

Abstract
Objective
Angiotensin II type 1 receptor (AT1) blockers (ARBs) reduce the bacterial endotoxin lipopolysaccharide (LPS)-induced innate immune response in human circulating monocytes expressing few AT1. To clarify the mechanisms of anti-inflammatory effects of ARBs with different peroxisome proliferator-activated receptor-γ (PPARγ)-activating potencies, we focused our study on telmisartan, an ARB with the highest PPARγ-stimulating activity.
Methods
Human circulating monocytes and monocytic THP-1 (human acute monocytic leukemia cell line) cells were exposed to 50 ng/ml LPS with or without pre-incubation with telmisartan. AT1 mRNA and protein expressions were determined by real-time PCR and membrane receptor binding assay, respectively. The expression of pro-inflammatory factors was determined by real-time PCR, western blot analysis and ELISA. PPARγ activation was measured by electrophoretic mobility shift assay and its role was determined by pharmacological inhibition and PPARγ gene silencing.
Results
In human monocytes, telmisartan significantly attenuated the LPS-induced expression of pro-inflammatory factors, the release of pro-inflammatory cytokines and prostaglandin E2, nuclear factor-κB activation and reactive oxygen species formation. In THP-1 cells, telmisartan significantly reduced LPS-induced tumor necrosis factor-α, inhibitor of κB-α, monocyte chemotactic protein-1 (MCP-1) and lectin-like oxidized low-density lipoprotein receptor-1 gene expression and MCP-1-directed migration. Telmisartan also stimulated the expression of the PPARγ target genes cluster of differentiation 36 and ATP-binding cassette subfamily G member 1 in monocytes. The anti-inflammatory effects of telmisartan were prevented by both PPARγ antagonism and PPARγ gene silencing. Anti-inflammatory effects of ARBs correlated with their PPARγ agonist potency.
Conclusion
Our observations demonstrate that in human monocytes, ARBs inhibit the LPS-induced pro-inflammatory response to a major extent through the PPARγ activation pathway and may be beneficial for the treatment of cardiovascular and metabolic disorders in which inflammation plays a major role.
Keywords: angiotensin receptor blockers, cytokines, inflammation, peroxisome proliferator-activated receptor-γ, transcription factors
Introduction
Excessive angiotensin II (Ang II) type 1 receptor (AT1) stimulation is associated with hypertension, and AT1 blockers (ARBs) were developed for the treatment of high blood pressure to antagonize increased Ang II-dependent vasoconstriction [1]. ARBs protect end organs not only because they ameliorate hypertension, but also as a consequence of beneficial effects on associated inflammatory and metabolic alterations beyond their effect on blood pressure control [2,3]. The protective effect of ARBs is enhanced with increasing doses beyond full AT1 blockade [4], suggesting beneficial mechanisms additional to AT1 blockade.
Most of the ARBs are biphenyl-tetrazole derivatives with similar but not identical pharmacological profiles [5]. The biphenyl-nontetrazole telmisartan is structurally unique and, in addition to its AT1 blocking properties, functions as a partial agonist of the peroxisome proliferator-activated receptor-γ (PPARγ) [6], an intracellular nuclear hormone receptor regulating multiple pathways involved in carbohydrate and lipid metabolism [7] and the control of expression of pro-inflammatory genes [8,9]. Full PPARγ agonists reduce inflammation and metabolic alterations associated with cardiovascular disease [10]. However, their associated toxicity limits their therapeutic value [11], and the identification of telmisartan as a well tolerated bifunctional molecule to treat both hemodynamic and metabolic alterations generated a high degree of enthusiasm [6]. It was later recognized that other ARBs, such as candesartan, activate PPARγ in vitro and in vivo [12,13].
Anti-inflammatory effects of ARBs, first established in the peripheral vasculature [14] were later demonstrated in the cerebral vasculature [15] and in stress-induced gastric ulcerations [16]. These observations suggest that ARBs may exert general anti-inflammatory effects beyond those associated with cardiovascular and metabolic disease. Evidence in support of this hypothesis was obtained in normotensive rodents in which candesartan reduced the peripheral and brain acute inflammation following systemic administration of the bacterial endotoxin lipopolysaccharide (LPS) [17-19]. Anti-inflammatory effects of candesartan were also demonstrated in cultured circulating human monocytes expressing few AT1 [20].
We hypothesized that, in human circulating monocytes with poor AT1 expression, the anti-inflammatory effects of ARBs may be associated with PPARγ activation. We compared ARBs with different PPARγ-activating potencies and focused on the effect of telmisartan, an ARB with the highest PPARγ-stimulating activity. We studied LPS-induced inflammation in monocytes as a relevant model, because these cells are critical in the development of insulin resistance, diabetes and arteriosclerosis; plasma LPS is elevated in obesity and diabetes [21]; and low-grade chronic inflammation with increased expression of monocyte-derived inflammatory factors is an established component of vascular inflammatory disease in hypertension [14].
Materials and methods
Detailed methods can be found in Supplemental Materials and Methods sections (http://links.lww.com/HJH/A136).
Human peripheral blood monocytes
Human peripheral blood mononuclear cells were isolated by density gradient centrifugation of heparinized blood, collected by leukaphoresis of healthy volunteers at the Department of Transfusion Medicine, National Institutes of Health (NIH) and prepared as described [22] (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
THP-1 cells
THP-1 cells, a human acute monocytic leukemia cell line, were grown in Royal Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum, 10 mmol/l N-2-hydroxyethlpiperazine-N′-2-ethanesulfonic acid, 1 mmol/l sodium pyruvate, 0.05 mmol/l 2-mercaptoethanol and 100 U/ml penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2–95% air at a density of 1–5 × 105 cells/ml. Before each experiment, the cells were starved overnight in a serum-free medium. All experiments were performed on nondifferentiated THP-1 cells.
Angiotensin II receptor binding assay
Untreated THP-1 cells (10 million) or human monocytes (150 million) were harvested and homogenized in ice-cold buffer containing 50 mmol/l Tris-HCl pH 7.5, 1 mmol/l ethylene glycol tetraacetic acid and 5 mmol/l MgCl2. A piece of fresh rat kidney cortex (100 mg) to be used as a positive control was homogenized in the same buffer. Crude membrane fraction was pelleted by centrifugation at 20 000g for 20 min at 4°C and the pellets were resuspended in 200 μl of the same buffer. Protein content was assessed by the Bradford reagent. The binding assay was performed as previously described [23], with 0.25 nmol/l [125I]Sar1-Ile8-Ang II as a receptor ligand.
Real-time PCR
Human monocytes or THP-1 cells were pre-incubated for 2 h with ARBs or vehicle (dimethylsulfoxide) followed by exposure to 50 ng/ml LPS or physiological saline, composed of 0.9% w/v sodium chloride, for additional 2 h, and reverse transcriptase-PCR measurements were performed as described [20] (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
Western blot analysis
Human monocytes were pre-incubated for 2 h with telmisartan or vehicle followed by addition of 50 ng/ml LPS or physiological saline, composed of 0.9% w/v sodium chloride, for different times, as indicated in the figures. Nuclear and whole-cell protein extracts were subjected to the separation by SDS-PAGE and analyzed as described in Supplemental Materials and Methods (http://links.lww.com/HJH/A136).
Measurement of prostaglandin E2 production
Human monocytes were incubated for 24 h with 50 ng/ml LPS alone or in a combination with different concentrations of telmisartan. The cell supernatants were collected at the end of incubation for prostaglandin E2 (PGE2) measurement by PGE2 enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, Michigan, USA) according to the manufacturer’s instructions.
Determination of cytokine release
Human monocytes were pre-incubated for 2 h with telmisartan or vehicle followed by addition of 50 ng/ml LPS or physiological saline, composed of 0.9% w/v sodium chloride. The cell supernatants were collected 4 h after the addition of LPS, and tumor necrosis factor-α (TNFα) and interleukin (IL)-6 concentrations were measured by human ELISA kits (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions.
Detection of intracellular reactive oxygen species
The levels of intracellular reactive oxygen species (ROS) were determined by the change in the fluorescence, resulting from the oxidation of the fluorescent probe dichlorodihydrofluorescein diacetate using OxiSelect ROS Assay Kit (Cell Biolabs, San Diego, California, USA) according to the manufacturer’s instructions as described [20] (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
Electrophoretic mobility shift assay
Monocytes or THP-1 cells were incubated for 4 h with vehicle, 10 μmol/l telmisartan or 10 μmol/l troglitazone. Nuclear protein extracts were prepared using Nuclear Extraction kit (Pierce, Rockford, Illinois, USA), according to the manufacturer’s instructions. Electrophoretic mobility shift assay (EMSA) was carried out using Light-Shift Chemiluminescent EMSA kit (Pierce) with double-stranded DNA probe 5′-GGTAAAGGTCA AAGGTCAATCGGC-3′ [24] labeled with biotin at the 5′-end (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
Peroxisome proliferator-activated receptor-γ small interfering RNA transfection
THP-1 cells were transfected with 40 nmol/l human PPARγ-specific small interfering RNA (siRNA) or scrambled negative control siRNA (Invitrogen) using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions and analyzed 48 and 72 h after the transfection (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
Cell migration assay
The cell migration assay was performed in THP-1 cells using a CytoSelect Cell Migration Assay kit (Cell Biolabs) (see Supplemental Materials and Methods, http://links.lww.com/HJH/A136).
Statistical analysis
Statistical significance was determined using GraphPad Prism 5 Software (GraphPad Software, San Diego, California, USA). Wilcoxon matched paired test was used to compare nonnormalized values from human monocytes, and two-tailed Student’s t-test was used to compare normalized results from human monocytes or THP-1 cells. Multiple group comparisons were performed by one-way analysis of variance followed by Newman–Keuls post test. Spearman’s correlation coefficient (r) was calculated to analyze the influence of age or AT1 expression on the telmisartan effect. Differences were considered statistically significant at P value less than 0.05. Values are expressed as the mean ± SEM.
Results
Telmisartan inhibited the lipopolysaccharide-induced inflammation in human monocytes and THP-1 cells
Telmisartan dose dependently inhibited LPS-induced TNFα and IL-6 release (Fig. 1a), and LPS-induced TNFα, IL-1β and IL-6 mRNA expression in human monocytes (Fig. 1b). The effective concentration of telmisartan was as low as 0.1 μmol/l with maximum efficacy achieved at 10 μmol/l (Fig. 1). Telmisartan did not show toxicity at any dose used, as evaluated by the release of lactate dehydrogenase (data not shown), and we used telmisartan at a 10 μmol/l concentration for subsequent experiments.
Fig. 1.
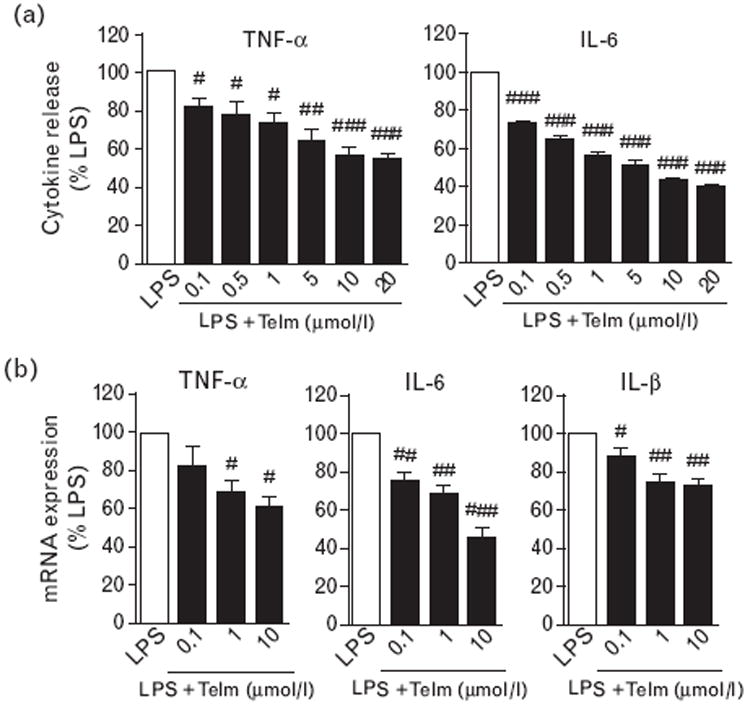
Telmisartan (Telm) inhibited lipopolysaccharide (LPS)-induced pro-inflammatory cytokine release and mRNA expression in human monocytes. Monocytes were incubated with 50 ng/ml LPS alone or after 2 h pre-incubation with different doses of Telm. (a) Cumulative release of pro-inflammatory cytokines 4 h after LPS addition as determined by ELISA. (b) Expression of cytokine mRNA after 2 h incubation with LPS as determined by real-time PCR. Results are means ± SEM from three independent experiments and are expressed as a percentage of LPS-induced response. IL, interleukin; TNF, tumor necrosis factor. #P<0.05; ##P<0.01; ###P<0.001 compared with LPS.
We analyzed a total of 39 samples from individual donors (36 males and three females), randomly selected for the different studies. Despite high individual variability to LPS stimulation, the effects of telmisartan were consistent in all analyzed samples (Fig. S1, http://links.lww.com/HJH/A136). There was no correlation between the age of the donors and the response to LPS or telmisartan (Fig. S2, http://links.lww.com/HJH/A136).
Telmisartan dose dependently decreased LPS-stimulated intercellular adhesion molecule 1 (ICAM-1), monocyte chemotactic protein-1 (MCP-1) and lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) gene expression (Fig. 2a), decreased the LPS-induced upregulation of cyclooxygenase-2 (COX-2) mRNA and protein expression and reduced PGE2 release (Fig. 2a and b). A similar dose-dependent effect of telmisartan on the LPS-induced gene expression of TNFα, inhibitor of κB-α (IκB-α), MCP-1 and LOX-1 was observed in THP-1 cells (Fig. S3, http://links.lww.com/HJH/A136).
Fig. 2.
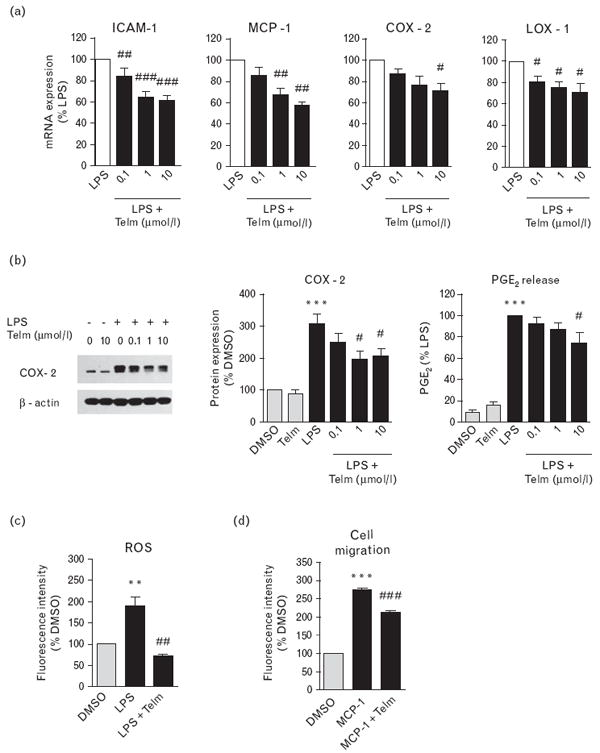
Telmisartan (Telm) inhibited lipopolysaccharide (LPS)-induced pro-inflammatory responses in human monocytes. Human monocytes were pre-incubated for 2 h with vehicle (DMSO) or Telm and subsequently exposed to 50 ng/ml LPS. (a) Expression of pro-inflammatory marker mRNA 2 h after LPS addition. (b) Cyclooxygenase-2 (COX-2) protein expression and cumulative prostaglandin E2 (PGE2) release in monocytes exposed to LPS for 24 h as determined by western blot and EIA, respectively. COX-2 protein expression was determined by western blot and quantified by densitometry. Values are presented as percentage of DMSO-treated group, as means ± SEM from at least three independent experiments. ***P<0.001 vs. DMSO; #P<0.05 vs. LPS. Picture shows a representative western blot. (c) Reactive oxygen species (ROS) production was determined by dichlorodihydrofluorescein diacetate probe oxidation in monocytes pretreated with vehicle or 10 μmol/l Telm and then exposed to LPS for 2 h. (d) Cell migration assay was performed on THP-1 monocytic cells pretreated with vehicle or 10 μmol/l Telm and subsequently stimulated with 100 ng/ml monocyte chemotactic protein-1 (MCP-1) for 4 h. Results are means ± SEM from at least three independent experiments. **P<0.01 vs. DMSO; ***P<0.001 vs. DMSO; #P<0.05 vs. LPS; ##P<0.01 vs. LPS; ###P<0.001 vs. LPS or MCP-1. ICAM-1, intercellular adhesion molecule 1; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1.
Telmisartan abolished the LPS-induced ROS generation (Fig. 2c) and significantly reduced migration of monocytic THP-1 cells in response to MCP-1 (Fig. 2d).
Anti-inflammatory effects of telmisartan involve inhibition of mitogen-activated protein kinase and nuclear factor-κB activation
Telmisartan attenuated the LPS-induced phosphorylation of p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinases 1/2 (ERK1/2) (Fig. 3a).
Fig. 3.
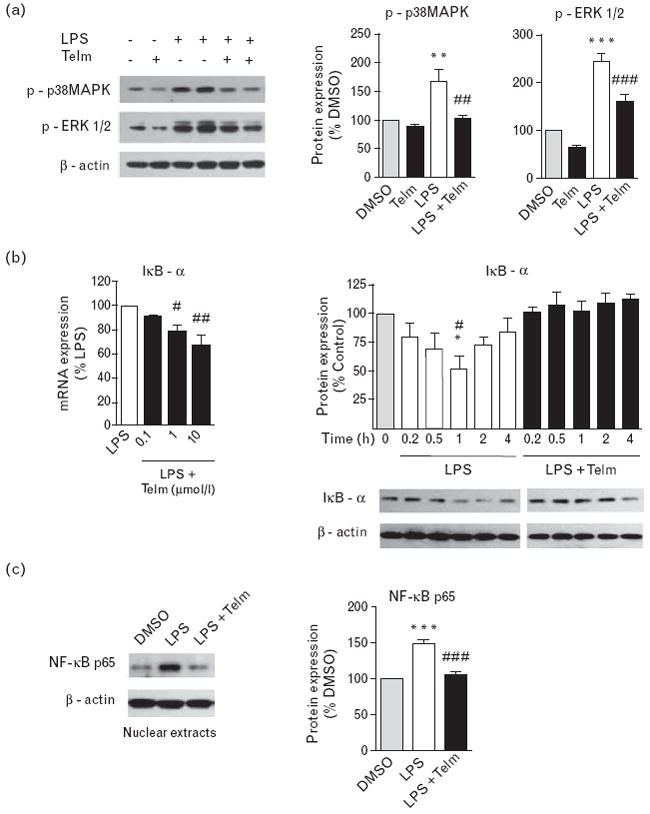
Anti-inflammatory effects of telmisartan (Telm) in human monocytes involved inhibition of mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) activation. Human monocytes were pre-incubated for 2 h with vehicle (DMSO) or 10 μmol/l Telm and subsequently exposed to lipopolysaccharide (LPS) (50 ng/ml). (a) Representative western blot image showing inhibitory effect of Telm on LPS-induced phosphorylation of p38 MAPK and extracellular signal-regulated kinases 1/2 (ERK1/2) in monocytes incubated for 30 min with LPS. Quantitative densitometric values are presented as percentage of DMSO-treated group, and as means ± SEM from at least three independent experiments. **P<0.01 vs. DMSO; ***P<0.001 vs. DMSO; ##P<0.01 vs. LPS; ###P<0.001 vs. LPS. (b) Telm dose dependently inhibited inhibitor of κB-α (IκB-α) mRNA expression induced by 2 h incubation with LPS, and prevented LPS-induced IκB-α protein degradation time dependently. IκB-α protein levels are presented as means ± SEM from three independent experiments. *P<0.05 vs. control (0 h); #P<0.05 vs. LPS with Telm group. Pictures are representative western blots. (c) Nuclear proteins extracted from human monocytes treated with LPS with or without Telm were used to measure NF-κB p65 protein expression by western blot. Representative western blot is shown. NF-κB p65 protein values are presented as means ± SEM from three independent experiments. ***P<0.001 vs. DMSO; ###P<0.001 vs. LPS.
LPS-induced activation of nuclear factor-κB (NF-κB) was determined by increase in IκB-α mRNA expression, enhanced IκB-α protein degradation and the translocation of NF-κB p65 subunit into the nucleus (Fig. 3b and c). All these effects of LPS were significantly attenuated by telmisartan (Fig. 3b and c).
Telmisartan activated peroxisome proliferator-activated receptor-γ in monocytes and THP-1 cells
We demonstrated PPARγ activation by telmisartan in monocytes and THP-1 cells by EMSA with a DNA oligoprobe containing the PPARγ response element AGGTCAXAGGTCA. Pre-incubation with both cold probe and 2 μg of anti-PPARγ antibody reduced PPARγ–DNA binding, indicating that the observed shift was PPARγ-specific (Fig. 4a and b).
Fig. 4.
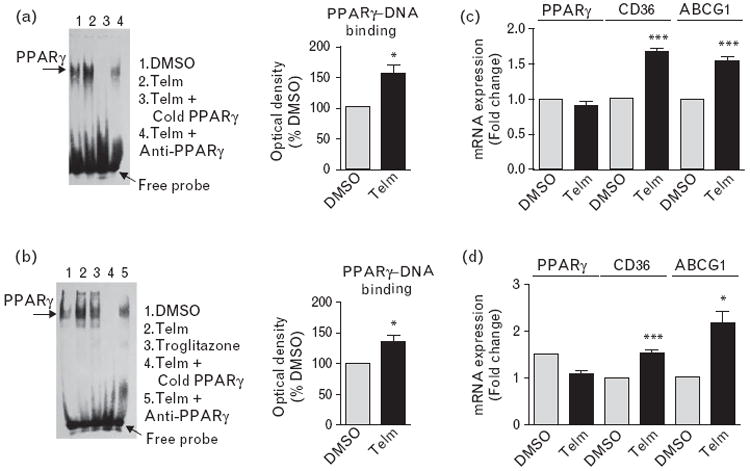
Telmisartan (Telm) activated peroxisome proliferator-activated receptor-γ (PPARγ) and increased PPARγ target gene expression in human monocytes and THP-1 cells. Primary human monocytes or THP-1 cells were incubated for 4 h with vehicle (DMSO) or 10 μmol/l Telm. PPARγ activation was analyzed by electrophoretic mobility shift assay. Representative pictures show PPARγ–DNA binding in human monocytes (a) and THP-1 cells (b), and the intensity of each band was measured by densitometry for quantitative analysis. Anti-PPARγ antibody (2 μg) and a 100-fold excess of cold probe were used to determine the specificity of the shift. Pre-incubation of nuclear extracts from Telm-treated cells with an anti-PPARγ antibody led to a profound decrease in PPARγ–DNA binding activity, demonstrating the reaction specificity. Expression of mRNA of PPARγ and PPARγ target genes cluster of differentiation 36 (CD36) and ATP-binding cassette subfamily G member 1 (ABCG1) in monocytes (c) and THP-1 cells (d) was determined by real-time PCR. Results are means ± SEM of three independent experiments. *P<0.05 vs. DMSO; ***P<0.001 vs. DMSO.
Telmisartan significantly increased the expression of the PPARγ target genes cluster of differentiation 36 (CD36) and ATP-binding cassette subfamily G member 1 (ABCG1) in both human monocytes and THP-1 cells (Fig. 4c and d), but did not affect PPARγ gene expression (Fig. 4c and d).
Peroxisome proliferator-activated receptor-γ gene silencing abrogated telmisartan effect on lipopolysaccharide-induced expression of pro-inflammatory factors in THP-1 cells
Transfection of THP-1 cells with 40 nmol/l human PPARγ siRNA (siRNA) time dependently reduced PPARγ protein expression in THP-1 cells (Fig. 5a) and its mRNA expression 72 h after transfection (Fig. 5b). Transfection with the negative control siRNA was ineffective (Fig. 5a and b). Seventy-two hours after transfection, the PPARγ siRNA selectively eliminated the telmisartan-induced increase in CD36 and ABCG1 mRNA expression (Fig. 5c), as well as the inhibitory effect of telmisartan on the LPS-induced TNFα and IκB-α mRNA expression (Fig. 5d).
Fig. 5.
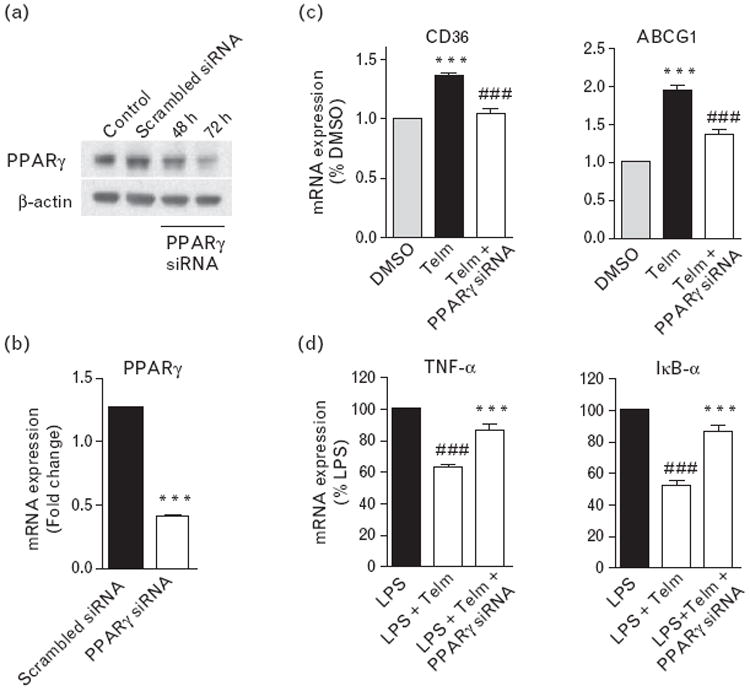
Peroxisome proliferator-activated receptor-γ (PPARγ) gene silencing eliminated the effect of telmisartan (Telm) on lipopolysaccharide (LPS)-induced gene expression in THP-1 cells. Human PPARγ-specific or nontargeting scrambled control small interfering RNA (siRNA) were transfected into THP-1 cells. PPARγ protein levels were measured at 48 and 72 h posttransfection by western blot (a). PPARγ mRNA expression (b) and cluster of differentiation 36 (CD36) and ATP-binding cassette subfamily G member 1 (ABCG1) mRNA expression (c) were measured at 72 h of posttransfection by real-time PCR. ***P<0.001 vs. scrambled siRNA or DMSO; ###P<0.001 vs. Telm. The inflammatory markers tumor necrosis factor-α (TNFα) and inhibitor of κB-α (IκB-α) were measured in cells treated for 2 h with LPS (50 ng/ml) alone or in a combination with 10 μmol/l Telm with and without PPARγ siRNA transfection (d). Results are means ± SEM from three independent experiments. ###P<0.001 vs. LPS; ***P<0.001 vs. LPS with Telm.
A peroxisome proliferator-activated receptor-γ antagonist prevents telmisartan amelioration of lipopolysaccharide-induced tumor necrosis factor-α release
Both telmisartan and PPARγ agonist troglitazone significantly attenuated LPS-induced release of TNFα (Fig. 6). The effect of both drugs was prevented to a similar extent by the PPARγ antagonist T0070907 (Fig. 6).
Fig. 6.
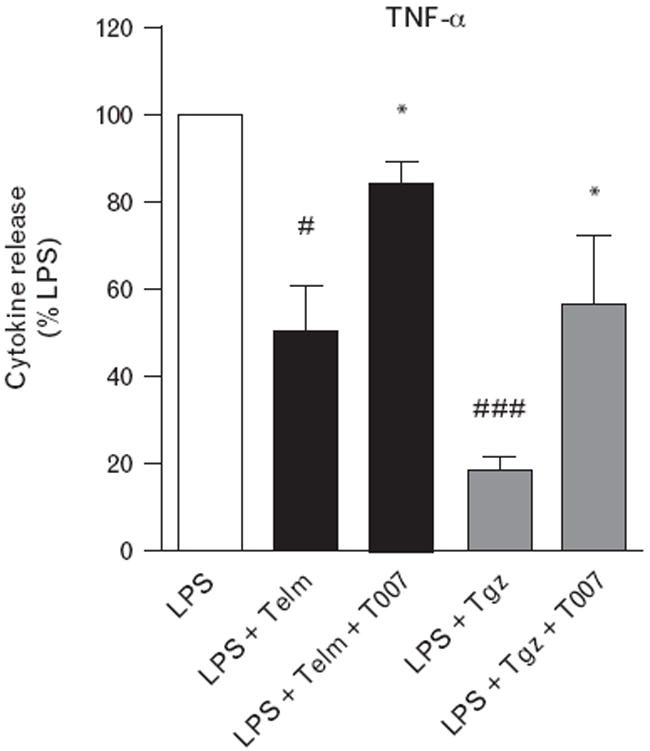
Peroxisome proliferator-activated receptor-γ (PPARγ) antagonist prevents telmisartan (Telm) amelioration of lipopolysaccharide (LPS)-induced tumor necrosis factor-α (TNFα) release. Monocytes pretreated for 1 h with 1 μmol/l PPARγ antagonist T0070907 (T007) were incubated for 1 h with 10 μmol/l Telm or 10 μmol/l PPARγ agonist troglitazone (Tgz) followed by 4 h incubation in the presence of 50 ng/ml LPS. Cumulative TNFα release was determined in culture medium by ELISA. Results are means ± SEM of three independent experiments and are expressed as a percentage of LPS-induced response. #P<0.05 vs. LPS; ###P<0.001 vs. LPS; *P<0.05 vs. LPS with Telm or LPS with Tgz.
Effect of different angiotensin II type 1 receptor blockers on lipopolysaccharide-induced tumor necrosis factor-α release in human monocytes
Incubation with 10 μmol/l telmisartan significantly reduced LPS-stimulated TNFα release approximately by 50% (P<0.001) (Fig. S4, http://links.lww.com/HJH/A136). Candesartan, at the same concentration, inhibited LPS-induced TNFα release about 30% (P<0.05), whereas 10 μmol/l losartan decreased TNFα release only by about 15%, and the change did not reach statistical significance (Fig. S4, http://links.lww.com/HJH/A136).
Effect of different angiotensin II type 1 receptor blockers on lipopolysaccharide-induced tumor necrosis factor-α mRNA expression in THP-1 cells
Incubation with 10 μmol/l telmisartan, candesartan or losartan significantly reduced LPS-stimulated TNFα mRNA expression in THP-1 cells (Fig. S5A, http://links.lww.com/HJH/A136). The order of potency was telmisartan > candesartan > losartan. The anti-inflammatory effects of all ARBs were abrogated by simultaneous incubation in the presence of the PPARγ antagonist T0070907 (Fig. S5A, http://links.lww.com/HJH/A136). The effect of telmisartan was of the same magnitude than those of the PPARγ agonist troglitazone (Fig. S5A, http://links.lww.com/HJH/A136).
Effect of different angiotensin II type 1 receptor blockers on mRNA expression of peroxisome proliferator-activated receptor-γ target gene cluster of differentiation 36 in THP-1 cells
The increase in CD36 mRNA expression produced by telmisartan was of the same magnitude than that of the PPARγ agonist troglitazone and was abrogated by the PPARγ antagonist T0070907 (Fig. S5B, http://links.lww.com/HJH/A136). All ARBs tested significantly induced CD36 mRNA expression, with an order of potency telmisartan > candesartan > losartan, and these effects were completely prevented by the PPARγ antagonist T0070907 (Fig. S5B, http://links.lww.com/HJH/A136).
Expression of angiotensin II type 1 receptor in human monocytes and THP-1 cells
Very low levels of AT1 mRNA expression were detected in nine of 20 monocyte samples studied by real-time PCR, and in most cases values were close to the limit of detection (Fig. S6A and B, http://links.lww.com/HJH/A136). There was no detectable AT1 mRNA expression in THP-1 cells (results not shown). AT1 binding was undetectable in membrane preparations from circulating human monocytes or THP-1 cells (Fig. S6C, http://links.lww.com/HJH/A136). There was no correlation between the level of AT1 mRNA expression and the effect of telmisartan on LPS-induced TNFα release (Fig. S6D, http://links.lww.com/HJH/A136).
Discussion
We demonstrate here that in circulating human monocytes obtained from a randomly selected healthy human population, and in human monocytic THP-1 cells, telmisartan significantly reduced the LPS-induced innate immune response through mechanisms involving PPARγ activation.
In agreement with previous studies [20,25,26], and in the human THP-1 monocytic cell line, LPS induced an acute immune response with stimulation of multiple inflammatory pathways. As described [20], monocyte responses to LPS were consistent in all preparations, although with large differences in individual responses. Such differences maybe related to sex, age, medications, inflammatory status or genetic variability, but these factors were not the object of our study, and we did not find age correlation with the response to LPS. The concentration of LPS used is comparable to the LPS levels found in obesity, insulin resistance and diabetes, characterized by low-grade chronic inflammatory conditions, and was substantially below the levels encountered during sepsis [21].
Telmisartan significantly reduced, in both human monocytes and THP-1 cells, all LPS-induced pro-inflammatory effects studied. In addition, telmisartan decreased the LPS-induced expression of LOX-1, a target of LPS and a major factor in the development of atherosclerosis [20,27].
LPS stimulates multiple pro-inflammatory genes in part through NF-κB activation with participation of ERK1/2 and p38 MAPK [25]. We report that LPS increased IκB-α mRNA expression and NF-κB nuclear translocation, indicative of NF-κB activation [28,29], and a reduction of NF-κB activation by telmisartan. In addition telmisartan attenuated LPS-induced MAPK activation and ROS formation, intimately associated with NF-κB activation [21,25-33].
We demonstrate that the anti-inflammatory effect of telmisartan is to a great extent the result of PPARγ activation. Telmisartan was more potent than candesartan, as reported earlier [20], whereas losartan was less effective in primary human monocytes, probably due to variations of LPS response in different preparations. The effects of telmisartan compared well with those of the PPARγ agonist troglitazone [34]. All ARBs tested inhibited LPS-induced pro-inflammatory gene expression in THP-1 cells, which is consistent with the recent report of anti-inflammatory effects of losartan through a PPARγ-dependent mechanism [35]. This order of potency parallels the reported PPARγ agonist activity of the ARBs tested [6,12]. The anti-inflammatory effects of telmisartan were apparent even at low concentrations in the 0.1–10 μmol/l range, in the range of peak and steady-state telmisartan concentrations in humans [36]. The higher potency of telmisartan is explained by its unique structural characteristics and high lipophilicity, favoring its incorporation into the cell [6,12] and by its strong hydrophobic interactions at unique sites within the PPARγ ligand domain [6,12]. We demonstrated that telmisartan activates PPARγ in human monocytes and THP-1 cells, enhancing mRNA expression of the PPARγ target genes CD36 and ABCG1 [37,38]. PPARγ antagonists and PPARγ gene silencing eliminated the anti-inflammatory effects and PPARγ target gene expression produced by telmisartan. In differentiated macrophages, Ang II promotes inflammation through AT1 stimulation [39] and mechanisms similar to those involved in the LPS effects [25]. As previously reported [20], we could not detect AT1 binding in human monocytes or THP-1 cells (present results). We did not study AT1 protein expression because we and others [40] have not been able to validate any of the commercially available AT1 antibodies (results not shown). The expression of AT1 mRNA in human circulating monocytes was close to the lower limit of detection by real-time PCR, which could only be clearly demonstrated in some of the preparations studied and could not be found in THP-1 cells. In addition, non-differentiated human monocytes do not produce Ang II [41], and Ang II did not stimulate inflammatory factors in our preparations of human circulating monocytes [20].
We demonstrate that PPARγ activation is important for the anti-inflammatory effects of ARBs in human monocytes. Our results do not totally exclude a participation of AT1 on the anti-inflammatory effects of ARBs, independent or associated with PPARγ agonist effects. There is cross-talk between AT1 and PPARγ activation [42]; PPARγ agonists reduce AT1-mediated inflammation and hypertension in vivo [43], and downregulate AT1 expression [44], whereas Ang II downregulates PPARγ mRNA expression [45].
Perspectives and significance
Reducing excessive Ang II AT1 activation ameliorates hypertension, metabolic syndrome and diabetes [14]. Some ARBs, most significantly telmisartan, activate the nuclear receptor PPARγ [6,12], a most relevant property contributing to additional therapeutic benefit in these conditions [6,10,46].
We demonstrate, in human monocytes, that PPARγ activation plays a fundamental role in the anti-inflammatory effects of ARBs. The PPARγ agonist properties of telmisartan may contribute to ameliorate the chronic low-grade systemic inflammation characteristic of hypertension, diabetes and dyslipidemia [21] and is associated with increases in plasma LPS concentrations with an origin in the gut microbial community [47,48]. This may also be important for the treatment of chronic vascular and renal inflammatory diseases exaggerated by excess production of inflammatory factors originated in peripheral monocytes [14,21]. The relevance of our findings is supported by the recent report of enhanced PPARγ mRNA expression and attenuation that of MCP-1 in peripheral monocytes of telmisartan-treated patients with essential hypertension [49].
Telmisartan is a well tolerated compound without the significant cardiovascular toxicity of full PPARγ agonists [50]. Its therapeutic properties may extend to brain disorders such as stroke and dementia [51] in which amelioration of the inflammatory response in circulating monocytes leads to decreased macrophage infiltration into the brain parenchyma, reduction of neuronal injury and protection of cognition [18,52,53].
Supplementary Material
Acknowledgments
The authors thank Astra-Zeneca, Moőlndal, Sweden, for their supply of candesartan. Candesartan was a gift from Astra-Zeneca, R&D, Moőlndal, Sweden.
This study was supported by the Division of Intramural Research Programs, Department of Health and Human Services, National Institute of Mental Health, National Institutes of Health, USA.
Abbreviations
- ABCG1
ATP-binding cassette subfamily G member 1
- Ang II
angiotensin II
- ARB
Ang II receptor blocker
- AT1
Ang II type 1 receptor
- CD36
cluster of differentiation 36
- COX-2
cyclooxygenase-2
- EMSA
electrophoretic mobility shift assay
- ERK1/2
extracellular signal-regulated kinases 1/2
- ICAM-1
intercellular adhesion molecule 1
- IL
interleukin
- IκB-α
inhibitor of κB-α
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemotactic protein-1
- NF-κB
nuclear factor-κB
- PGE2
prostaglandin E2
- PPARγ
peroxisome proliferator-activated receptor-γ
- ROS
reactive oxygen species
- THP-1
human acute monocytic leukemia cell line
- TNFα
tumor necrosis factor-α
Footnotes
Conflicts of interest
There are no conflicts of interest.
Disclaimers: none.
References
- 1.Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- 2.Chrysant SG, Chrysant GS, Chrysant C, Shiraz M. The treatment of cardiovascular disease continuum: focus on prevention and RAS blockade. Curr Clin Pharmacol. 2010;5:89–95. doi: 10.2174/157488410791110742. [DOI] [PubMed] [Google Scholar]
- 3.Bakris G. Are there effects of renin-angiotensin system antagonists beyond blood pressure control? Am J Cardiol. 2010;105:21A–29A. doi: 10.1016/j.amjcard.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Konstam MA, Neaton JD, Dickstein K, Drexler H, Komajda M, Martinez FA, et al. Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double-blind trial. Lancet. 2009;374:1840–1848. doi: 10.1016/S0140-6736(09)61913-9. [DOI] [PubMed] [Google Scholar]
- 5.Neldam S. Choosing an angiotensin-receptor blocker: blood pressure lowering, cardiovascular protection or both? Future Cardiol. 2010;6:129–135. doi: 10.2217/fca.09.61. [DOI] [PubMed] [Google Scholar]
- 6.Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, et al. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- 7.Rotman N, Wahli W. PPAR modulation of kinase-linked receptor signaling in physiology and disease. Physiology (Bethesda) 2010;25:176–185. doi: 10.1152/physiol.00018.2010. [DOI] [PubMed] [Google Scholar]
- 8.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 9.Wang P, Anderson PO, Chen S, Paulsson KM, Sjögren HO, Li S. Inhibition of the transcription factors AP-1 and NF-kappaB in CD4 T cells by peroxisome proliferator-activated receptor gamma ligands. Int Immunopharmacol. 2001;1:803–812. doi: 10.1016/s1567-5769(01)00015-7. [DOI] [PubMed] [Google Scholar]
- 10.Duan SZ, Usher MG, Mortensen RM. PPARs: the vasculature, inflammation and hypertension. Curr Opin Nephrol Hypertens. 2009;18:128–133. doi: 10.1097/MNH.0b013e328325803b. [DOI] [PubMed] [Google Scholar]
- 11.Villacorta L, Schopfer FJ, Zhang J, Freeman BA, Chen YE. PPARgamma and its ligands: therapeutic implications in cardiovascular disease. Clin Sci (Lond) 2009;116:205–218. doi: 10.1042/CS20080195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erbe DV, Gartrell K, Zhang YL, Suri V, Kirincich SJ, Will S, et al. Molecular activation of PPARgamma by angiotensin II type 1-receptor antagonists. Vascul Pharmacol. 2006;45:154–162. doi: 10.1016/j.vph.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, Saavedra JM. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol. 2006;552:112–122. doi: 10.1016/j.ejphar.2006.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) 2007;112:375–384. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Ando H, Macova M, Dou J, Saavedra JM. Angiotensin II AT1 receptor blockade abolishes brain microvascular inflammation and heat shock protein responses in hypertensive rats. J Cereb Blood Flow Metab. 2005;25:878–886. doi: 10.1038/sj.jcbfm.9600082. [DOI] [PubMed] [Google Scholar]
- 16.Bregonzio C, Armando I, Ando H, Jezova M, Baiardi G, Saavedra JM. Anti-inflammatory effects of angiotensin II AT1 receptor antagonism prevent stress-induced gastric injury. Am J Physiol Gastrointest Liver Physiol. 2003;285:G414–G423. doi: 10.1152/ajpgi.00058.2003. [DOI] [PubMed] [Google Scholar]
- 17.Sánchez-Lemus E, Benicky J, Pavel J, Saavedra JM. In vivo angiotensin II AT1 receptor blockade selectively inhibits LPS-induced innate immune response and ACTH release in rat pituitary gland. Brain Behav Immun. 2009;23:945–957. doi: 10.1016/j.bbi.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT(1) receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36:1–18. doi: 10.1016/j.psyneuen.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benicky J, Sánchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larrayoz IM, Pang T, Benicky J, Pavel J, Sánchez-Lemus E, Saavedra JM. Candesartan reduces the innate immune response to lipopolysaccharide in human monocytes. J Hypertens. 2009;27:2365–2376. doi: 10.1097/HJH.0b013e3283314bc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, et al. Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler Thromb Vasc Biol. 2010;30:802–808. doi: 10.1161/ATVBAHA.109.201681. [DOI] [PubMed] [Google Scholar]
- 22.Egidy G, Friedman J, Viswanathan M, Wahl LM, Saavedra JM. CGP-42112 partially activates human monocytes and reduces their stimulation by lipopolysaccharides. Am J Physiol. 1997;273:C826–C833. doi: 10.1152/ajpcell.1997.273.3.C826. [DOI] [PubMed] [Google Scholar]
- 23.Heemskerk FM, Saavedra JM. Quantitative autoradiography of angiotensin II AT2 receptors with [125I]CGP 42112. Brain Res. 1995;677:29–38. doi: 10.1016/0006-8993(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 24.Klotz L, Schmidt M, Giese T, Sastre M, Knolle P, Klockgether T, Heneka MT. Proinflammatory stimulation and pioglitazone treatment regulate peroxisome proliferator-activated receptor gamma levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J Immunol. 2005;175:4948–4955. doi: 10.4049/jimmunol.175.8.4948. [DOI] [PubMed] [Google Scholar]
- 25.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Wahl LM. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF-kappa B activity in lipopolysaccharide-activated human primary monocytes. J Immunol. 2005;175:5423–5429. doi: 10.4049/jimmunol.175.8.5423. [DOI] [PubMed] [Google Scholar]
- 27.Morawietz H. LOX-1 and atherosclerosis: proof of concept in LOX-1-knockout mice. Circ Res. 2007;100:1534–1536. doi: 10.1161/CIRCRESAHA.107.101105. [DOI] [PubMed] [Google Scholar]
- 28.Dandona P, Kumar V, Aljada A, Ghanim H, Syed T, Hofmayer D, et al. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an antiinflammatory action. J Clin Endocrinol Metab. 2003;88:4496–4501. doi: 10.1210/jc.2002-021836. [DOI] [PubMed] [Google Scholar]
- 29.Velasco M, Díaz-Guerra MJ, Martin-Sanz P, Alvarez A, Boscaá L. Rapid upregulation of IkappaBbeta and abrogation of NF-kappaB activity in peritoneal macrophages stimulated with lipopolysaccharide. J Biol Chem. 1997;272:23025–23030. doi: 10.1074/jbc.272.37.23025. [DOI] [PubMed] [Google Scholar]
- 30.Xing B, Xin T, Hunter RL, Bing G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J Neuroinflammation. 2008;5:4. doi: 10.1186/1742-2094-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-γ and nuclear factor-κB. J Biol Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 32.Lombardi A, Cantini G, Piscitelli E, Gelmini S, Francalanci M, Mello T, et al. A new mechanism involving ERK contributes to rosiglitazone inhibition of tumor necrosis factor-α and interferon-γ inflammatory effects in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:718–724. doi: 10.1161/ATVBAHA.107.160713. [DOI] [PubMed] [Google Scholar]
- 33.Tian Q, Miyazaki R, Ichiki T, Imayama I, Inanaga K, Ohtsubo H, et al. Inhibition of tumor necrosis factor-alpha-induced interleukin-6 expression by telmisartan through cross-talk of peroxisome proliferator-activated receptor-gamma with nuclear factor kappaB and CCAAT/enhancer-binding protein-beta. Hypertension. 2009;53:798–804. doi: 10.1161/HYPERTENSIONAHA.108.126656. [DOI] [PubMed] [Google Scholar]
- 34.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 35.An J, Nakajima T, Kuba K, Kimura A. Losartan inhibits LPS-induced inflammatory signaling through a PPARgamma-dependent mechanism in human THP-1 macrophages. Hypertens Res. 2010;33:831–835. doi: 10.1038/hr.2010.79. [DOI] [PubMed] [Google Scholar]
- 36.Stangier J, Su CA, Roth W. Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J Int Med Res. 2000;28:149–167. doi: 10.1177/147323000002800401. [DOI] [PubMed] [Google Scholar]
- 37.Hodgkinson CP, Ye S. Microarray analysis of peroxisome proliferator-activated receptor-gamma induced changes in gene expression in macrophages. Biochem Biophys Res Commun. 2003;308:505–510. doi: 10.1016/s0006-291x(03)01416-5. [DOI] [PubMed] [Google Scholar]
- 38.Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, Iadecola C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41:898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 40.Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP, 3rd, Howatt DA, Subranmanian V, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor-/- mice. Circ Res. 2011;108:574–581. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim MP, Zhou M, Wahl LM. Angiotensin II increases human monocyte matrix metalloproteinase-1 through the AT2 receptor and prostaglandin E2: implications for atherosclerotic plaque rupture. J Leukoc Biol. 2005;78:195–201. doi: 10.1189/jlb.1204715. [DOI] [PubMed] [Google Scholar]
- 42.Xiao J, Leung JC, Chan LY, Tang SC, Lai KN. Crosstalk between peroxisome proliferator-activated receptor-gamma and angiotensin II in renal tubular epithelial cells in IgA nephropathy. Clin Immunol. 2009;132:266–276. doi: 10.1016/j.clim.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Ji Y, Liu J, Wang Z, Liu N, Gou W. PPARgamma agonist, rosiglitazone, regulates angiotensin II-induced vascular inflammation through the TLR4-dependent signaling pathway. Lab Invest. 2009;89:887–902. doi: 10.1038/labinvest.2009.45. [DOI] [PubMed] [Google Scholar]
- 44.Zhao SM, Shen LH, Li HW, Wang L, Chen H, Wang YL, Guo CY. Down-regulation of the expression of angiotensin II type 1 receptor in neonatal rat cardiac fibroblast by activation of PPARgamma signal pathway. Chin J Physiol. 2008;51:357–362. [PubMed] [Google Scholar]
- 45.Tham DM, Martin-McNulty B, Wang YX, Wilson DW, Vergona R, Sullivan ME, et al. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics. 2002;11:21–30. doi: 10.1152/physiolgenomics.00062.2002. [DOI] [PubMed] [Google Scholar]
- 46.Leibovitz E, Schiffrin EL. PPAR activation: a new target for the treatment of hypertension. J Cardiovasc Pharmacol. 2007;50:120–125. doi: 10.1097/FJC.0b013e318062153b. [DOI] [PubMed] [Google Scholar]
- 47.Cani PD, Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Ther. 2011;130:202–212. doi: 10.1016/j.pharmthera.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Ghanim H, Sia CL, Korzeniewski K, Lohano T, Abuaysheh S, Marumganti A, et al. A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal. J Clin Endocrinol Metab. 2011;96:1409–1414. doi: 10.1210/jc.2010-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marketou ME, Kontaraki JE, Tsakountakis NA, Zacharis EA, Kochiadakis GE, Arfanakis DA, et al. Differential effect of telmisartan and amlodipine on monocyte chemoattractant protein-1 and peroxisome proliferator-activated receptor-gamma gene expression in peripheral monocytes in patients with essential hypertension. Am J Cardiol. 2011;107:59–63. doi: 10.1016/j.amjcard.2010.08.048. [DOI] [PubMed] [Google Scholar]
- 50.Stafylas PC, Sarafidis PA, Lasaridis AN. The controversial effects of thiazolidinediones on cardiovascular morbidity and mortality. Int J Cardiol. 2009;131:298–304. doi: 10.1016/j.ijcard.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, et al. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension. 2009;54:782–787. doi: 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- 53.Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.