structure of the group 2a coronavirus MHV complexed with its receptor, murine CEACAM1a, emphasizes again the broad diversity and flexibility of CoV Spike glycoproteins. The core structure of the MHV RBD is hypothesized to have been derived from a host sugar-binding protein (galectin) and subsequently modified to allow mCEACAM1a binding, thus enhancing MHV affinity for host cells [43]. Other coronaviruses encode a second putative viral attachment protein, the hemagglutinin esterase (HE), which was likely derived from influenza C strains by recombinatory mechanisms [46]. Coronaviruses selected in vitro to broaden host range oftentimes mutate to bind heparin sulfate for docking and entry [47]. It is notable that OC43 and BCoV have carbohydrate (sialic acid) binding capacities, as well as broader host ranges [48]. The capacity to bind carbohydrates for docking and entry may provide an additional pathway for coronavirus host range expansion, cross-species transmission, and disease emergence, and requires further study.
The key determinant of SARS coronavirus host specificity is the Spike glycoprotein, an envelope-anchored trimeric protein responsible for binding human ACE2 as the principle receptor for virus docking and entry. SARS-CoV S glycoprotein also binds C-type lectins like DC-Sign and/or L-Sign as a coreceptor, an interaction which is blocked by mannose binding lectin [49, 50]. Importantly, SARS-CoV docking and entry is also highly dependent upon transmembrane protease/serine subfamily member 2 (TMPRSS2) S and ACE2 cleavage, especially in airway and alveolar sites, and cathepsin L cleavage and subsequent S2 fusion activation [51, 52, 53]. Several studies in the past two years have worked to clarify the plasticity of this protein, with particular emphasis on the RBD. The Spike glycoprotein underwent rapid evolution during the human epidemic [40], was the most significantly variable protein across civet and human isolates [22], and shows evidence of positive selection during both interspecies and intraspecies transmission events [10, 22, 40, 54]. The SARS Spike can recognize and use bat, civet, mouse, and raccoon dog ACE2 receptor molecules for docking and entry, indicating that SARS trafficked along receptor ortholog networks to move between species [34, 55, 56]. As several alphacoronaviridae also use APN from different species, these data suggest a common theme in coronavirus host range switching: recognizing receptor orthologs from different species [36]. Additionally, the role of different ortholog proteases for facilitating coronavirus S glycoprotein cleavage and entry processes remains undefined, and could significantly contribute to the efficiency of virus cross-species transmission processes.
SARS-CoV replicates but does not produce clinical disease in mice. Two experimental adaptations of SARS-CoV to murine hosts by serial passage independently identified a substitution in the Spike gene at residue 436 which alone has been shown to enhance infectivity and pathogenesis in mice, and is predicted to allow stronger binding to the murine ACE2 receptor [29, 57, 58•]. However, substitutions outside of Spike are necessary for the full lethal disease phenotype in MA15, and presumably also in v2163 [57]. For example, two other proteins, nsp9 and nsp13, contained mutations in both mouse-adapted strains, MA15 and v2163. Additionally, single substitutions in the M gene are common to MA15 and adaptation to persistent infection of human tubular kidney cells, suggesting the M protein influences tropism or pathogenesis by facilitating the efficiency of particle egress [59]. The substitutions common to both mouse-adapted strains suggest potential SARS-CoV virulence factors in the later stages of adaption to a novel host, and indicate potential mutation driven emergence pathways. The mouse-adapted viruses may not represent true cross-species transmission events, as SARS could already replicate in the mouse lung, but it is notable that the most conserved change in both mouse-adapted strains enhances receptor binding at the same Spike residue. Further, serological studies indicate multiple cross-species transmissions into humans in the years before the epidemic, suggesting that the virulence factors contributing to the later stages of adaptation to novel hosts, in Spike or elsewhere, are critically important [23].
The RBD (aa318–510) is the strongest determinant of host range for SARS-CoV and other coronaviruses [29]. Single substitutions within the RBD can significantly affect the binding affinity of Spike to its receptor [60]. Indeed, a minimum of 1–2 substitutions in the RBD are sufficient to allow the virus to alter host receptor specificity [61]. Experimental adaptation of civet-Spiked SARS virus to human ACE2 receptor by Sheahan et al. demonstrated the minimal requirements for host range expansion. In these studies, a civet-Spiked SARS-CoV was incapable of propagating in Vero cells until a human-tropic substitution was introduced at residue 479. When the civet-Spiked virus included the K479T substitution it was capable of propagating on Vero cells and further capable of replicating on human airway epithelial cells (HAE) and hACE2-expressing DBT cells, demonstrating that single substitutions are capable of expanding the virus host range. Interestingly, when the K479T-civet-SARS was experimentally selected for enhanced replication on human airway epithelial cells, the substitutions that improved replication did not exactly replicate the substitutions seen in the epidemic strains. Rather, an initial substitution at 479 was necessary for the civet-SARS to use primate ACE2 and propagate in Vero cells, but the adaptive mutations following passage on human airway epithelial cells (HAE) selected for substitutions at two different contact interface sites at residues 442 and 472, rather than the 487 site identified in the epidemic strain [61, 62]. This suggests that multiple genetic pathways exist which can improve S RBD–human ACE2 receptor interactions, providing the virus with multiple strategies to adapt to new host species [56]. It is interesting to note that this alternative pathway for recognizing hACE2 ablated interactions with the cACE2 receptor, supporting the hypothesis that epidemic SARS-CoV strains were coselected to efficiently recognize both civet and human ACE2 receptors.
Antibodies that neutralize SARS-CoV predominantly bind to the RBD of Spike. Rockx et al. selected and sequenced a number of different escape mutants to a panel of 23 human monoclonal antibodies, the majority of which contained single substitutions along the RBD interface with ACE2 [63••]. All but one escape site mapped within 4 angstroms of contact interface residues, and yet all viruses grew to comparable peak titers in Vero and hACE2-restricted DBT cells. However, growth on civet-ACE2-restricted DBT cells was restricted for all escape viruses, suggesting that escape from antibody neutralization can alter Spike–receptor binding and, consequently, host range [63••]. That antibody escape variants can stably adopt substitutions in the Spike–ACE2 receptor interface suggests that the host response to an infection may select for host range variants by a mutation-driven mechanism.
Extensive structural modeling tools are available to predict receptor binding, antibody neutralization, or the stability of substitutions within the RBD of the SCoV Spike. Three coronavirus Spike-RBDs have been complexed with receptors to date, allowing for prediction and validation of the structural determinants of binding to host and orthologous receptors (Figure 3 ). Application of mathematical modeling to Spike–receptor and Spike–antibody structural models allowed for the prediction of escape substitutions with a high probability of fixation in a viral population [64]. These predictions are partially in accordance with published data, predicting selection with antibody 80R would select for a substitution at D480 of Spike, as seen in vitro following SARS-CoV escape from 80R neutralization [64, 65].
Figure 3.

Crystal structures of coronavirus receptor binding domains (RBDs) complexed with their receptors. To date, the crystal structures of three coronavirus Spike RBD–receptor complexes have been solved: (a) the RBDs of SARS complexed with human ACE2 (pdb 2AJF) [73], (b) NL63 complexed with human ACE2 (3KBH) [71••], and (c) MHV complexed with murine CEACAM11a (3R4D) [43].
Plasticity of the Spike glycoprotein
The coronavirus Spike glycoprotein is remarkably plastic, capable of accommodating mutations and deletions up to 479 (MHV) or 681 nucleotides (PRCoV) while retaining receptor binding and entry functions [66, 67, 68]. To date, large deletions in the SARS-CoV S glycoprotein have not been reported. The S protein is divided into discrete domains: an N-terminal domain, RBD, two heptad repeats, a transmembrane anchor, and an intracellular tail [43]. Discrete regions can be exchanged between strains while preserving both protein function and antibody binding [29, 36]. Multiple coincident substitutions as well as contact interface site substitutions can be tolerated to allow escape from antibody neutralization while maintaining receptor specificity [42, 60, 69, 70]. This flexibility allows for multiple genetic pathways from the use of zoonotic receptors to the human ACE2 receptor [56].
Diversity and flexibility of the Spike glycoprotein is characteristic of coronaviruses beyond SARS-CoV. The lack of a clear ACE2 receptor binding motif (RBM) in the horseshoe Bat-SL-CoV Spike, and the inability to use hACE2 as a receptor, led to an early hypothesis that the human-SCoV emerged from Bat-SL-CoV following a recombination event, perhaps with a NL63-like CoV, as NL63 also uses ACE2 as a receptor. Such a recombination event would have allowed direct acquisition of an ACE2 binding motif and the resulting cross-species transmission [35]. Alternatively, SARS-CoV used batACE2 for docking and entry and introduction into human/civet populations selected for mutations that enhanced interaction with the civet or humanACE2 receptor. The recently published crystal structure of NL63-CoV complexed with the ACE2 receptor shows no structural homology with the SARS-CoV RBM or the core RBD (Figure 3) [71••, 72]. This suggests that convergent evolution, rather than recombination-mediated transfer, lead to the common use of ACE2 by NL63 and SARS-CoV [72].
Early data suggested that the RBD of SARS-CoV and perhaps HCoV-NL63 were derived by recombination processes, rather than mutation driven evolution. While these ideas remain highly speculative, these data suggested that the S glycoprotein RBDs and/or fusion cores of CoVs may be interchangeable between distant strains. In support of this hypothesis, the consensus bat SARS-like genome HKU3 was replication competent, but was not sufficient for sequential rounds of infection, presumably because of the lack of appropriate receptors for docking and entry. The insertion of the SARS RBD into the HKU3 Spike allowed for the production of progeny virus that grew to high titer in ACE2-expressing DBT cells, and was capable of replicating in human airway epithelial cells and mouse lungs, although it grew with reduced efficiency in the latter [29]. Thus, under certain conditions, recombination processes can result in bat CoV host shifting. Further, the bat-SARS-like coronavirus with the SARS RBD was capable of replicating in mouse lungs, although with greatly reduced efficiency. It is notable that attempts to isolate CoV from bats have repeatedly failed, limiting our ability to study adaptive mechanisms or pathogenesis of CoV in host species, but that synthetic biology provides alternative sources of these viruses. The construction of a synthetic bat SARS-like coronavirus provided strong evidence that the interspecies movement of coronaviruses, specifically SARS-like coronaviruses, resides strongly in the RBD [29]. While previous studies had indicated that small changes in the Spike glycoprotein could alter host specificity of coronaviruses, the sufficiency of a discrete RBD change in the context of a divergent 30 kb genome demonstrates the RBD is a minimum determinant of species tropism. Further, it suggests a potential mechanism of host range expansion, suggesting both recombination and single substitution events allow for infection of novel hosts. Determining receptor specificities for these novel bat coronaviruses offer considerable opportunity to enrich our understanding of coronavirus–receptor interactions, identify new receptors that coronaviruses use for docking and entry, and provide novel models for studying the ease and mechanism of cross-species transmission.
Conclusions
Fundamental insights into the molecular mechanisms and pathways that govern virus cross-species transmission are central to protecting global health. Coronaviruses readily traffic between host species and the Spike glycoprotein is the most extensively characterized viral determinant of host range expansion. Binding of the coronavirus Spike to the host receptor is the minimum determinant of infectivity and species specificity, and many recent studies have demonstrated the ability of S RBD to mutate and engage ortholog receptors or escape antibody neutralization [61, 63••]. We need to know more about the breath of novel coronavirus receptors that are used in nature and the mechanisms governing ortholog receptor recognition. Importantly, the coronavirus RBD interface is a robust iterative model for predicting structure–function relationships between mutation-driven host range expansion, virus–receptor interactions, and antibody binding and neutralization. The SARS S-RBD model captures highly regulated variables that recapitulate real-life biological processes critical for coronavirus cross-species transmission and host immune response (Figure 4 ). The SARS RBD–receptor–neutralizing antibody interface provides considerable opportunity for predicting and studying the role of mutations in cross-species transmission and immunity. In addition, recent work has also expanded our appreciation of how intragenic recombination may influence coronavirus host range, as evidenced by targeted recombination, recombination between different bat coronaviruses, and identification of the RBD as a minimum determinant of host range expansion [29, 39•]. While the precise ancestor and route of emergence for SARS-CoV remains unidentified, extensive sampling and phylogenetic studies of bat CoVs has raised the possibility that the epidemic strain may have jumped directly to humans before jumping to civets. Thus, future coronavirus epidemics may be more frequent than appreciated as compared with a two-step emergence model that required an intermediate host. Additionally, while it remains unclear whether recombination and/or mutation of Spike mediated the emergence of SARS, both mechanisms can readily impact coronavirus host range. Future studies are needed to clarify the potential roles of host proteases or antibody mediated selection in cross-species transmission, and to clarify whether modulation of RNA proof-reading activity could impact viral adaptation to a novel host. Further, structural and mathematical modeling tools offer novel predictive capabilities that, when integrated with experimental studies, will assist in predicting the ease of cross-species transmission, the mechanisms of emergence, and contribute to improvements in therapeutic design.
Figure 4.
Experimental evolution at the SARS S glycoprotein RBD–ligand interface. The SARS RBD is heterogeneous and includes defined sequence variation at specific residues that engage the ACE2 receptor from different species (Parts 1 and 2). Bioinformatics can be used to predict and then test the impact of targeted mutations on variant virus–receptor interactions. Iterative rounds of mutation driven selection are also possible using recombinant viruses encoding targeted mutations and variant ACE2 receptors for docking and entry. The model allows a deep structural understanding of the potential pathways and molecular mechanisms that govern cross-species transmission and pathogenesis. The biological impact of host shifting on antigenicity can be predicted using structural models of antibody–RBD interfaces, and then studied using a panel of well characterized human and mouse monoclonal antibodies targeting the different SARS-CoV RBD domains (Part 3). In parallel, neutralizing monoclonal antibodies can be used to select for escape mutations (Part 4), allowing for iterative rounds of prediction and testing on how these mutations impact host range and ACE2 recognition.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
This work was supported by grants from the National Institutes of Health (U54 AI057157-08 and RO1 RO1AI085524) and the UNC-CH Medical Science Training Program (T32GM008719 (MEB)).
References
- 1.Cleri D.J., Ricketti A.J., Vernaleo J.R. Severe acute respiratory syndrome (SARS) Infect Dis Clin North Am. 2010;24:175–202. doi: 10.1016/j.idc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pepin K.M., Lass S., Pulliam J.R.C., Read A.F., Lloyd-Smith J.O. Identifying genetic markers of adaptation for surveillance of viral host jumps. Nat Rev Microbiol. 2010;8:802–813. doi: 10.1038/nrmicro2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau S.K.P., Woo P.C.Y., Li K.S.M., Huang Y., Tsoi H.-W., Wong B.H.L., Wong S.S.Y., Leung S.-Y., Chan K.-H., Yuen K.-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drexler J.F., Gloza-Rausch F., Glende J., Corman V.M., Muth D., Goettsche M., Seebens A., Niedrig M., Pfefferle S., Yordanov S. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010;84:11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gloza-Rausch F., Ipsen A., Seebens A., Göttsche M., Panning M., Felix Drexler J., Petersen N., Annan A., Grywna K., Müller M. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poon L.L.M., Chu D.K.W., Chan K.H., Wong O.K., Ellis T.M., Leung Y.H.C., Lau S.K.P., Woo P.C.Y., Suen K.Y., Yuen K.Y. Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 8.Tang X., Zhang J., Zhang S., Wang P., Fan X., Li L., Li G., Dong B., Liu W., Cheung C. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80:7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfefferle S., Oppong S., Drexler J.F., Gloza-Rausch F., Ipsen A., Seebens A., Müller M.A., Annan A., Vallo P., Adu-Sarkodie Y. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis. 2009;15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo P.C.Y., Lau S.K.P., Huang Y., Yuen K.-Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med. 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 11.Deming D., Baric R. Genetics and reverse genetics of nidoviruses. In: Perlman S., Gallagher T., Snijder E., editors. Nidoviruses. ASM Press; 2008. p. 459. [Google Scholar]
- 12.Duffy S., Shackelton L.A., Holmes E.C. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008;9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 13.Eckerle L.D., Becker M.M., Halpin R.A., Li K., Venter E., Lu X., Scherbakova S., Graham R.L., Baric R.S., Stockwell T.B. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8:270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu K., Baric R.S. Map locations of mouse hepatitis virus temperature-sensitive mutants: confirmation of variable rates of recombination. J Virol. 1994;68:7458–7466. doi: 10.1128/jvi.68.11.7458-7466.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes E. The evolution and emergence of RNA viruses. In: Harvey P.H., May R.M., editors. Oxford Series in Ecology and Evolution. Oxford University Press Inc.; 2009. [Google Scholar]
- 17.Pasternak A., Spaan W., Snijder E. Nidovirus transcription: how to make sense…? J Gen Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- 18.Rest J.S., Mindell D.P. SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host-shifting. Infect Genet Evol. 2003;3:219–225. doi: 10.1016/j.meegid.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vijgen L., Keyaerts E., Moës E., Thoelen I., Wollants E., Lemey P., Vandamme A.-M., Van Ranst M. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol. 2005;79:1595–1604. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocherhans R., Bridgen A., Ackermann M., Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X.M., Herbst W., Kousoulas K.G., Storz J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J Med Virol. 1994;44:152–161. doi: 10.1002/jmv.1890440207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song H.-D., Tu C.-C., Zhang G.-W., Wang S.-Y., Zheng K., Lei L.-C., Chen Q.-X., Gao Y.-W., Zhou H.-Q., Xiang H. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng B.J., Wong K.H., Zhou J., Wong K.L., Young B.W., Lu L.W., Lee S.S. SARS-related virus predating SARS outbreak, Hong Kong. Emerg Infect Dis. 2004;10:176–178. doi: 10.3201/eid1002.030533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan Y., Zheng B., He Y., Liu X., Zhuang Z., Cheung C., Luo S., Li P., Zhang L., Guan Y. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 25.Lau S.K.P., Poon R.W.S., Wong B.H.L., Wang M., Huang Y., Xu H., Guo R., Li K.S.M., Gao K., Chan K.-H. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol. 2010;84:11385–11394. doi: 10.1128/JVI.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Vijgen L., Keyaerts E., Moës E., Maes P., Duson G., Van Ranst M. Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol. 2005;43:5452–5456. doi: 10.1128/JCM.43.11.5452-5456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quan P.-L., Firth C., Street C., Henriquez J.A., Petrosov A., Tashmukhamedova A., Hutchison S.K., Egholm M., Osinubi M.O.V., Niezgoda M. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. MBio. 2010;1:1–9. doi: 10.1128/mBio.00208-10. Article Number: e00208–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vijaykrishna D., Smith G., Zhang J., Peiris J., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J Virol. 2007;81:4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker M.M., Graham R.L., Donaldson E.F., Rockx B., Sims A.C., Sheahan T., Pickles R.J., Corti D., Johnston R.E., Baric R.S. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc Natl Acad Sci U S A. 2008;105:19944–19949. doi: 10.1073/pnas.0808116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson L.J., Tong S. Update on SARS research and other possibly zoonotic coronaviruses. Int J Antimicrob Agents. 2010;36(Suppl. 1):S21–S25. doi: 10.1016/j.ijantimicag.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donaldson E.F., Haskew A.N., Gates J.E., Huynh J., Moore C.J., Frieman M.B. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol. 2010;84:13004–13018. doi: 10.1128/JVI.01255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Lau S.K.P., Li K.S.M., Huang Y., Shek C.-T., Tse H., Wang M., Choi G.K.Y., Xu H., Lam C.S.F., Guo R. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. 2010;84:2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. An extensive study of bat coronaviruses in China reported the co-infection of the same bat with both SARSr-Rh-BatCoV and HKU2, a beta and alphacoronavirus, respectively. Further, they provide evidence that bats can host CoVs with minimal mortality, and they show evidence of frequent recombination between the strains of BatCoV, contributing to the diversity and flexibility of this coronavirus reservoir.
- 33.Ren W., Qu X., Li W., Han Z., Yu M., Zhou P., Zhang S.-Y., Wang L.-F., Deng H., Shi Z. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J Virol. 2008;82:1899–1907. doi: 10.1128/JVI.01085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou Y., Peng C., Yu M., Li Y., Han Z., Li F., Wang L.-F., Shi Z. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch Virol. 2010;155:1563–1569. doi: 10.1007/s00705-010-0729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W., Wong S.-K., Li F., Kuhn J.H., Huang I.-C., Choe H., Farzan M. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol. 2006;80:4211–4219. doi: 10.1128/JVI.80.9.4211-4219.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham R.L., Baric R.S. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. 2010;84:3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M., Tachedjian M., Crameri G., Shi Z., Wang L.-F. Identification of key amino acid residues required for horseshoe bat angiotensin-I converting enzyme 2 to function as a receptor for severe acute respiratory syndrome coronavirus. J Gen Virol. 2010;91:1708–1712. doi: 10.1099/vir.0.020172-0. [DOI] [PubMed] [Google Scholar]
- 38.Janies D., Habib F., Alexandrov B., Hill A., Pol D. Evolution of genomes, host shifts and the geographic spread of SARS-CoV and related coronaviruses. Cladistics. 2008;24:111–130. doi: 10.1111/j.1096-0031.2008.00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Yuan J., Hon C.-C., Li Y., Wang D., Xu G., Zhang H., Zhou P., Poon L.L.M., Lam T.T.-Y., Leung F.C.-C. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J Gen Virol. 2010;91:1058–1062. doi: 10.1099/vir.0.016378-0. [DOI] [PubMed] [Google Scholar]; Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. The authors sampled R. sinicus bats and identifying two novel strains of SL-CoV, Rs672, and Rs806. Notably, in addition to evidence of recombination in strain Rs672, the sequence showed a closer phylogenetic relationship to human strains than to other bat CoV sequences. Suggesting possibility that Rs672, or a closely related strain, may be ancestor.
- 40.Consortium C.S.M.E. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 41.Rockx B., Sheahan T., Donaldson E., Harkema J., Sims A., Heise M., Pickles R., Cameron M., Kelvin D., Baric R. Synthetic reconstruction of zoonotic and early human severe acute respiratory syndrome coronavirus isolates that produce fatal disease in aged mice. J Virol. 2007;81:7410–7423. doi: 10.1128/JVI.00505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rockx B., Corti D., Donaldson E., Sheahan T., Stadler K., Lanzavecchia A., Baric R. Structural basis for potent cross-neutralizing human monoclonal antibody protection against lethal human and zoonotic severe acute respiratory syndrome coronavirus challenge. J Virol. 2008;82:3220–3235. doi: 10.1128/JVI.02377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng G., Sun D., Rajashankar K.R., Qian Z., Holmes K.V., Li F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc Natl Acad Sci U S A. 2011;108:10696–10701. doi: 10.1073/pnas.1104306108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwegmann-Wessels C., Herrler G. Sialic acids as receptor determinants for coronaviruses. Glycoconj J. 2006;23:51–58. doi: 10.1007/s10719-006-5437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultze B., Krempl C., Ballesteros M.L., Shaw L., Schauer R., Enjuanes L., Herrler G. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J Virol. 1996;70:5634–5637. doi: 10.1128/jvi.70.8.5634-5637.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng Q., Langereis M.A., van Vliet A.L.W., Huizinga E.G., de Groot R.J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc Natl Acad Sci U S A. 2008;105:9065. doi: 10.1073/pnas.0800502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Haan C.A., Li Z., te Lintelo E., Bosch B.J., JHaijema B.J., Rottier P.J.M. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J Virol. 2005;79:14451–14456. doi: 10.1128/JVI.79.22.14451-14456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwegmann-Weßels C., Herrler G. Sialic acids as receptor determinants for coronaviruses. Glycoconj J. 2006;23:51–58. doi: 10.1007/s10719-006-5437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Thackray L.B., Young M.D., Mason R.J. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y., Lu K., Pfefferle S., Bertram S., Glowacka I., Drosten C., Pöhlmann S., Simmons G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J Virol. 2010;84:8753–8764. doi: 10.1128/JVI.00554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosch B.J., Bartelink W., Rottier P.J.M. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol. 2008;82:8887–8890. doi: 10.1128/JVI.00415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S., Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glowacka I., Bertram S., Müller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C.-Y., Wei J.-F., He S.-H. Adaptive evolution of the spike gene of SARS coronavirus: changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006;6:88. doi: 10.1186/1471-2180-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu L., Zhang Y., Liu Y., Chen Z., Deng H., Ma Z., Wang H., Hu Z., Deng F. Angiotensin-converting enzyme 2 (ACE2) from raccoon dog can serve as an efficient receptor for the spike protein of severe acute respiratory syndrome coronavirus. J Gen Virol. 2009;90:2695–2703. doi: 10.1099/vir.0.013490-0. [DOI] [PubMed] [Google Scholar]
- 56.Sheahan T., Rockx B., Donaldson E., Corti D., Baric R. Pathways of cross-species transmission of synthetically reconstructed zoonotic severe acute respiratory syndrome coronavirus. J Virol. 2008;82:8721–8732. doi: 10.1128/JVI.00818-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts A., Deming D., Paddock C., Cheng A., Yount B., Vogel L., Herman B., Sheahan T., Heise M., Genrich G. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58•.Day C.W., Baric R., Cai S.X., Frieman M., Kumaki Y., Morrey J.D., Smee D.F., Barnard D.L. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395:210–222. doi: 10.1016/j.virol.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. An independently derived mouse-adapted virus shows compelling commonalities to the MA15 virus, particularly in Spike. Additional potential virulence factors outside of Spike, such as Nsp9 and Nsp13, should be further characterized.
- 59.Pacciarini F., Ghezzi S., Canducci F., Sims A., Sampaolo M., Ferioli E., Clementi M., Poli G., Conaldi P.G., Baric R. Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. J Virol. 2008;82:5137–5144. doi: 10.1128/JVI.00096-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W., Zhang C., Sui J., Kuhn J., Moore M., Luo S., Wong S., Huang I., Xu K., Vasilieva N. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheahan T., Rockx B., Donaldson E., Sims A., Pickles R., Corti D., Baric R. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J Virol. 2008;82:2274–2285. doi: 10.1128/JVI.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li F., Li W., Farzan M., Harrison S. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 63••.Rockx B., Donaldson E., Frieman M., Sheahan T., Corti D., Lanzavecchia A., Baric R.S. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J Infect Dis. 2010;201:946–955. doi: 10.1086/651022. [DOI] [PMC free article] [PubMed] [Google Scholar]; Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. Monoclonal antibody neutralization of SARS-CoV can select for stable escape sites along the RBD contact interface with ACE2. It is interesting that given the location of the escape substitutions, the escape viruses suffered no loss in viability on Vero or hACE2-DBT cells, but had reduced growth on civet-ACE2-DBT cells, suggesting that host antibody pressure may select for host range variants in coronavirus emergence.
- 64.Watabe T., Kishino H. Structural considerations in the fitness landscape of a virus. Mol Biol Evol. 2010;27:1782–1791. doi: 10.1093/molbev/msq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sui J., Aird D.R., Tamin A., Murakami A., Yan M., Yammanuru A., Jing H., Kan B., Liu X., Zhu Q. Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog. 2008;4:e1000197. doi: 10.1371/journal.ppat.1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaughn E.M., Halbur P.G., Paul P.S. Three new isolates of porcine respiratory coronavirus with various pathogenicities and spike (S) gene deletions. J Clin Microbiol. 1994;32:1809–1812. doi: 10.1128/jcm.32.7.1809-1812.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gallagher T.M., Parker S.E., Buchmeier M.J. Neutralization-resistant variants of a neurotropic coronavirus are generated by deletions within the amino-terminal half of the spike glycoprotein. J Virol. 1990;64:731–741. doi: 10.1128/jvi.64.2.731-741.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rowe C.L., Baker S.C., Nathan M.J., Fleming J.O. Evolution of mouse hepatitis virus: detection and characterization of spike deletion variants during persistent infection. J Virol. 1997;71:2959–2969. doi: 10.1128/jvi.71.4.2959-2969.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qu X.-X., Hao P., Song X.-J., Jiang S.-M., Liu Y.-X., Wang P.-G., Rao X., Song H.-D., Wang S.-Y., Zuo Y. Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy. J Biol Chem. 2005;280:29588–29595. doi: 10.1074/jbc.M500662200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang X., Li G., Vasilakis N., Zhang Y., Shi Z., Zhong Y., Wang L., Zhang S. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evol Biol. 2009;9:52. doi: 10.1186/1471-2148-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71••.Wu K., Li W., Peng G., Li F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc Natl Acad Sci U S A. 2009;106:19970–19974. doi: 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. The crystal structure of NL63 complexed with its ACE2 receptor identified a common binding hotspot with SARS-CoV despite no structural homology between the two viral RBDs.
- 72.Wu K., Chen L., Peng G., Zhou W., Pennell C.A., Mansky L.M., Geraghty R.J., Li F. A virus-binding hotspot on human angiotensin-converting enzyme 2 is critical for the binding of two different coronaviruses. J Virol. 2011;106:19970–19974. doi: 10.1128/JVI.02274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]