

Abstract
Binge administration of the psychostimulant drug, methamphetamine (mAMPH), produces long-lasting structural and functional abnormalities in the striatum. mAMPH binges produce non-exocytotic release of dopamine (DA), and mAMPH-induced activation of excitatory afferent inputs to cortex and striatum is evidenced by elevated extracellular glutamate (GLU) in both regions. The mAMPH-induced increases in DA and GLU neurotransmission are thought to combine to injure striatal DA nerve terminals of mAMPH-exposed brains. Systemic pretreatment with either competitive or noncompetitive N-methyl-D-aspartic acid (NMDA) antagonists protects against mAMPH-induced striatal DA terminal damage, but the locus of these antagonists’ effects has not been determined. Here, we applied either the NMDA receptor antagonist, (DL)-amino-5-phosphonovaleric acid (AP5), or the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist, dinitroquinoxaline-2,3-dione (DNQX), directly to the dura mater over frontoparietal cortex to assess their effects on mAMPH-induced cortical and striatal immediate-early gene (c-fos) expression. In a separate experiment we applied AP5 or DNQX epidurally in the same cortical location of rats during a binge regimen of mAMPH, and assessed mAMPH-induced striatal dopamine transporter (DAT) depletions one week later. Our results indicate that both ionotropic glutamate receptor antagonists reduced the mAMPH-induced Fos expression in cerebral cortex regions near the site of epidural application and reduced Fos immunoreactivity in striatal regions innervated by the affected cortical regions. Also, epidural application of the same concentration of either antagonist during a binge mAMPH regimen blunted the mAMPH-induced striatal DAT depletions with a topography similar to its effects on Fos expression. These findings demonstrate that mAMPH-induced dopaminergic injury depends upon cortical NMDA and AMPA receptor activation and suggest the involvement of the corticostriatal projections in mAMPH neurotoxicity.
Keywords: neurotoxicity, DNQX, AP5, cerebral cortex, striatum, glutamate
INTRODUCTION
mAMPH is a commonly abused psychostimulant drug that produces long-lasting brain injury and cognitive impairments. Studies of human mAMPH abusers have demonstrated significant neurocognitive deficits that include impairments in executive and psychomotor functions, learning, memory and attention (Chang et al., 2007; Scott et al., 2007; Johanson et al., 2006; Paulus et al., 2003; Volkow et al., 2001). These impairments occur together with structural and functional deficits in the cerebral cortex and striatum. In particular, neuroimaging studies of mAMPH abusers reveal abnormal regional glucose metabolism in the anterior cingulate, orbital frontal and parietal cortices, as well as reductions of striatal dopamine transporters DAT; Kim et al., 2005, London et al., 2004; Volkow et al., 2001). These human studies are consistent with animal experiments showing persistent reductions in regional cerebral glucose metabolism and long-lasting decreases in several markers of striatal dopaminergic terminal integrity, such as dopamine (DA) content, tyrosine hydroxylase activity, and DAT (Huang et al., 1999; Pontieri et al., 1991; Hotchkiss et al., 1979; Ricaurte et al., 1982).
Acutely, mAMPH administration leads to a leakage of DA from intraneuronal vesicular storage sites into the cytosol, followed by reverse transport of cytosolic DA into the extracellular space via the plasmalemmal uptake carrier, DAT (Schmidt and Gibb, 1985; Sulzer et al., 2005; Fleckenstein et al., 1997). mAMPH-induced elevations in extracellular DA concentration lead to altered activity in striatal efferent pathways. Striatal projection neurons that send efferent fibers to the output nuclei of the basal ganglia influence the cerebral cortex through recurrent circuitry (Gerfen, 1990; Alexander and Crutcher, 1990; Parent and Hazrati, 1995). The striatum receives dense glutamatergic input from all regions of cerebral cortex, as well as from intralaminar and ventral thalamic nuclei, via recurrent circuits (McGeorge and Faull, 1989; Mengual et al., 1999; McFarland and Haber, 2001; Smith et al., 2004). The mAMPH-induced increases in striatal DA and GLU neurotransmission are thought to damage DA nerve terminals through the combined influences of oxidative stress and GLU-mediated excitotoxicity (Sonsalla et al., 1991; Nash and Yamamoto, 1992; LaVoie and Hastings, 1999).
Both the striatum and cerebral cortex are richly populated with glutamatergic synapses (Monoghan et al., 1989; Greenamyre and Young, 1989; Albin et al., 1992), where they contribute to neuronal activation during mAMPH exposure. Ionotropic GLU receptors of the AMPA and NMDA classes are needed for mAMPH-induced immediate-early gene (IEG) induction within striatal and cerebral cortical neurons (Wang and McGinty, 1996; Gross and Marshall, 2009). Systemic pretreatment with competitive or noncompetitive NMDA antagonists during binge mAMPH administration protects against mAMPH-induced striatal DA terminal damage (Sonsalla et al., 1991; Weihmuller et al., 1992; Stephans and Yamamoto, 1994). Microdialysis studies show that mAMPH-induced increases in striatal extracellular GLU concentrations are necessary for the resulting damage to striatal DA terminals (Stephans and Yamamoto, 1994). It has been argued that increased striatal dopaminergic neurotransmission causes a secondary rise in GLU in the striatum via the recurrent cortico-striato-nigro-thalamo-cortical loop circuit. Direct evidence for the involvement of this circuitry in mAMPH-induced dopaminergic injury comes from Mark et al. (2004), who found that interfering with gamma-amino-butyric acid neurotransmission in the substantial nigra during binge mAMPH administration prevented the mAMPH-induced rise in striatal GLU concentrations as well as subsequent striatal dopaminergic toxicity.
Although the systemic administration of GLU receptor antagonists has provided evidence for involvement of these receptors in mAMPH-induced neurotoxicity, where the antagonists act in the aforementioned circuitry remains to be clarified. The present investigation tested the role of cerebral cortical ionotropic GLU receptors in mAMPH-induced injury to striatal DA terminals. We employed a ‘cortical well’ to administer ionotropic GLU antagonists epidurally, thereby blocking GLU receptors in a region of cortex underneath the well. This blockade was achieved in a minimally invasive manner, by slow diffusion of the antagonist through the cortical layers, with the corpus callosum acting as a diffusional barrier. This epidural application method has been previously used to disinhibit cortical regions, resulting in secondary activation of anatomically restricted populations of striatal neurons (Berretta et al., 1997, 1999; Trevitt and Marshall, 2002). In the first experiment, we demonstrated that frontoparietal epidural application of the NMDA receptor antagonist, AP5, or the AMPA receptor antagonist, DNQX, effectively blocked cortical expression of the IEG, c-fos, near the locus of epidural application. We also found that the blockade of GLU neurotransmission in these cortical areas decreased excitatory input to anterior striatum, as assess by reductions in striatal c-fos activation.. Subsequently, we evaluated the effects of epidural application of these same GLU receptor antagonists on the DAT depletions arising from a single-day neurotoxic-binge mAMPH regimen. The results of these experiments suggest that cortical NMDA and AMPA receptor activation during mAMPH administration each contributes to augmented glutamatergic neurotransmission at cortico-striatal synapses as well as to the consequent mAMPH-induced striatal DAT depletions.
EXPERIMENTAL PROCEDURES
Subjects
Male Sprague-Dawley rats (275–325 g) were obtained from Charles Rivers Labs (Hollister, Ca.) and individually housed, with food and water ad libitum, under a standard 12 hr-light/12 hr-dark cycle (lights on 6 am-6 pm). Animals remained in the holding room for four days prior to surgery and were handled every day following surgery. All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Approval to conduct the experiments described was obtained from the UC Irvine animal subjects review board (IACUC). All efforts were made to minimize the number of animals used and any potential stress and discomfort.
Implantation of cortical well
Rats were anesthetized with Equithesin (10 mM sodium pentobarbital, 256.8 mM chloral hydrate, 86 mM MgSO4, 10.5% propylene glycol, and 12% ethanol, administered at 4.2 mg/kg, i.p.) and placed in a Kopf stereotaxic apparatus. A 2 mm by 4 mm bone flap, extending anterior +2.0 mm and lateral +/− 2.0 mm relative to bregma (Paxinos and Watson, 2003) was removed (Figure 1). This positioning of the skull defect was chosen because it exposes cingulate, motor, and somatosensory cortical regions to the applied GLU antagonists. Prior research has shown that these cortical regions send strong, topographically-organized excitatory projections to the anterior striatum (e.g., e.g., McGeorge and Faull, 1989). Further, the regions of the striatum targeted by these cortical projection axons have been found to be vulnerable to mAMPH-induced DAT loss (Eisch et al, 1992). The cortical wells were constructed using 0.65 ml capacity polypropylene microcentrifuge tubes (GeneMate, ISC BioExpress, Kaysville, UT). Each cortical well had a diameter exceeding the maximum diameter of the skull defect, and its height was approximately 1 cm above the skull. Without disturbing the dura mater, the cortical well was fitted around the skull opening with dental cement, filled with 200 µl of 0.9% saline, and capped. Also during surgery, a temperature transponder (IPTT-300, BioMedicData Systems, Inc., Seaford, DE) was implanted subcutaneously (s.c.) near the animal’s hindquarters. The rats were allowed to recover for 3–4 days following surgery before the day of the mAMPH or saline administration day.
Figure 1.

Schematic diagrams depicting anterior-posterior and medial-lateral extent of cortical well position. Bone diagram (left) illustrating cortical well placement (circular dotted line). A 2 mm by 4 mm bone flap (black rectangle), extending anterior +2.0 mm and lateral +/− 2.0 mm (right), relative to bregma was removed to expose underlying dura mater. Glutamate antagonists applied in cortical well diffused through skull defect and across dura mater. Schematics derived from Paxinos & Watson (2003).
Epidural GLU antagonist and mAMPH-induced Fos
Drugs and injection procedures
The 0.9% saline solution was removed from each well, and 200 µl of the AMPA receptor antagonist DNQX [1 mg/ml phosphate-buffered saline (PBS)] or the NMDA receptor antagonist AP5 (5 mg/ml 0.9% saline) was epidurally applied via the cortical well. The antagonist doses chosen were based on preliminary experiments in which Fos immunoreactivity was consistently reduced in cortical and striatal target regions. Other rats received the vehicle (PBS or 0.9% saline) solutions in the cortical wells. The rats were placed into large Plexiglas boxes with 5–7 animals per box. Approximately 6.5 hrs following initial epidural antagonist application (a time corresponding to when animals in the mAMPH neurotoxicity experiment received their fourth mAMPH or saline injection) animals were given a single injection of mAMPH [(+)-mAMPH hydrochloride; Sigma, St. Louis, MO)] (4 mg/kg free base, s.c; in saline) or saline (1 ml/kg) and euthanized 90 min later.
Tissue preparation
Rats were anesthetized with sodium pentobarbital (5 ml/kg, i.p.) and transcardially perfused. The perfused brains were processed for Fos immunohistochemical analysis as previously described (Gross and Marshall, 2009). Forty µm thick coronal sections were cut on a freezing microtome at three antero-posterior levels (see ‘Fos quantification’ below).
Fos immunohistochemistry
Sections were initially rinsed in 0.1 M PBS, followed by inhibition of endogenous peroxidase activity with 0.5% H2O2 in 70% methanol. Nonspecific antibody binding was blocked using 5% dry milk in PBS containing 0.2% Triton X-100. Sections were incubated with a rabbit anti-Fos polyclonal primary antibody [(Santa Cruz Biotechnology, Inc.; sc-52; Santa Cruz, Ca.) (1:8500 dilution in 1% dry milk in PBS with 0.1% Triton X-100)] for 48 hr at 4 °C. Sections were incubated in a solution containing biotinylated anti-rabbit IgG made in goat (1:200 dilution in 1% dry milk in PBS), and then incubated in a solution containing an avidin-biotin-peroxidase complex (Vectastain Elite ABC), and immunostaining was visualized by peroxidase reaction with a stabilized, metal-enhanced diaminobenzidine (DAB) complex (10% solution; Pierce Chemical Co.; Rockford, IL). The tissue was stored in Tris buffer until mounted onto gelatin-chrom-alum subbed slides.
Fos quantification
Levels of Fos immunoreactivity were determined by counting Fos-immunolabeled nuclei in the cerebral cortex and striatum at three antero-posterior levels corresponding to bregma AP +3.0 (‘prelimbic level’), +1.0 (‘striatal level’), and −2.0 mm (‘hippocampal level’). Because the three levels sampled correspond to cortical regions anterior to, posterior to, and at the center of the skull defect, separate analyses of the influences of GLU antagonists on Fos at these three levels was expected to provide information concerning the diffusion of GLU antagonist influences both anterior and posterior to the defect. The three levels sampled were: bregma AP +3.0 mm (1 mm anterior to front edge of the epidural opening in skull), AP +1.0 mm (center of epidural opening), and AP −2.0 mm (2 mm posterior to the back edge of epidural opening). Fos-positive nuclei were visualized at 100 X magnification using an Olympus BX60 microscope and were counted using computer-assisted image analysis employing a Roper Cool-Snap digital camera and a grain counting module of an MCID image analysis system (Imaging Research; Chalfont St. Giles, UK). Fos immunoreactivity levels were quantified using the “mean proportional area” measure of this module, which represents the proportion of the sampling area containing Fos-positive nuclei, as defined by density and size criteria. Here, values are expressed as the mean proportional area X 100. Because the cortical well extended over both hemispheres equally, effects on IEG expression were expected to be bilaterally equivalent; correspondingly, Fos immunoreactivity in one hemisphere per section was used for analysis.
Epidural glutamate antagonist and mAMPH-induced DAT depletion
Drugs and injection procedures
Rats were moved to a procedure room where the temperature was maintained between 23–24 °C. The 0.9% saline solution was removed from each well, and 200 µl of DNQX (1 mg/ml PBS) or AP5 (5 mg/ml of saline), was added to the cortical well 30 minutes prior to the first of four mAMPH injections. Vehicle (200 µl) for DNQX and AP5 was PBS and 0.9% saline, respectively. The rats were placed into large Plexiglas boxes (16×16x16 in) with 5–7 animals per box.
Methamphetamine injections
Thirty minutes after adding the GLU receptor antagonist (or vehicle), the animals received a systemic injection of mAMPH (4 mg/kg free base, s.c; in saline) or saline (1 ml/kg). Rats were given injections of mAMPH or saline solution at two hour intervals for a total of four injections. Each animal’s temperature was measured hourly using a wireless hand-held scanner (Smartprobe, BioMedicData Systems, Inc., 2010) during the mAMPH administration day.
DAT autoradiography
One week following the binge mAMPH administration, rats were anesthetized with sodium pentobarbital (5 ml/kg, i.p.) and decapitated, and their brains were removed and frozen at −20 °C for use in DAT autoradiography. Twenty µm-thick coronal sections were cut with a cryostat at the level of the striatum and stored at −20 °C until used. Tissue sections were taken at a level (AP +1.9 mm, relative to bregma) corresponding to the most anterior plane of fusion of the corpus callosum across the midline, and continuing posteriorly for 1 mm. At this AP level, the striatum has been found to be highly vulnerable to mAMPH-induced depletions of DA and DAT (Eisch et al, 1992), and is innervated by cortical neurons located in regions directly underneath the skull defect (McGeorge & Faull, 1989) and therefore subject to influence by NMDA antagonists added to the cortical wells.. Sections were incubated with 50 pM [125I]RTI-55 for autoradiographic quantification of binding to DAT in the striatum. To block [125I]RTI-55 from binding to the 5-HT transporter, 100 nM fluoxetine was included in all incubation solutions. Autoradiographic slides and 14C standard slides containing known amounts of radioactivity were apposed to Hyperfilm (GE Healthcare) for 48 hrs before development. Quantification of [125I]RTI-55 binding to DAT was performed using an MCID image analyzer. Image densities were converted to binding levels (nCi/g) using a calibration curve based on images of the standard slides packed with each film. To investigate the regional specificity of mAMPH-induced striatal DAT depletions, DAT levels were determined by sampling whole caudate-putamen (CPu), dorsal CPu [dorsolateral (DL), dorsocentral (DC), and dorsomedial (DM)], ventral CPu [ventrolateral(VL), ventrocentral (VC), and ventromedial (VM)], and nucleus accumbens core (NAcC) and shell (NAcSh) regions. Results from the two hemispheres of each animal were combined for analysis. Neuroprotective influences of epidural DNQX or AP5 were calculated for each striatal region by calculating the proportion of the mAMPH-induced DAT loss that was prevented by DNQX or AP5 administration.
Statistics
Three-way ANOVAs were used to determine main effects of systemic mAMPH treatment, epidural treatment, and brain region on DAT binding and Fos expression as well as to examine interactions between these factors. Three-way ANOVAs were also used to determine main effects of systemic mAMPH treatment, epidural treatment, and time on body temperature as well as to examine interactions between these factors. The between-subjects factors were systemic injection (mAMPH vs. saline) and epidural treatment (e.g., DNQX, AP5, or vehicle). The within-subjects factors were brain region for the DAT binding and Fos expression data, and time for the body temperature data. Where the three-way ANOVAs permitted, one-way ANOVAs were performed on the DAT binding and Fos expression data to test for group differences within the different cortical and striatal regions, and on body temperature data to test for group differences at the four time points (1, 3, 5, and 7 hrs post first mAMPH injection). One-way ANOVAs were used to evaluate three comparisons, (1) epidural vehicle / saline injection vs. epidural vehicle / mAMPH injection, to test for effects of systemic mAMPH administration, (2) epidural vehicle / mAMPH injection vs. epidural antagonist [e.g. DNQX (1 mg/ml) or AP5 (5 mg/ml)] / mAMPH injection to test for effects of epidural antagonist treatment on mAMPH-induced striatal DAT depletion, Fos immunoreactivity and hyperthermia, and (3) epidural vehicle / saline injection vs. epidural antagonist /saline-injection, to test for effects of antagonist alone. P values less than .05 were considered statistically significant. Data are presented as mean ± SEM and analyzed using SPSS 13.0 (Chicago, IL).
RESULTS
Epidural GLU antagonists and mAMPH-induced Fos
Effects of epidural DNQX application
A single systemic injection of mAMPH (4 mg/kg) induced a substantial increase in cortical and striatal Fos immunoreactive nuclei. Frontoparietal DNQX application significantly reduced this mAMPH-induced increase Fos in cortical regions underneath and in close proximity to the skull defect through which epidurally-applied substances diffused into cerebral cortex. Three-way ANOVA yielded a significant main effects for systemic injection, F(1,16) = 803.01, p < .001, and epidural treatment, F(1,16) = 47.49, p < .001, and significant interactions were found (region x systemic injection, F(14,16) = 24.48, p < .001; region x epidural treatment, F(14,16) = 6.03, p < .001, region x epidural treatment x systemic injection, F(14,16) = 6.17, p < .001; and epidural treatment x systemic injection, F(1,16) = 51.67, p < .01).
One-way ANOVAs showed that Fos immunoreactivity was increased in all regions examined of the PBS / mAMPH group compared to the PBS / saline group, p’s <.05. Additionally, DNQX-induced antagonism of mAMPH-stimulated Fos was statistically significant (p’s < .05) for cortical areas directly under the skull defect (‘striatal level’) and 1 mm in front of the anterior edge of defect (‘prelimbic level’), but not 2 mm behind its posterior edge (‘hippocampal level’; Figures 2,3). These effects of mAMPH and DNQX on Fos immunoreactivity were observed throughout cortical laminae (Fig. 4 C,D). At the middle and anterior levels of analysis, DNQX effects were greater in magnitude for medial than lateral cortical and striatal regions. For example, at the middle level, DNQX application decreased Fos immunoreactivity by ~80% in secondary motor cortex, but only by ~15% in somatosensory cortex, and ~40% in medial striatum compared to ~30% in lateral striatum. In all regions, Fos expression in the cortex and striatum of the DNQX / saline and PBS / saline groups did not differ (p’s>.05 in all cases).
Figure 2.

Representative photomicrographs depicting Fos-positive immunoreactive nuclei in SAL/mAMPH (left panel), DNQX/mAMPH (middle panel) and AP5/mAMPH (right panel) at three anterior-posterior levels. At the anterior “prelimbic” level, representative cortical regions include cingulate (Cg) and somatosensory (Ssx) cortex. At the middle “striatal” level, representative regions include secondary (Mtx2) motor cortex, and dorsomedial (DM) striatum. At the posterior “hippocampal” level, a representative cortical region is represented by motor cortex (Mtx). Scale bar = 200 µm.
Figure 3.

Levels of cortical and striatal Fos immunoreactivity at three antero-posterior levels after epidural application of vehicle, DNQX (top), or AP5 (bottom), with systemic saline (SAL) or mAMPH injection. At the anterior “prelimbic” level, sampled cortical regions include prelimbic (PrL), cingulate (Cg), motor (Mtx), and somatosensory (Ssx) cortex, represented in schematic insert as 1, 2, 3, 4. At the middle “striatal” level, sampled cortical regions include cingulate, secondary motor (Mtx2), primary motor (Mtx1) and somatosensory cortex, and identified as 1, 2, 3, 4. Striatal subregions sampled in dorsal [dorsomedial (DM), dorsolateral (DL)], and ventral [ventromedial (VM), ventrolateral (VL)] striatum, identified as 5, 6, 7, 8. At the posterior “hippocampal” level, sampled cortical regions include cingulate, motor and somatosensory cortex, identified as 1, 2, 3. Statistically significant differences between the vehicle/SAL and vehicle/mAMPH groups are denoted as: ^ p < .05, ^^ p < .01, ^^^ p < .001. Significant differences between the vehicle/mAMPH and GLU antagonist/mAMPH groups are represented as * p < .05, ** p < .01, *** p < .001, and trends (t) noted when p < 0.1.
Figure 4.

Photomicrographs and schematic representations illustrating extent of functional influence of epidurally applied glutamate receptor antagonists. (A) Schematic representation of cortical zone of functional influence of both DNQX and AP5 at concentrations employed in present experiments. Using a dorsal view of the rat cerebral cortex, the area of skull defect is depicted as dashed rectangle, and the zone of cortical influence of the glutamate antagonists is depicted as gray zone that included skull defect and cortex 1.5 mm beyond the border of the defect. Area of cortical influence is derived from analysis of significant Fos effects with respect to Paxinos and Watson (1993) coordinates. (B) Schematic illustration of size and positioning of photomicrographs shown in Panels C and D. (C,D) Photomicrographs of Fos immunoreactivity in frontoparietal cortex of representative rats with epidural application of PBS (C) or DNQX (D; 1 mg/ml).
Effects of epidural AP5 application
The frontoparietal application of AP5, like that of DNQX, significantly reduced the mAMPH-induced Fos immunoreactivity in cortical regions immediately anterior to and underneath the cortical well, and in most sampled striatal regions (Figures 2, 3). Three-way ANOVA yielded significant main effects for systemic injection, F(1,10) = 718.16, p < .001 and epidural treatment, F(1,10) = 48.82.01, p < .001, and significant interactions were found (region x systemic injection, F(14,10) = 21.85, p < .001; region x epidural treatment, F(14,10) = 5.22, p < .001, region x epidural treatment x systemic injection, F(14,10) = 5.41, p < .001; and epidural treatment x systemic injection, F(1,10) = 48.04, p < .001).
One-way ANOVAs showed that the effects of epidural AP5 application were similar to those of DNQX application, with the greatest effects of the AP5 treatment occurring in medial cortex and striatum. For example, at the middle level of analysis, Fos immunoreactivity was decreased ~65% in secondary motor cortex, and ~30% in somatosensory cortex; and ~35% in medial striatum compared to ~25% in lateral striatum. No significant effects on Fos expression were observed in the cortex at the most posterior level of analysis (Figure 3, bottom panels).
An estimate of the cortical zone of diffusion of the GLU antagonists could be made based on the finding that the effects of each antagonist was limited to an area extending 1–2 mm beyond the borders of the skull defect, along both the anterior-posterior axis and lateral to edge of this skull defect (Figure 4A).
Epidural GLU antagonists, mAMPH-induced DAT loss, and body temperature
Effects of epidural DNQX application
A. mAMPH effects on body temperature
Binge mAMPH treatment induced a significant elevation in body temperature that was not significantly changed by epidural DNQX treatment (Figure 5). Three-way ANOVA yielded a significant main effects for systemic injection (mAMPH vs. saline), F(1,37) = 215.38 p < .001, and a significant interaction were observed (time x systemic injection, F(3,37) = 5.85, p < .01). No main effect or interactions involving epidural application were found. One-way ANOVAs at each time point (i.e. 1, 3, 5, 7 hrs post first mAMPH injection) demonstrated a significant increase in temperature at all time points measured in the PBS / mAMPH group compared to PBS / saline group, p’s <.05.
Figure 5.

Mean (±SEM) body temperature of animals receiving epidural vehicle, DNQX (left panel), or AP5 (right panel) with systemic injections of vehicle or mAMPH. Statistically significant differences between the vehicle/SAL and vehicle/mAMPH groups are represented as * p < .05, ** p < .01, *** p < .001, and significant differences between the vehicle/mAMPH and glutamate antagonist/mAMPH groups are represented as: ^^ p < .01.
B. mAMPH effects on striatal DAT binding levels
The binge mAMPH administration induced substantial depletions of striatal [125I]RTI-55 binding, which were greatest in the ventral caudate-putamen, and the epidural application of DNQX partially protected against this mAMPH-induced striatal DAT loss (Figure 6, top and middle panels). Three-way ANOVA yielded a significant main effect for systemic injection, F(1,37) = 127.45, p < .001, and significant interactions were found (region x systemic injection, F(8,37) = 101.14, p < .001; region x epidural treatment x systemic injection, F(8,37) = 5.08, p < .001; epidural treatment x systemic injection, F(8,37) = 8.93, p < .01 ).
Figure 6.
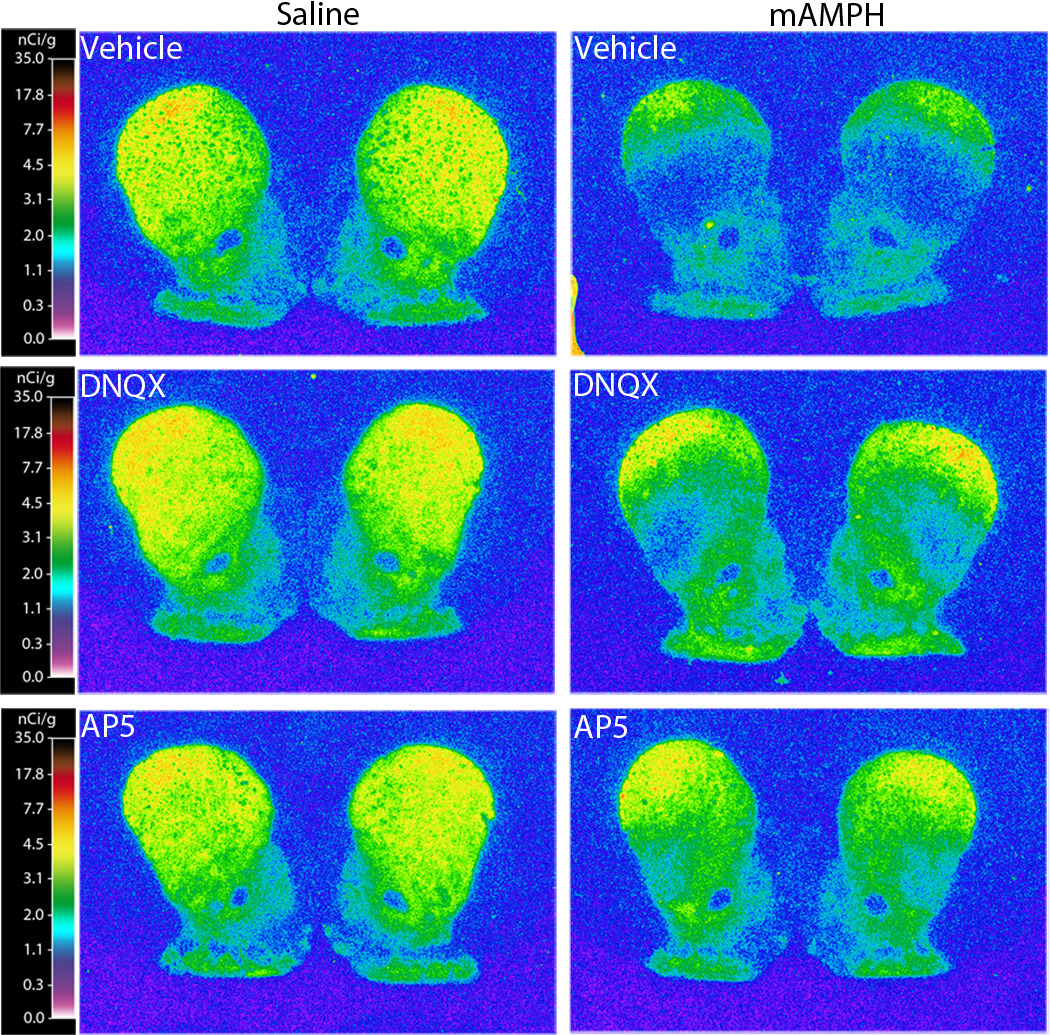
Representative striatal DAT autoradiograph images at the level of the anterior striatum. The [125I]RTI-55 autoradiographs depict DAT binding levels (nCi/g) in rats that received frontoparietal epidural application of vehicle (top), DNQX (middle), AP5 (bottom), followed by four systemic injections of either saline (1 ml/kg) or mAMPH (4 mg/kg).
One-way ANOVAs showed that DAT binding was decreased for all regions examined in the PBS / mAMPH group compared to the PBS / saline group, p’s <.05. Comparisons between the PBS / mAMPH group and DNQX / mAMPH group showed that epidural DNQX treatment significantly attenuated mAMPH-induced DAT depletions in all dorsal and ventral striatal subregions analyzed. The magnitude of this neuroprotective influence of DNQX application varied from region to region, with greatest normalization to control values occurring in the NAcSh (~60% neuroprotection) and NAcC (~50% neuroprotection) subregions, followed by dorsal striatal subregions (DM and DC, ~40%; DL, ~30% ), and ventral striatal subregions (VM, ~30%; VC and VL, ~20%) (Figure 7, top panel). Finally, the DAT levels of the DNQX / saline group and PBS / saline group did not differ for any region, p’s>.05.
Figure 7.

Mean (±SEM) striatal DAT binding density (nCi/g) of animals receiving epidural application of vehicle, DNQX (top), or AP5 (bottom) with systemic injections of vehicle or mAMPH. The schematic insert illustrates striatal dorsal and ventral subregions where DAT levels were quantified. Statistically significant differences between the vehicle/SAL and vehicle/mAMPH groups are represented as * p < .05, ** p < .01, *** p < .001, and significant differences between the vehicle/mAMPH and glutamate antagonist/mAMPH groups are represented as: ^ p < .05, ^^ p < .01, ^^^ p < .001.
Effects of epidural AP5 application
A. mAMPH effects on body temperature
Binge mAMPH treatment induced a significant elevation in body temperature that was significantly changed by epidural AP5 treatment (Figure 5, right panel). Three-way ANOVAs yielded significant main effects for systemic injection, F(1,30) = 121.51, p < .001, and significant interactions were observed (time x systemic injection, F(3,30) = 3.27, p <.05; time x epidural treatment x systemic injection, F(3,30) = 3.89, p < .05). One-way ANOVAs at each time point (i.e. 1, 3, 5, 7 hrs post first mAMPH injection) demonstrated that body temperatures of the saline / mAMPH group were higher at all time points compared to saline / saline group, p’s <.05. Oneway ANOVAs also showed that AP5 application did not significantly attenuate mAMPH-induced hyperthermia at the 1, 3, and 7 hr. time points (p > .05), but did significantly reduce body temperature at the 5 hr. time point (p < .01). Finally, at no time point was there a significant body temperature difference between the AP5 / saline group and the saline / saline group, p’s>.05.
B. mAMPH effects on striatal DAT binding levels
The binge mAMPH administration induced substantial depletions of striatal DAT binding, and the epidural application of AP5 partially protected against this mAMPH-induced striatal DAT loss (Figures 6 and 7). Significant main effects were found for epidural treatment, F(1,30) = 5.07, p < .05 and systemic injection, F(1,30) = 76.25, p < .001, and significant interactions were observed (epidural treatment x systemic injection, F(1,30) = 8.53, p < .01; region x epidural treatment x systemic injection, F(8,30) = 3.38, p < .01; and region x systemic injection, F(8,30) = 52.73, p < .001).
One-way ANOVAs revealed significant decreases in DAT levels in all regions examined in the saline / mAMPH group compared to the saline / saline group, p’s <.05. Also, the epidural AP5 application significantly attenuated mAMPH-induced DAT depletions in all dorsal and ventral striatal subregions analyzed. The magnitude of this neuroprotective influence of AP5 application varied from region to region, with greatest normalization to control values occurring in the NAcSh (~100% neuroprotection) and NAcC (~70% neuroprotection) subregions, followed by dorsal striatal subregions (DC, ~60%; DM and DL, ~50%), and ventral striatal subregions (VM, ~40%; VC and VL, ~30%) (Figure 7, bottom panel). Finally, the DAT levels of the AP5 / saline group did not differ from those of the saline / saline group, p’s >.05 in all regions.
DISCUSSION
The present experiments point to the conclusions that epidural application of ionotropic GLU receptor antagonists can block mAMPH-induced activation of corticostriatal projections, and that this blockade reduces mAMPH-induced injury to striatal dopaminergic nerve terminals. To determine the extent to which epidural application of GLU receptor antagonists affected activation of specific cortical and striatal regions, immunohistochemical analysis of the IEG product, Fos, was performed. Frontoparietal epidural application of DNQX or AP5 attenuated the mAMPH-induced increases in Fos immunoreactivity in cortical regions located under and near the skull defect, but not in more distant locations. Additionally, epidural application of these drugs reduced mAMPH-induced Fos in dorsal and ventral striatal subregions. These findings led to the prediction that a similar antagonism of either class of ionotropic GLU receptor during an otherwise neurotoxic regimen of mAMPH would attenuate the subsequent loss of striatal dopaminergic terminal markers. This prediction was supported by a second set of experiments, showing frontoparietal epidural application of DNQX or AP5 during a binge mAMPH regimen attenuated mAMPH-induced DAT loss in a topographically-organized manner within both dorsal and ventral striatal subregions.
Cortical GLU receptor involvement in mAMPH-induced activation of striatal neuron activity
These experiments show that a focal blockade of cortical ionotropic GLU receptors can attenuate mAMPH-induced striatal IEG expression in both cortex and striatum. The skull defect underneath the cortical well extended 2 mm laterally on each side of the midline. Reflecting this medial positioning, epidural application of DNQX or AP5 attenuated mAMPH-induced cortical Fos expression in a medial-to-lateral gradient, evidenced by significant reductions in the anterior cingulate, prelimbic, and primary and secondary motor cortices, and only nonsignificant reductions in the primary somatosensory cortex.
A medial-to-lateral gradient of GLU antagonist effects on Fos expression was also observed in the striatum, where greater Fos reductions occurred in dorsomedial and ventromedial, compared to dorsolateral and ventrolateral, CPu subregions. The striatum receives topographically organized afferent fibers from the cerebral cortex, whereby medially-positioned limbic and association cortical areas maintain functionally distinct projections into medial and ventral striatal regions referred to as association and limbic striatal territories (Alexander and Crutcher, 1990; Graybiel, 1990; Parent and Hazrati, 1995). Importantly, this regional pattern of interference with mAMPH-induced striatal Fos expression consequent to cortical GLU receptor blockade is inconsistent with a direct drug diffusion to striatum, since such a diffusional pattern would predict diminishing effects of the epidural GLU antagonists at greater distances from the site of their application. Instead, we observed greater effects of each GLU antagonist on levels of Fos expression in the ventrolateral striatum compared to the dorsolateral striatum. Notably, these anteroventral striatal subregions receive extensive inputs from medial cortical areas, including cingulate and secondary motor cortex (McGeorge & Faull, 1989). Collectively, these observations support the notion that epidural GLU receptor antagonists alter striatal function by reducing activity in corticostriatal projections rather than by direct diffusion to striatum.
The present experiments confirm the importance of cortical GLU neurotransmission for mAMPH-induced activation of corticostriatal projections. Although previous studies showed that systemic injection of an AMPA or NMDA receptor antagonist decreased psychostimulant-evoked IEG expression in the sensorimotor cortex and striatum (Dragunow et al., 1991; Wang et al, 1994a,b; Konradi et al., 1996), this systemic blockade left unresolved the locus of these effects. Extensive prior research supports the importance of cortical activity in the control of striatal IEG expression. For instance, disinhibition of cortical neurons by epidural application of picrotoxin induces striatal immediate-early gene activation (Berretta et al., 1997, 1999; Trevitt et al., 2005). Also, transection of corticostriatal projections reduces amphetamine-induced striatal c-fos expression (Cenci and Bjorklund, 1993). Here, cortical blockade of either AMPA or NMDA receptors during mAMPH administration had similar effects on cortical and striatal IEG expression, presumably because both AMPA and NMDA receptors make significant contributions to thalamo-cortical and cortico-cortical excitatory transmission (Salt et al., 1995; Armstrong-James et al., 1993). Overall, our results are consistent with a model whereby mAMPH administration induces increased GLU neurotransmission in the cerebral cortex which, through both AMPA and NMDA cortical receptors, contributes to increased excitatory transmission at corticostriatal synapses.
Cortical GLU receptor involvement in mAMPH-induced loss of striatal DAT
Previous studies regarding the role played by GLU receptor activation in mAMPH-induced neurotoxicity have shown that systemic pretreatment with competitive and noncompetitive NMDA receptor antagonists protect against mAMPH-induced striatal DA terminal damage (Sonsalla et al., 1991; Weihmuller et al., 1992). The noncompetitive NMDA antagonist, MK-801 has been found to protect against several other forms of brain injury, including those resulting from cerebral ischemia/hypoxia (Globus et al., 1988) and hypoglycemia (Westerberg et al., 1988). The findings that MK-801 and other NMDA antagonists are protective in these excitotoxic models and also in mAMPH-induced DA terminal damage suggests an excitotoxic component to mAMPH-induced damage.
The present experiments provide evidence that cortical NMDA and AMPA classes of ionotropic GLU receptors contribute to mAMPH-induced striatal DAT depletion. As previously discussed, all striatal subregions receive dense glutamatergic innervation from cortical areas in a topographically organized manner. Here, epidural GLU antagonist application attenuated mAMPH-induced striatal DAT depletion in a medial to lateral gradient evidenced by greatest neuroprotection, relative to control values, in the dorsomedial, dorsocentral, and ventromedial striatal subregions (including the NAcC and NAcSh), which receive extensive cortical excitatory input from prelimbic, cingulate, and secondary motor areas.
In the present experiments, the magnitude of the mAMPH-induced DAT loss in CPu was very extensive (65–70% DAT losses in the two experiments), and we also noted less extensive but significant DAT depletions in the nucleus accumbens. This accumbens damage is in contrast with several previous experiments that reported no significant mAMPH-induced loss of DA terminal markers in nucleus accumbens, even under dosing conditions that reduced DA, DAT or TH in CPu (Eisch et al, 1992; Belcher et al, 2005, 2008; Thomas et al, 2008). However, those studies also reported lower CPu DAT depletions than those seen in the present experiments. Overall, the available evidence suggests that relative to the CPu, the nucleus accumbens is spared from mAMPH-induced dopaminergic damage, but that this sparing is not absolute . At high levels of mAMPH-induced injury, damage to the nucleus accumbens, although less than that in the CPU, will become significant. We also report, as has been previously described (Broening et al., 1997; Burrows and Meschul, 1997), that the NAcC was more vulnerable to mAMPH-induced dopaminergic damage than NAcSh. Thus, the rat striatum exhibits marked heterogeneity to the effects of mAMPH, with the loss of DAT, DA, and TH in ventral CPu exceeding that seen for dorsal CPu, and with the NAcSh exhibiting relative, but not absolute sparing.
The hyperthermia that occurs during mAMPH administration can be an important contributing factor to the development of striatal DA terminal injury (Bowyer et al., 1994; Ali et al., 1994), and systemic NMDA receptor antagonist administration during mAMPH exposure blunts this hyperthermia (Albers and Sonsalla, 1995). In the present experiment, epidural DNQX provided neuroprotection against mAMPH-induced striatal DAT depletions while having no effect on mAMPH-induced hyperthermia. Epidural application of AP5 also protected against mAMPH-induced striatal DAT depletion while affecting mAMPH-induced hyperthermia at one of the four time points measured. This finding raises the question of how cortical NMDA receptors could contribute to body temperature, as measured subcutaneously. The Fos data shown in Figure 3 show that epidural AP5 had significant effects on the function of medial prefrontal cortex (mPFC). The mPFC is considered to be an autonomic motor area because of its dense innervations to hypothalamic nuclei that both receive temperature information (Shibata et al, 1988) and play a role in controlling cardiovascular responses (Chiba et al., 2001; Bennett, 2011). Prefrontal cortical stimulation can affect core and extremity body temperatures (see Van Eden and Buijs, 2000), and decortication or damage to frontal pole has been found to alter thermoregulatory responses of rodents in some studies (Blass, 1969; Monda et al, 1994) but not others (Osaka, 2003).
Separate from the potential contribution of PFC glutamatergic transmission to body temperature, the similarities between epidural DNQX and AP5 in the pattern and magnitude of the protection against mAMPH-induced DAT loss strongly suggest that the transitory effect of AP5 on body temperature does not explain the neuroprotective effects of the NMDA antagonist. Instead, the effects that these two ionotropic GLU receptor antagonists had in common, including their significant reduction of mAMPH-induced cortical and striatal Fos expression, point to the interpretation that their interference with corticofugal circuitry, and particularly corticostriatal GLU transmission, is the critical factor in their ability to blunt mAMPH-induced striatal DAT loss.
Summary and Conclusions
Epidural application of either the AMPA receptor antagonist, DNQX, or the NMDA receptor antagonist, AP5, attenuated the mAMPH-induced increases in Fos protein expression in cortical regions located under the ‘cortical well’ as well as in dorsal and ventral striatal subregions. The administration of a binge mAMPH regimen resulted in significant striatal dopaminergic terminal loss, as measured by DAT depletion, and this neurotoxicity was attenuated by frontoparietal epidural application of either GLU receptor antagonist. Each of these influences of epidural GLU receptor antagonism occurred heterogeneously through striatal subregions, in keeping with the topography of the projections from medial frontal cortical areas whose activity was most affected by the GLU antagonist applications.
Highlights.
Blockade of NMDA and AMPA receptors in frontal cortex blunts the cortical and striatal responses to systemic methamphetamine.
Application of glutamate receptor antagonists reduces methamphetamine-evoked c-fos induction in cortex and striatum.
Application of these antagonists attenuates methamphetamine-induced neurotoxicity of the striatal dopaminergic innervation.
Striatal regions most affected by cortical glutamate antagonism receive inputs from the most heavily affected cortical areas.
Corticostriatal neurotransmission plays an important role in methamphetamine’s activating and neurotoxic influences.
ACKNOWLEDGEMENTS
This work was supported by PHS grant DA-12204 to J.F.M. The authors would also like to extend their appreciation to Dr. Steven J. O’Dell for his insightful comments on the manuscript.
Abbreviations
- AMPA
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AP5
(DL)-amino-5-phosphonovaleric acid
- CG
Cingulate cortex
- CPu
Caudate putamen
- DL
Dorsolateral
- DC
Dorsocentral
- DM
Dorsomedial
- VL
Ventrolateral
- VC
Ventrocentral
- VM
Ventromedial
- DA
Dopamine
- DAB
Diaminobenzidine
- DAT
Dopamine transporter
- DNQX
Dinitroquinoxaline-2,3-dione
- GLU
Glutamate
- IEG
Immediate early gene
- mAMPH
Methamphetamine
- Mtx1
Primary motor cortex
- Mtx2
Secondary motor cortex
- NAcC
Nucleus accumbens core
- NAcSh
Nucleus accumbens core
- NMDA
N-methyl-D-aspartic acid
- PBS
Phosphate buffer saline
- PrL
Prelimbic cortex
- s.c
Subcutaneously
- SSX
Somatosensory cortex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- Albin RL, Makowie RL, Hollingsworth ZR, Dure LS, Penney JB, Young AB. Excitatory amino acid binding sites in the basal ganglia of the rat: a quantitative autoradiographic study. Neuroscience. 1992;46:35–48. doi: 10.1016/0306-4522(92)90006-n. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. doi: 10.1016/0166-2236(90)90107-l. [DOI] [PubMed] [Google Scholar]
- Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Welker E, Callahan CA. The contribution of NMDA and non- NMDA receptors to fast and slow transmission of sensory information in the rat SI barrel cortex. J Neurosci. 1993;13(5):2149–2160. doi: 10.1523/JNEUROSCI.13-05-02149.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher AM, O’Dell SJ, Marshall JF. Impaired object recognition memory following methamphetamine-, but not p-chloroamphetamine- or d-amphetamine-induced neurotoxicity. Psychopharmacology. 2005;30:2026–2034. doi: 10.1038/sj.npp.1300771. [DOI] [PubMed] [Google Scholar]
- Belcher AM, Feinstein EM, O’Dell SJ, Marshall JF. Methamphetamine influences on recognition memory: comparison of escalating and single-day dosing regimens. Neuropsychopharoacology. 2008;33:1453–1463. doi: 10.1038/sj.npp.1301510. [DOI] [PubMed] [Google Scholar]
- Bennett MR. The prefrontal-limbic network in depression: Modulation by hypothalamus, basal ganglia and midbrain. Prog Neurobiol. 2011;93(4):468–487. doi: 10.1016/j.pneurobio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Berretta S, Parthasarathy HB, Graybiel AM. Local release of GABAergic inhibition in the motor cortex induces immediate-early gene expression in indirect pathway neurons of the striatum. J Neurosci. 1997;17:4752–4763. doi: 10.1523/JNEUROSCI.17-12-04752.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta S, Sachs Z, Graybiel AM. Cortically driven Fos induction in the striatum is amplified by local dopamine D2-class receptor blockade. Eur J Neurosci. 1999;11:4309–4319. doi: 10.1046/j.1460-9568.1999.00866.x. [DOI] [PubMed] [Google Scholar]
- Blass EM. Thermaregulatory adjustments in rats after removal of the frontal poles of the brain. J Comp Physiol Psychol. 1969;69:83–90. doi: 10.1037/h0027924. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- Broening HW, Pu C, Vorhees CV. Methamphetamine selectively damages dopaminergic innervation to the nucleus accumbens core while sparing the shell. Synapse. 1997;27:153–160. doi: 10.1002/(SICI)1098-2396(199710)27:2<153::AID-SYN6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Burrows KB, Meshul CK. Methamphetamine alters presynaptic glutamate immunoreactivity in the caudate nucleus and motor cortex. Synapse. 1997;27(2):133–144. doi: 10.1002/(SICI)1098-2396(199710)27:2<133::AID-SYN4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Chang L, Alicata D, Ernst T, Volkow N. Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction. 2007;102 suppl 1:16–32. doi: 10.1111/j.1360-0443.2006.01782.x. [DOI] [PubMed] [Google Scholar]
- Chiba T, Kayahara T, Nakano K. Efferent projections of infralimbic and prelimbic areas of the medial prefrontal cortex in the Japanese monkey, Macaca fuscata. Brain Res. 2001;888(1):83–101. doi: 10.1016/s0006-8993(00)03013-4. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Bjorklund A. Transection of corticostriatal afferents reduces amphetamine-and apomorphine-induced striatal Fos expression and turning behaviour in unilaterally 6-hydroxydopamine-lesioned rats. Eur J Neurosci. 1993;5(8):1062–1070. doi: 10.1111/j.1460-9568.1993.tb00959.x. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Logan B, Laverty R. 3,4-Methylenedioxymethamphetamine induces Fos-like proteins in rat basal ganglia: reversal with MK 801. Eur J Pharmacol. 1991;206:255–258. doi: 10.1016/s0922-4106(05)80027-6. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Gaffney M, Weihmuller FB, O’Dell SJ, Marshall JF. Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res. 1992;598(1–2):321–326. doi: 10.1016/0006-8993(92)90201-j. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Gibb JW, Hanson GR. A rapid and reversible change in dopamine transporters induced by methamphetamine. Eur J Pharmacol. 1997;323(23):R9–R10. doi: 10.1016/s0014-2999(97)00148-9. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Globus MY, Busto R, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of ischemia on the in vivo release of striatal dopamine, glutamate, and gamma-aminobutyric acid studied by intracerebral microdialysis. J Neurochem. 1988;51(5):1455–1464. doi: 10.1111/j.1471-4159.1988.tb01111.x. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13(7):244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Young AB. Synaptic localization of striatal NMDA, quisqualate and kainate receptors. Neurosci Lett. 1989;101:133–137. doi: 10.1016/0304-3940(89)90519-3. [DOI] [PubMed] [Google Scholar]
- Gross NB, Marshall JF. Striatal dopamine and glutamate receptors modulate methamphetamine-induced cortical Fos expression. Neuroscience. 2009;16:1114–1125. doi: 10.1016/j.neuroscience.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Tsai SJ, Su TW, Sim CB. Effects of repeated high-dose methamphetamine on local cerebral glucose utilization in rats. Neuropsychopharmacology. 1999;21:427–434. doi: 10.1016/S0893-133X(99)00029-9. [DOI] [PubMed] [Google Scholar]
- Hotchkiss AJ, Morgan ME, Gibb JW. The long-term effects of multiple doses of methamphetamine on neostriatal tryptophan hydroxylase, tyrosine hydroxylase, choline acetyltransferase and glutamate decarboxylase activities. Life Sci. 1979;25(16):1373–1378. doi: 10.1016/0024-3205(79)90414-4. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Frey KA, Lundahl LH, Keenan P, Lockhart N, Roll J, Galloway GP, Koeppe RA, Kilbourn MR, Robbins T, Schuster CR. Cognitive function and nigrostriatal markers in abstinent methamphetamine abusers. Psychopharmacology (Berl) 2006;185(3):327–338. doi: 10.1007/s00213-006-0330-6. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Lyoo IK, Hwang J, Sung YH, Lee HY, Lee DS, Jeong DU, Renshaw PF. Frontal glucose hypometabolism in abstinent methamphetamine users. Neuropsychopharmacology. 2005;30(7):1383–1391. doi: 10.1038/sj.npp.1300699. [DOI] [PubMed] [Google Scholar]
- Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19(4):1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ED, Simon SL, Berman SM, Mandelkern MA, Lichtman AM, Bramen J, Shinn AK, Miotto K, Learn J, Dong Y, Matochik JA, Kurian V, Newton T, Woods R, Rawson R, Ling W. Mood disturbances and regional cerebral metabolic abnormalities in recently abstinent methamphetamine abusers. Arch Gen Psychiatry. 2004;61:73–84. doi: 10.1001/archpsyc.61.1.73. [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Haber SN. Organization of thalamostriatal terminals from the ventral motor nuclei in the macaque. J Comp Neurol. 2001;429:321–336. doi: 10.1002/1096-9861(20000108)429:2<321::aid-cne11>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- McGeorge AJ, Faull RL. The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29:503–537. doi: 10.1016/0306-4522(89)90128-0. [DOI] [PubMed] [Google Scholar]
- Mengual E, de las Heras S, Erro E, Lanciego JL, Gimenez-Amaya JM. Thalamic interaction between the input and the output systems of the basal ganglia. J Chem Neuroanat. 1999;16:187–200. doi: 10.1016/s0891-0618(99)00010-1. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- Monda M, Amaro S, De Luca B. Non-shivering thermogenesis during prostaglandinE1 fever in rats: role of the cerebral cortex. Brain Res. 1994;651:148–154. doi: 10.1016/0006-8993(94)90691-2. [DOI] [PubMed] [Google Scholar]
- Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- Osaka T. Thermogenesis elicited by skin cooling in anaesthetized rats: lack of contribution of the cerebral cortex. J Physiol. 2003;555:503–513. doi: 10.1113/jphysiol.2003.053215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20:91–127. doi: 10.1016/0165-0173(94)00007-c. [DOI] [PubMed] [Google Scholar]
- Paulus MP, Hozack N, Frank L, Brown GG, Schuckit MA. Decision making by methamphetamine-dependent subjects is associated with error-rate-independent decrease in prefrontal and parietal activation. Biol Psychiatry. 2003;53(1):65–74. doi: 10.1016/s0006-3223(02)01442-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain Atlas in Stereotaxic Coordinates. Ed. 5. Sydney, Australia: Academic Press; 2003. [Google Scholar]
- Pontieri FE, Crane AM, Seiden LS, Kleven MS, Porrino LJ. Metabolic mapping of the effects of intravenous methamphetamine administration in freely moving rats. Psychopharmacology (Berl) 1991;102:175–182. doi: 10.1007/BF02245919. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235(1):93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Salt TE, Meier CL, Seno N, Krucker T, Herrling PL. Thalamocortical and corticocortical excitatory postsynaptic potentials mediated by excitatory amino acid receptors in the cat motor cortex in vivo. Neuroscience. 1995;64(2):433–442. doi: 10.1016/0306-4522(94)00357-b. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: effects of amfonelic acid. Eur J Pharmacol. 1985;109:73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- Shibata M, Hori T, Kiyohara T, Nakashima T. Convergence of skin and hypothalamic temperature signals on the sulcal prefrontal cortex in the rat. Brain Res. 1988;443:17–46. doi: 10.1016/0006-8993(88)91596-x. [DOI] [PubMed] [Google Scholar]
- Smith Y, Raju DV, Pare JF, Sidibe M. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004;27:520–527. doi: 10.1016/j.tins.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Riordan DE, Heikkila RE. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J Pharmacol Exp Ther. 1991;256(2):506–512. [PubMed] [Google Scholar]
- Stephans SE, BK Yamamoto. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17(3):203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Trevitt JT, Morrow J, Marshall JF. Dopamine manipulation alters immediate-early gene response of striatal parvalbumin interneurons to cortical stimulation. Brain Res. 2005;1035:41–50. doi: 10.1016/j.brainres.2004.11.039. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. The newly synthesized pool of dopamine determines the severityof methamphetamine-induced neurotoxicity. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2007.05155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eden CG, Buijs RM. Functional neuroanatomy of the prefrontal cortex: autonomic interactions. In: Uylings HBM, Van Eden CG, De Bruin JPC, Feenstra MGP, Pennartz CMA, editors. Progress in Brain Research. vol. 126. New York: Elsevier; 2000. pp. 49–62. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association ofdopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Daunais JB, McGinty JF. NMDA receptors mediate amphetamine-induced upregulation of zif/268 and preprodynorphin mRNA expression in rat striatum. Synapse. 1994a;18:343–353. doi: 10.1002/syn.890180410. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Daunais JB, McGinty JF. Role of kainate/AMPA receptors in induction of striatal zif/268 and preprodynorphin mRNA by a single injection of amphetamine. Brain Res Mol Brain Res. 1994b;27:118–126. doi: 10.1016/0169-328x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- Wang JQ, McGinty JF. Acute methamphetamine-induced zif/268, preprodynorphin, and preproenkephalin mRNA expression in rat striatum depends on activation of NMDA and kainate/AMPA receptors. Brain Res Bull. 1996;39:349–357. doi: 10.1016/0361-9230(96)00002-0. [DOI] [PubMed] [Google Scholar]
- Weihmuller FB, O’Dell SJ, Marshall JF. MK-801 protection against methamphetamine-induced striatal dopamine terminal injury is associated with attenuated dopamine overflow. Synapse. 1992;11(2):155–163. doi: 10.1002/syn.890110209. [DOI] [PubMed] [Google Scholar]
- Westerberg E, Kehr J. The NMDA antagonist MK-801 reduces extracellular amino acid levels during hypoglycemia and prevents striatal damage. Neuosci Res Commun. 1988;3:151–158. [Google Scholar]