

Abstract
Toll-like receptors (TLR) are pivotal in macrophage activation. The molecular mechanisms controlling TLR signaling and macrophage activation are not completely understood. Zc3h12d is originally identified as a possible tumor suppressor gene. However, its function remains unknown. We here report that Zc3h12d negatively regulates TLR signaling and macrophage activation. Zc3h12d was enriched in spleen, lung and lymph node. In macrophages, the expression of Zc3h12d was remarkably induced by TLR ligands through JNK and NF-κB signal pathways. On the other hand, overexpression of Zc3h12d significantly inhibited TLR2 and TLR4 activation-induced JNK, ERK and NF-κB signaling as well as macrophage inflammation. Similar to Zc3h12a/MCPIP1, Zc3h12d also decreased the global cellular protein ubiquitination. These findings suggest that Zc3h12d is a novel negative feedback regulator of TLR signaling and macrophage activation and thus may play a role in host immunity and inflammatory diseases.
Keywords: Macrophage, Toll-like receptor, Signal transduction, JNK, NF-κB
1. Introduction
Toll-like receptors (TLRs) are the archetypal pattern recognition receptors in sensing exogenous pathogens. Activation of TLRs is a first line of defense of the immune system, leading to the activation and recruitment of neutrophils and macrophages to sites of infection and enhances antimicrobial activity. The TLR signaling through different intracellular molecules, such as MAP kinases and IκB kinases which are conserved signaling elements for many receptors, leads to increased expression of a distinct set of proinflammatory gene [1, 2]. However, how these pathways being precisely regulated remains largely unknown.
Macrophages express all the TLRs thus far identified. These receptors collectively provide robust surveillance mechanisms in response to vascular infection as well as oxidized low density lipoprotein and injured tissue components in atherosclerotic lesions [3, 4]. Stimulation by both endogenous and exogenous ligands of macrophage TLR2 and TLR4 has been implicated in inflammation, foam cell formation, and macrophage differentiation [5, 6]. Immediately following engagement with the ligand, TLRs are activated to induce a largely defined signal transduction cascade through recruiting differential adapter proteins to the cytosolic TIR domain. Subsequently, this leads to the activation of NF-κB signal transduction pathway, mitogen-activated protein kinase, and interferon regulatory factor 3 in a specific manner determined by the nature of the ligand and the engaged TLR [7].
Zc3h12d (also known as TFL and p34) is originally reported as a putative tumor suppressor that is deregulated in transformed follicular lymphoma [8]. Zc3h12d belongs to a recently identified CCCH-zinc finger containing protein family including Zc3h12a(also known as MCPIP1), Zc3h12b, Zc3h12c and Zc3h12d [9, 10]. As an initially characterized member, Zc3h12a/MCPIP1 was significantly induced by LPS through the TLR4 signal pathway and feedback inhibited TLR4-activated JNK and NF-κB signaling by deubiquitinating TNF receptor-associated factor family [11]. However, the role of Zc3h12d in TLR-induced macrophage inflammation is essentially unknown. Here we report that Zc3h12d was enriched in lymphoid and inflamed tissues such as spleen, lung and lymph node. Zc3h12d was remarkably induced by TLR2, TLR5 and TLR9 activation in macrophages. Overexpression of Zc3h12d significantly inhibited TLR-induced JNK, ERK and NF-κB activation of macrophages. These findings suggest that Zc3h12d is a novel regulator of TLR signaling and macrophage activation and thus may play a role in host immunity and inflammatory diseases.
2. Materials and methods
2.1. Mice
Male C57BL/6 wild-type mice (~6–8 wk old) were purchased from The Jackson Laboratories (Bar Harbor, ME) and housed in a temperature-controlled environment with 12 hr light/dark cycles at University of Missouri Kansas City Laboratory Animal Research Center. These studies were reviewed and approved by the Institutional Animal Care and Use Committee of University of Missouri Kansas City.
2.2. Cells
Raw264.7 and HEK293 cells were obtained from the American Type Culture Collection (ATCC). These cells were grown as a monolayer in DMEM (Invitrogen) containing 10% FBS, 2 mM L-glutamine, with 100 U/ml penicillin and streptomycin in 5.0% CO2. HEK293-TLR2 cell line was obtained from ATCC approved by BEI Resources and cultured as above.
2.3. Plasmids
The plasmids for Zc3h12d-58kd (also called p58TFL) and Zc3h12d-36kd (also called p36TFL) were generated as described previously [12]. Zc3h12d-106K (also called HA-p34-A) and Zc3h12d-106R (also called HA-p34-G) were kindly provided by Dr. M. You (University of Washington, St. Louis) [13]. MCPIP1-GFP was described previously. Flag-Zc3h12d was generated by PCR and inserted into pCMV-Mat-Flag-1 vector (Sigma) by Hind III and Kpn I. NF-κB and AP-1 reporters were purchased from Strategene. HA-ubiquitin was generated by T. Dawson (John Hopkins University, Baltimore) and obtained through Addgene [14].
2.4. Reagents
Rabbit anti-Zc3h12d polyclonal antibody and mouse anti-Zc3h12d monoclonal antibody were generated by immunizing GFT-tagged human recombinant Zc3h12d-36kd. Rabbit anti-Zc3h12d polyclonal antibody (ab100862) was purchased from Abcam. Goat anti-Zc3h12a polyclonal antibody (sc-136750) was purchased from Santa Cruz Biotech. Phosphor-ERK1/2, phosphor-JNK, phosphor-P38, phosphor-IKKβ, JNK, ERK1/2, P38, IKKβ, actin and HA-tag antibodies were purchased from Cell Signaling Technology Inc. LPS, TNFα, SP600125, SN50, U0126, SB203580 and calphostin C were purchased from Sigma. The ligands for TLR1-9 were purchased from Invivogen.
2.5. Transfection
Transient transfection of plamids into cells were per formed as described previously [10].
2.6. Protein isolation and Western blot
Protein isolation and Western blot were essentially performed as described previously [10].
2.7. RNA isolation and Northern blot
RNA isolation and Northern blot were essentially performed as described previously [10].
2.8. Quantitative real-time PCR
After removal of genomic DNA using DNAse I (Ambion), 2.4 μg of total RNA from RAW264.7 cells was reverse transcribed to cDNA using a commercial kit (Applied Biosystems). Quantitative real-time PCR was performed with iCycler Thermal Cycler (Bio-Rad, Herculer, CA) using 2×SYBR Green master mix (Bio-Rad) as described previously. Quantification was performed by the dCT method, with β-actin used for normalization.
2.9. Immunohistochemistry
6–8 week-old male C57BL/6 wild-type were sacrificed, and their spleen were harvested, fixed in 4% formaldehyde, processed in Leica TP1050 Fully Enclosed Vacuum Tissue Processor, and embedded in paraffin. The paraffin-embedded tissue sections (5 μm) were incubated overnight at 4 in Rabbit anti-Zc3h12d polyclonal antibody (1:100 dilution, abcam). Biotin conjugated goat anti-rabbit IgG (Jackson ImmunoResearch) was used as the secondary antibody at 1:1000 dilution for 1 h at room temperature followed by peroxidase-conjugated streptavidin (1:1000 dilution, Jackson ImmunoResearch) for 20 min at Room temperature. Brown color was developed with DAB reagent (sigma). The slides were mounted, examined and photographed using Olympus BX60 microscopy. The negative control slides were treated in an identical manner except that the primary antibodies were omitted (buffer substitution).
2.10. Statistics
Data were expressed as mean±SD. For comparison between two groups, the unpaired Student’s test was used. For multiple comparisons, analysis of variance followed by unpaired Student’s test was used. A value of p<0.05 was considered significant.
3. Results
3.1. Zc3h12d is highly enriched in lymphoid and inflamed tissues
There are two alternative spliced forms of human Zc3h12d identified [12]. One encodes 527 amino acids (Zc3h12d-58kd); the other encodes 321 amino acids(Zc3h12d -36kd). The last 23 amino acids of Zc3h12d-36kd are distinct from the carboxyl 229-residue sequence of Zc3h12d-58kd (Fig. 1A). In the mouse tissue, we detected both isoforms of Zc3h12d. As shown in Fig. 1B, Zc3h12d-58kd is broadly distributed but highly enriched in spleen, lung and the lymph nodes of the tested tissues. It is also expressed in other examined tissues including heart, liver, intestine, kidney, thymus and muscle, but not in the brain. Interestingly, Zc3h12d-36kd is only expressed in spleen, thymus and lymph node. In the tested spleen, Zc3h12d was selectively expressed in red pulp but not white pulp (Fig. 1C), which is consistent with the elevated expression of Zc3h12d in monocytes as red pulp reserve 50% of the body’s monocyte population. In macrophages, Zc3h12d-58kd was predominantly expressed compared to Zc3h12d-36kd (Huang et al., unpublished data). A previous study showed no functional difference between the two spliced forms [12].
Fig. 1.

Tissue distribution of mouse Zc3h12d. (A) Schematic representation of the structure of human Zc3h12d-58 and Zc3h12d-36 protein. The conserved domain and distinct regions have been boxed and colored. (B) A range of total protein (50 μg/lane) extracted from adult mouse tissues was examined by Western blot analysis with mouse anti-Zc3h12d monoclonal antibody. (C) Representative photomicrographs of immunochemical staining of mouse spleen with anti-Zc3h12d antibody. Three segments showed similar results. Bar=100 μm.
3.2. Zc3h12d was induced by TLR2, TLR5 and TLR9 ligands in macrophages
Macrophages express all the TLRs thus far identified. These receptors collectively provide robust surveillance mechanisms in response to vascular infection as well as oxidized LDL and injured tissue components in atherosclerotic lesions [3, 4]. To investigate whether Zc3h12d expression is inducible during TLR-triggered macrophage activation, we incubated Raw264.7 cells with the ligands for TLR1-9. As shown in Fig. 2A, Zc3h12d expression was markedly induced by TLR2, TLR5 and TLR9. Further experiments clearly showed that Zc3h12d was induced by TLR2, TLR4, TLR5 and TLR9 in a time-dependent manner (Fig. 2B). Moreover, Fig. 2C showed that Zc3h12d was dose-dependently induced by Pam3CSK4 (TLR2 ligand). These results indicate that Zc3h12d may be involved in the regulation of specific TLR signaling and macrophage activation.
Fig. 2.

Zc3h12d was markedly induced by TLR ligands. (A) Raw264.7 cells were incubated with the ligands for TLR1/2 (Pam3CSK4, 0.2μg/ml), TLR2 (HKLM, 10 μmol/L), TLR3 (Poly (I:C), 2 μg/ml), TLR4 (LPS, 0.2 μg/ml), TLR5 (Flagellin, 0.2 μg/ml), TLR6 (FSL-1, 0.2 μg/ml), TLR7/8 (ssRNA40, 0.2 μg/ml) and TLR9 (ODN1826, 0.75 μM) respectively for 8 hours. The cell lysates were subjected to Western blot analysis with Zc3h12d and actin antibodies. The quantitative data from three independent experiments were analyzed by Gel-Pro Analyzer software and presented as fold changes at the bottom of the image. *p<0.05 vs control. (B) Raw264.7 cells were incubated with indicated TLR ligands for different time points and subjected to Western blot analysis. (C) Raw264.7 cells were incubated with different concentrations of Pam3CSK4 as indicated for 8 hours and subjected to Western blot analysis. The bands of Western blot were quantified by Gel-Pro Analyzer software and presented as fold changes at the bottom of each image.
3.3. Zc3h12d negatively regulates LPS-induced inflammatory gene expression in macrophages
We previously reported that Zc3h12a/MCPIP1 acts as a negative regulator of macrophage activation [10]. To determine whether Zc3h12d also regulates macrophage inflammation, we investigated the effect of Zc3h12d on LPS-stimulated gene expression in macrophages. As shown in Fig. 3, LPS dramatically induced inflammatory cytokine (TNFα, IL-1β and IL-6) and iNOS expression by several folds to thousands folds in Raw264.7 cells. Interestingly, Zc3h12d overexpression significantly blunted the induction of TNFα, IL-1β, IL-6 and iNOS by LPS. These results suggest that Zc3h12d may also function as a suppressor for macrophage activation as Zc3h12a/MCPIP1 does.
Fig. 3.

Overexpression of Zc3h12d inhibits LPS-induced proinflammatory cytokine expression. Raw264.7 cells were transiently transfected with GFP or Zc3h12d-GFP plasmids by electroporation (Amaxa). The transfected cells were quiescent for 24 hours by incubated with macrophage serum-free medium and then treated with PBS or 1 μg/ml LPS for 8 hours. Total RNA was isolated and the expression of TNFα, IL-1β, IL-6 and iNOS was determined by quantitative PCR. Data were normalized by β-actin and represented as Mean±SD, n=3. *p<0.05, **P<0.001.
3.4. Zc3h12d inhibited TLR4-induced AP-1 and NF-κB activation
Many inflammatory genes are coordinately regulated by NF-κB and AP-1. To examine whether Zc3h12d affect the NF-κB and AP-1 activation, we evaluated minimal promoters containing binding sites for NF-κB and AP1 to determine whether they are targets for negative regulation by Zc3h12d. As shown in Fig. 4, forced Zc3h12d expression dose-dependently inhibited LPS-induced NF-κB and AP-1 activation. These results suggest that Zc3h12d may target the signaling pathways that activate AP -1 and NF-κB.
Fig. 4.

Zc3h12d represses NF-κB- and AP-1-dependent gene expression. Raw264.7 cells were co-transfected NF-κB-TK-Luc (A) or AP1-TK-Luc (B) as indicated with increasing amounts of HA-Zc3h12d (0, 25, 100 and 400 ng/per well) by Fugene 6. After quiescent for 16 hours, cells were treated with or without LPS (100 ng/ml) and collected for analysis of reporter gene activity 24 hours later. The luciferarse activity was measured and normalized by β-Gal activity. The data represent mean±SD, n=6. *P<0.01 vs stimulated without HA-Zc3h12d transfected group.
3.5. Zc3h12d expression inhibited TLR2-induced phosphorylation of JNK and ERK
Next, we examined the effect of Zc3h12d on TLR2-induced phosphorylation of JNK, ERK and p38 in macrophage. Raw264.7 cells were transfected with an empty vector or Zc3h12d plasmids. The transfected cells were stimulated with TLR2 ligand (Pam3CSK4) for different times as indicated. As shown in Fig. 5A, overexpression of Zc3h12d significantly attenuated TLR2-induced JNK and ERK phosphorylation, but does not significantly affected p38 kinase phosphorylation. The quantitative data from two independent experiments were shown in Fig. 5B. To further confirm the inhibition of Zc3h12d on TLR2 signaling, HEK293 cells that stably express TLR2 were transfected with Zc3h12d or an empty vector. The transfected cells were stimulated with Pam3CSK4 for different times as indicated. As shown in Fig. 5C&D, Pam3CSK4 induced JNK phosphorylation in time-dependent manner. Overexpression of Zc3h12d significantly attenuated Pam3CSK4-induced JNK phosphorylation. These results suggest that Zc3h12d negatively regulates TLR-induced JNK and ERK activation.
Fig. 5.

Zc3h12d expression inhibited TLR2-induced phosphorylation of JNK and ERK. Raw264.7 cells were transfected with Zc3h12d or an empty vector. After 24 hours quiescent, cells were stimulated with Pam3CSK4 (0.2 μg/ml) (A) for different time points as indicated. Whole cell extract was isolated and subjected to analysis by Western blot with indicated antibodies. The bands of Western blot were quantified by Gel-Pro Analyzer. The data from two independent experiments were averaged and presented as fold changes in (B). (C) A HEK293 cell line that stably expressed TLR2 were incubated with Pam3CSK4 (0.2 μg/ml) for different time points as indicated. The cell lysates were subjected to Western blot analysis with the antibodies as indicated. (D)The quantitative data from three independent experiments of (C) were analyzed by Gel-Pro Analyzer software and presented as fold changes. *p<0.05.
3.6. Zc3h12d expression attenuated global cellular ubiquitination
We previously reported that Zc3h12a/MCPIP1 negatively regulated JNK and NF-κB signaling by deubiquitination [11]. To examine whether Zc3h12d also affect global cellular ubiquitination as MCPIP1 does, HEK293 cells were transfected with HA-tagged ubiquitin and Zc3h12d-GFP or MCPIP1-GFP or control vector. The transfected cells were stimulated with TNFα for 30 min before harvest. As shown in Fig. 6, immunoblotting of whole cell lysates with HA antibody revealed that cells expressing Zc3h12d or MCPIP1 contained much fewer ubiquitinated proteins. These results suggest that Zc3h12d may also act as a deubiquitinase to modulate TLR signaling.
Fig. 6.
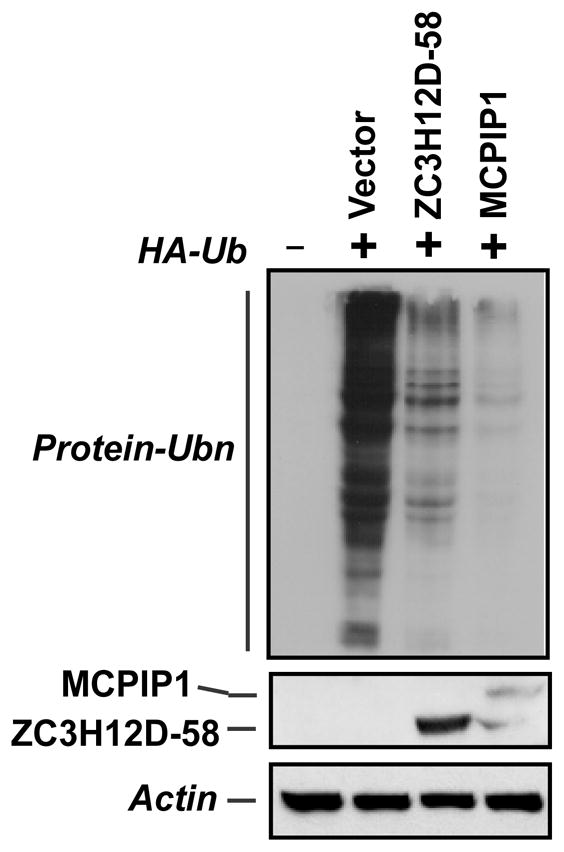
Zc3h12d attenuated global cellular ubiquitination. Raw264.7 cells were co-transfected with HA-Ub and Zc3h12d-GFP or MCPIP1-GFP as indicated. 24 h post-transfection, cells were stimulated with LPS (1 μg/ml) for 30 min and then collected for analysis of HA-Ub conjugation by Western blot with HA antibody. Proper expression of Zc3h12d-GFP and MCPIP1-GFP was also observed with anti-GFP.
3.7. Effect of Zc3h12d-K106R mutant on global cellular ubiquitination and LPS -induced JNK activation
It was previously reported that a single nucleotide polymorphism A/G in exon 3 of human Zc3h12d gene, which alters the residue 106 from lysine (K) to arginine (R), was associated with lung cancer [13]. Moreover, Zc3h12d-106K showed tumor suppressor activity, whereas Zc3h12d-106R lost the tumor suppressor activity [13]. We checked the protein sequence of Zc3h12d from different species. The area contains 106K is highly conserved across all of species as shown in Fig. 7A. It would be interesting to examine the effect of the Zc3h12d-K106R mutant on global cellular ubiquitination and JNK signaling. HEK293 cells were transfected with HA-tagged ubiquitin and Flag-Zc3h12d-58, Flag-MCPIP1, HA-Zc3h12d-106K or HA-Zc3h12d-106R. The transfected cells were stimulated with TNFα for 30 min before harvest. As shown in Fig. 7B, similar to MCPIP1, both Zc3h12d-58 and Zc3h12d-106K significantly attenuated global protein ubiquitination. However, Zc3h12d-106R lost the suppressor activity on protein ubiquitination. Consistently, Zc3h12d-106K inhibited LPS-induced JNK phosphorylation in macrophages, whereas Zc3h12d-106R did not (Fig. 7C). These results suggest that the two spliced isoforms of Zc3h12d seems to have similar effect on cellular ubiquitination and the mutation at residue 106 of Zc3h12d that is associated with lung cancer impairs its effect on cellular ubiquitination and JNK signaling.
Fig. 7.

Effect of Zc3h12d-K106R mutant on global cellular ubiquitination and LPS-induced JNK activation. (A) Alignment of the K106 region of Zc3h12d protein. The access numbers for Zc3h12d of human, monkey, cattle, turkey, hamster, dog, mouse and pig are AAI57833, XP 001087043, DAA26045, XP 003204160, EGV97157, XP 541146, NP 766373 and XP 003121206 respectively. Grey shadows indicate completely conserved of sequenses. (B) HEK293 cells were co-transfected with HA-Ub and Flag-Zc3h12d-58 or Flag-MCPIP1, HA-Zc3h12d-106K, HA-Zc3h12d-106R as indicated. 24 h post-transfection, cells were stimulated with TNFα (10 ng/ml) for 30 min and then collected for analysis of HA-Ub conjugation by Western blot with HA antibody. Proper expression of Flag-Zc3h12d-58 and Flag-MCPIP1 was observed with anti-Flag. The expression of HA-Zc3h12d-106K and HA-Zc3h12d-106R was observed with anti-HA. (C) Raw264.7 cells were transfected with Zc3h12d-1066R, Zc3h12d-106K or an empty vector. After 24 hours quiescent, cells were stimulated with LPS (1 μg/ml) (A) for different time points as indicated. Whole cell extract was isolated and subjected to analysis by Western blot with indicated antibodies.
3.8. Inhibition of JNK and NF-κB blocked TLR4-induced Zc3h12d expression in macrophages
To explore which signal pathway potentially mediates TLR4-induced Zc3h12d expression, Raw264.7 cells were pretreated with various signaling inhibitors as indicated in Fig. 8A, and then stimulated with or without TLR4 ligand (LPS, 1 μg/ml) for 8 hours. Zc3h12d mRNA level were analyzed by Northern blotting. As shown in Fig. 8A, JNK inhibitor (SP600125, 10 μmol/L) and NF-κB inhibitor (SN50, 50 μM) significantly blocked TLR2-induced Zc3h12d expression, but ERK and p38 kinase inhibitors had no effects. In contrast, PKC inhibitor increased TLR2-induced Zc3h12d expression. Western blot confirmed these results (Fig. 8B). These data suggest that TLR2 and TLR4 induced Zc3h12d expression probably through JNK and NF-κB signaling pathways.
Fig. 8.

Inhibition of JNK and NF-κB blocked TLR2 and TLR4-induced Zc3h12d expression. (A) Raw264.7 cells were pretreated with or without NF-κB inhibitor (SN50, 50μmol/L), ERK inhibitor (U0126, 10μmol/L), p38 inhibitor (SB203580, 25μmol/L), JNK inhibitor (SP600125, 10 μmol/L) and PKC inhibitor (Calphostin C, 10μmol/L) respectively, and then stimulated with PBS or LPS (1 μg/ml) for 8 hours. Total RNA was isolated and subjected to Northern blot analysis. (B) Raw264.7 cells were pretreated with or without various inhibitors as indicated, and then stimulated with PBS or Pam3CSK4 (0.2 μg/ml) for 16 hours. Whole cell extracts were isolated and subjected to Western blot analysis.
4. Discussion
Zc3h12d belongs to a novel CCCH-zinc finger containing protein family, which contains four members including Zc3h12a/MCPIP1, Zc3h12b, Zc3h12c and Zc3h12d [9, 10]. As the first characterized member, Zc3h12a/MCPIP1 was recently reported to function as an immune modifier and a negative regulator of macrophage inflammation [11, 15]. We and others have observed that Zc3h12a/Mcpip1 deficiency lead to multi-organ inflammation, severe anemia, autoimmune disease and premature death of mice [11, 15]. Further studies have suggested that Zc3h12a/MCPIP1 contain RNase and deubiquitinating enzyme activities and thus modulates inflammatory response and immune homeostasis by promoting inflammatory mRNA degradation and turning off the inflammatory signal transduction such as JNK and NF-κB signaling [11, 15]. Compared to Zc3h12a/MCPIP1, the other three members of this protein family are less characterized. Zc3h12d was originally reported as a putative tumor suppressor and is deregulated in transformed follicular lymphoma in human [8]. Interestingly, a single nucleotide polymorphism A/G in exon 3 of human Zc3h12d gene, which alters the residue 106 from lysine (K) to arginine (R), was associated with lung cancer [13]. Moreover, overexpression of the wild-type form (Zc3h12d-K)suppressed tumor cell growth both in vitro and in vivo. However, The mutant form (Zc3h12d-R) lost the tumor suppressor function [13]. Considering JNK and NF-κB signaling are over-activated in most cancers [16–19], it would be extremely interesting to examine whether Zc3h12d also function as a negative regulator of JNK and NF-κB signaling.
In this study, we observed that Zc3h12d was significantly induced by multiple TLR ligands through JNK and NF-κB signaling in macrophages. Expression of Zc3h12d feedback suppressed TLR-induced JNK, ERK and NF-κB signaling as well as macrophage inflammation. Similar to Zc3h12a/MCPIP1, Overexpression of Zc3h12d also significantly attenuated global protein ubiquitination, suggesting that Zc3h12d may also contain deubiquitinating enzymatic activity and can turn off the JNK, ERK and NF-κB signaling through deubiquitination.
Human Zc3h12d has two isoforms generated by alternative splicing: one encodes 527 amino acids designated as Zc3h12d-58kd; the other encodes 321 amino acids designated as Zc3h12d-36kd. The sequence difference between the two isoforms is that the last 23 amino acids of Zc3h12d-36kd are distinct from the carboxyl 229-residue sequence of Zc3h12d-58kd (Fig. 1A). In this study, we found that mouse Zc3h12d also has two isoforms. Zc3h12d-58kd is broadly distributed but highly enriched in spleen, lung and lymph node of mouse, whereas Zc3h12d-36kd is only expressed in spleen, thymus and lymph node (Fig. 1B). The difference between the two isoform that we observed so far is their cellular localization. Zc3h12d-58 forms a granule-like structure in the cytoplasm, whereas Zc3h12d-36 was localized in the cytoplasm as diffuse pattern (Qi et al. unpublished data). Both isoforms showed similar effect on TLR-induced JNK signaling and global protein ubiquitination.
Deletion of the long arm of chromosome 6 (6q) have been assumed to play an important role in the pathogenesis of tumors including lymphoma, lung cancer, breast cancer and ovarian cancer (20–22). A novel translocation t(2;6)(p12;q23) that appeared during transformation of follicular lymphoma to diffuse large B-cell lymphoma (23). Human Zc3h12d gene is located at the translocation breakpoint of t(2;6)(p12;q23), which may cause deregulation of Zc3h12d [8]. Indeed, Zc3h12d expression was prominent in normal human lymphocytes but defective in some leukemia/lymphoma cell lines [12]. Overexpression of Zc3h12d in cells inhibited G1 to S phase progression through suppression of retinoblastoma protein phosphorylation [12]. The results from our present study may suggest that Zc3h12d not only serves as a negative regulator of innate immunity but may also function as a tumor suppressor gene by controlling JNK, ERK and NF-κB activation.
In conclusion, we here report that Zc3h12d acts as a negative feedback regulator of TLR signaling and macrophage activation and may be implicated in host immunity and inflammatory diseases. However, the physiological function of Zc3h12d need to be further studied in vivo.
Highlights.
We studied the role of a novel CCCH-zinc finger protein Zc3h12d in toll-like receptor signaling.
Zc3h12d suppressed Toll-like receptor activation-induced JNK, ERK and NF-KB signaling.
Zc3h12d is a novel negative feedback regulator of Toll-like receptor signaling and macrophage inflammation.
Zc3h12a may play a role in host immunity and inflammatory diseases.
Acknowledgments
This work was supported by the American Heart Association BGIA Award (to M.F) and National Institute of Health Grant HL-098794 (to M.F). J. Liu was supported by National Institute of Health Grants CA-137126 and AR-055353. D. Fan was supported by National Institute of Health Grants HL-106325 and RR-016434. We thank Drs. M. You and T. Dawson for providing plasmids used in this work.
Abbreviation
- MCPIP1
MCP induced protein 1
- TLR
Toll-like receptor
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- JNK
c-Jun N-terminal kinase
- NF-κB
nuclear factor κB
- AP-1
activator protein 1
- Ub
ubiquitin
- GFP
green fluorescent protein
- TNF
tumor necrosis factor
- IL
interleukin
- iNOS
inducible nitrogen oxide synthase
- LPS
lipopolysaccharide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Medzhitov R. Nature Reviews Immunology. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 2.de Kleijn D, Pasterkamp G. Cardiovascular Research. 2003;60:58–67. doi: 10.1016/s0008-6363(03)00348-1. [DOI] [PubMed] [Google Scholar]
- 3.Schoneveld AH, Oude Nijhuis MM, van Middelaar B, Laman JD, de Kleijn DP, Pasterkamp G. Cardiovascular Research. 2005;66:162–169. doi: 10.1016/j.cardiores.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Oude Nijhuis MM, van Keulen JK, Pasterkamp G, Quax PH, de Kleijn DP. Current Pharmaceutical Design. 2007;13:983–994. doi: 10.2174/138161207780487593. [DOI] [PubMed] [Google Scholar]
- 5.Schoneveld AH, Hoefer I, Sluijter JP, Laman JD, de Kleijn DP, Pasterkamp G. Atherosclerosis. 2008;197(2008):95–104. doi: 10.1016/j.atherosclerosis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 6.den Dekker WK, Cheng C, Pasterkamp G, Duckers HJ. Atherosclerosis. 2010;209:314–20. doi: 10.1016/j.atherosclerosis.2009.09.075. [DOI] [PubMed] [Google Scholar]
- 7.Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. Cellular Molecular Life Science. 2008;65:2964–2978. doi: 10.1007/s00018-008-8064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minagawa K, Yamamoto K, Nishikawa S, Ito M, Sada A, Yakushijin K, Okamura A, Shimoyama M, Katayama Y, Matsui T. British Journal of Haematology. 2007;139:161–163. doi: 10.1111/j.1365-2141.2007.06752.x. [DOI] [PubMed] [Google Scholar]
- 9.Liang J, Song W, Tromp G, Kolattukudy PE, Fu M. PLoS ONE. 2008;3:e2880. doi: 10.1371/journal.pone.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattukudy PE, Fu M. The Journal of Biological Chemistry. 2008a;283:6337–6346. doi: 10.1074/jbc.M707861200. [DOI] [PubMed] [Google Scholar]
- 11.Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, Kolattukudy PE, Fu M. The Journal of Experimental Medicine. 2010;207:2959–2973. doi: 10.1084/jem.20092641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minagawa K, Katayama K, Nishikawa S, Yamamoto K, Sada A, Okamura A, Shimoyama M, Matsui T. Molecular Cancer Research. 2009;7:880–889. doi: 10.1158/1541-7786.MCR-08-0511. [DOI] [PubMed] [Google Scholar]
- 13.Wang M, Vikis HG, Wang Y, Jia D, Wang D, et al. Cancer Research. 2007;67:93–99. doi: 10.1158/0008-5472.CAN-06-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. Journal of Neuroscience. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S. Nature. 2009;458:1185–1190. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Lin A. Cell Research. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 17.Abdel-Wahab O, Levine RL. The Journal of Experimental Medicine. 2010;207:677–680. doi: 10.1084/jem.20100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis RE, Brown KD, Siebenlist U, Staudt LM. The Journal of Experimental Medicine. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staudt LM. The Journal of Experimental Medicine. 2000;191:207–212. doi: 10.1084/jem.191.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tilly H, Rossi A, Stamatoullas A, Lenormand B, Bigorgne C, Kunlin A, Monconduit M, Bastard C. Blood. 1994;84:1043–1049. [PubMed] [Google Scholar]
- 21.Zhang Y, Matthiesen P, Siebert R, Harder S, Theile M, Scherneck S, Schlegelberger B. Human Genetics. 1998;103:727–729. doi: 10.1007/s004390050899. [DOI] [PubMed] [Google Scholar]
- 22.Bailey-Wilson JE, Amos CI, Pinney SM, Petersen GM, De Andrade M, et al. American Journal of Human Genetics. 2004;75:460–474. doi: 10.1086/423857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto K, Okamura A, Minagawa K, Yakushijin K, Urahama N, Gomyo H, Shimoyama M, Itoh M, Matsui T. Cancer Genetics and Cytogenetics. 2003;147:128–133. doi: 10.1016/s0165-4608(03)00201-2. [DOI] [PubMed] [Google Scholar]