

Abstract
Intermittent nociceptive stimulation following a complete transection or contused spinal cord injury (SCI) has been shown to exert several short and long lasting negative consequences. These include maladaptive spinal plasticity, enhanced mechanical allodynia and impaired functional recovery of locomotor and bladder functions. The neurotrophin, brain derived neurotrophic factor (BDNF) has been shown to play an important role in adaptive plasticity and also to restore functions following SCI. This suggests that the negative behavioral effects of shock are most likely related to corresponding changes in BDNF spinal levels. In this study we investigated the cellular effects of nociceptive stimulation in contused adult rats focusing on BDNF, its receptor, TrkB, and the subsequent downstream signaling system. The goal was to determine whether the behavioral effect of stimulation is associated with concomitant cellular changes induced during the initial post-injury period.
Quantitative RT-PCR and western blotting were used to assess changes in the mRNA and/or protein levels of BDNF, TrkB and the downstream signaling proteins CAMKII and ERK1/2 at 1 hour, 24 hours and 7 days following administration of intermittent noxious shock to the tail of contused subjects. In addition, recovery of locomotor function (BBB score) was assessed daily for the first week post injury.
The results showed that, while nociceptive stimulation failed to induce any changes in gene expression at 1 hour, it significantly reduced the expression of BDNF, TrkB, ERK2 and CAMKII, at 24 hours. In general, changes in gene expression were spatially localized to the dorsal spinal cord. In addition, locomotor recovery was impaired by shock. Evidence is also provided suggesting that shock engages a neuronal circuitry without having any negative effects on neuronal survival at 24 hours. These results suggest that nociceptive activity following SCI decreases BDNF and TrkB levels, which may significantly contribute to diminished functional recovery.
Keywords: BDNF, spinal cord injury, functional recovery, spinal plasticity, nociceptive stimulation
Spinal cord injury (SCI) can result in a number of devastating consequences, which are evident at both cellular and behavioral levels. Common behavioral deficits include the loss of motor control, paralysis and pain. The extent to which these deficits occur depends largely on the cellular damage produced by the initial primary injury and the secondary processes, which occur from within hours to days post injury (Beattie et al., 2002; Whetstone et al., 2003; Rossignol et al., 2007). The contusion model of SCI provides a useful approach to examine not only the cellular processes occurring shortly after injury, but also the behavioral consequences of SCI. For example, a moderate contusion injury in rats produces near complete paralysis immediately post injury, followed by a period of recovery ranging from one to several weeks (Basso et al., 1995). Prior work suggests that long-term prognosis following SCI is well predicted by the extent of recovery observed over the first week of injury (Basso et al., 1996; Hook et al., 2004). As a result, much attention has focused on the biological changes that occur over this period of secondary injury.
Research from our laboratory has shown that pain (nociceptive) input to dermatomes below a contusion injury can profoundly affect long-term recovery. Just 6 minutes of intermittent nociceptive stimulation (applied to the leg or tail) produces both an acute disruption in locomotor performance and a long-term degradation in physiological function that leads to reduced tissue sparing, higher incidences of spasticity and mortality, delayed recovery of bladder function, and reduced weight gain 6 weeks after injury (Grau et al., 2004). These adverse effects are most pronounced if nociceptive stimulation is given within four days of injury and are only observed if the stimulation is uncontrollable (e.g., legshock given independent of leg position). An equivalent exposure to controllable stimulation (response-contingent stimulation; delivered when the leg is in an extended position) has no adverse effect on long-term recovery (Grau et al., 2004). These observations suggested that aberrant nociceptive input following SCI can have long lasting behavioral ramifications. Furthermore, that stimulation to the tail can influence the outcome of more distal spinal segments (lumbar), suggests that uncontrollable nociceptive stimulation following SCI engages a centrally-mediated mechanism that extends beyond the stimulated dermatomes. In addition, it suggests that the deleterious effects of stimulation depend on both peripheral (noxious stimulation) and central mechanisms.
To study how nociceptive input alters spinal function below an injury, we have also examined how electrical stimulation affects neurobiological mechanisms within the lumbar-sacral spinal cord in spinally transected rats. In this paradigm, rats undergo a complete transection at the second thoracic vertebra. In a typical experiment, intermittent stimulation, at an intensity that engages C (pain) fibers (Thompson et al., 1990; Hathway et al., 2009), is applied a day after injury and the impact of stimulation is tested over the next 24 hours (e.g. Crown and Grau, 2001). We found that intermittent stimulation inhibits a form of adaptive spinal plasticity (instrumental learning), the ability to learn a response-outcome relationship. Instead, it produces maladaptive plasticity, and enhances mechanical reactivity (allodynia) (Ferguson et al., 2006). In contrast, controllable stimulation facilitates spinal learning (Crown et al., 2002b). These earlier studies demonstrated the adaptive nature of the isolated spinal cord. At the cellular level, intermittent nociceptive stimulation down-regulates the expression of a number of genes involved in mediating various forms of plasticity, such as brain derived neurotrophic factor (BDNF), calcium-calmodulin kinase II (CAMKII) and CREB (Gómez-Pinilla et al., 2007).
The down regulation of BDNF by noxious stimulation is of special interest given the evidence suggesting that BDNF plays a key role in various forms of central plasticity (e.g. Patterson et al., 1996; Kerr et al., 1999; Garraway et al., 2003; Gómez-Pinilla et al., 2007). BDNF is also implicated in promoting axonal re-growth/elongation and in improving functional recovery following SCI (Xu et al., 1995; McTigue et al., 1998; Boyce et al., 2007). These studies, along with our prior observations, suggest that uncontrollable stimulation adversely affects recovery following a contusion injury by reducing the spinal expression levels of BDNF. It also implies a concomitant reduction in subsequent BDNF signaling targets, such as its receptor (tropomyosin-receptor-kinase [TrkB]), and downstream signaling proteins (CAMKII and extracellular signal-regulated kinases 1/2 [ERK1/2]) (Huie et al., unpublished results).
Most incidences of traumatic SCI are accompanied by tissue damage and noxious input from peripheral injury. Consequently, multiple cellular events can make significant contribution to maladaptive spinal plasticity such as, pain hypersensitivity and impaired behavioral recovery following SCI. These events include sensitization of spinal neurons (Hains et al., 2003; Lampert et al., 2008), excitotoxicity and cell death (Liu et al., 1999; Beattie et al., 2002; Xu et al., 2004; Kuzhandaivel et al., 2011), and increased glial activity (Frei et al., 2000; Hains and Waxman, 2006; Detloff et al., 2008). Despite these potential cellular events that can contribute to the long-term behavioral consequences of noxious stimulation, our previous studies have not identified the exact mechanism responsible.
Because BDNF plays an important role in adaptive plasticity, it can be proposed that actions, which produce maladaptive plasticity, decrease BDNF spinal levels and that deficits in BDNF levels contribute to the behavioral consequences of nociceptive stimulation. This study addresses this proposal by exploring the impact of intermittent nociceptive stimulation on key elements of the BDNF pathway in contused subjects, at three different time points during the first week, post injury. We further propose that changes in BDNF levels during this first week will be predictive of the long term recovery following SCI. Therefore, our goal is to assess the temporal and spatial changes in expression during the initial stages post contusion injury. By employing the contusion injury model, we can also assess the effect of noxious shock on the recovery of locomotion during this period and determine whether stimulation-induced behavioral effects are correlated with changes in gene expression.
Experimental procedures
Male Sprague-Dawley rats obtained from Harlan (Houston, TX) served as subjects. Rats were approximately 90-110 days old and weighed between 350 and 400 g. They were housed individually and maintained on a 12-hour light/dark cycle, with all behavioral testing performed during the light cycle. Food and water were available ad libitum. All experiments were carried out in accordance with NIH standards for the care and use of laboratory animals (NIH publications No. 80-23), and were approved by the University Laboratory Animal Care Committee at Texas A&M University. Every effort was made to minimize suffering and limit the number of animals used.
Surgery
Subjects were anesthetized with isoflurane (5%, gas). Once a stable level of anesthesia was achieved, the concentration of isoflurane was lowered to 2-3%. An area extending approximately 4.5 cm above and below the injury site was shaved and disinfected with iodine, and a 7.0 cm incision was made over the spinal cord. Next, two incisions were made on either side of the vertebral column, extending about 3 cm rostral and caudal to the T12-T13 segment. The dorsal spinous processes at T12-T13 were removed, and the spinal tissue exposed. The dura remained intact. For the contusion injury, the vertebral column was fixed within the MASCIS device (Gruner, 1992; Constantini & Young, 1994) and a moderate injury was produced by allowing the 10g impactor (outfitted with a 2.5 mm tip) to drop 12.5 mm. The wound was then closed with Michel clips.
To help prevent infection, subjects were treated with 105 units/kg Pfizerpen (penicillin G potassium) immediately after surgery and again 2 days later. For the first 24 hours after surgery, rats were placed in a recovery room maintained at 26.6° C. To compensate for fluid loss, subjects were given 2.5 ml of saline after surgery. Bladders were expressed twice daily (morning and evening) until the animals had empty bladders for three consecutive days, at the times of expressing.
Experiments
This study was conducted in three phases, each involving a separate cohort of subjects. The first experiment sought to establish the acute effect of injury (relative to sham-operated controls) and the impact of shock treatment on BDNF-TrkB signaling. In this experiment, the subjects were sacrificed one hour following noxious stimulation (1 hour group). Sixteen subjects were used in this experiment as follows: sham (n = 4), contusion-unshock (n = 6) and contusion-shock (n = 6). The second experiment was conducted to extend on the observations arising from experiment 1, by collecting tissue at 24 hours following noxious stimulation (24 hour group). In addition, to determine whether shock treatment had a differential effect across the dorsal/ventral regions of the spinal cord, the tissue was further divided to collect dorsal and ventral halves, which were subsequently processed for mRNA and protein, individually. We introduced this manipulation because concurrent work (reported in Huie et al; unpublished results), had revealed that controllable stimulation affected TrkB signaling in the dorsal, but not the ventral, cord. A total of 16 subjects [sham (n = 4), contusion-unshock (n = 6) and contusion-shock (n = 6)] were used in this experiment. A final experiment was conducted to extend on the observation that shock had significant effects at 24 hours. In this experiment, spinal cord tissue (dorsal and ventral) was collected 7 days after noxious shock (7 day group). Because our aim was to establish whether shock treatment had a lasting effect on BDNF-TrkB signaling in contused animals, we did not include uninjured rats in this experiment. Twelve subjects [contusion-unshock (n = 6) and contusion-shock (n = 6)] were used in this experiment. The data were collected and the results were analyzed individually for each of the three experiments. To conserve space, and to allow some comparison across time, the data are grouped by assay in the Results and figures.
Uncontrollable Intermittent Stimulation
Previously, we showed that just 6 minutes of intermittent shock to the tail exerts the same detrimental effects on adaptive plasticity as uncontrollable legshock (Crown et al., 2002a). In this study, we employed the tailshock paradigm to also assess the effect of nociceptive stimulation on locomotor recovery following SCI. Subjects were treated with electrical stimulation 24 hours after surgery (modified from Washburn et al., 2007). They were loosely restrained in Plexiglas tubes as previously described (Crown et al., 2002a; Grau et al., 2004). Electrical stimulation was applied to the tail using an electrode constructed from a modified fuse clip. The electrode was coated with electrode gel (Harvard Apparatus, Holliston, MA) and attached 2 cm from the tip of the tail with Orthaletic tape. Leads from the fuse clip were attached to a BRS/LVE shock generator (Model SG-903) and intermittent constant current 1.5-mA, AC (60 Hz) electrical stimulus was applied through the electrodes. Rats treated with uncontrollable intermittent stimulation received 180, 80-ms tail shocks on a variable time schedule with a mean inter-stimulus interval of 2 s (range 0.2 −3.8 s). Unshock subjects were placed in the restraining tubes for equal amount of time as shock subjects, had the electrodes attached, but did not receive the electrical stimuli.
Behavioral locomotor assessment
Twenty four hours after surgery, prior to shock, locomotor behavior was assessed using the Basso, Beattie, and Bresnahan (BBB) scale (Basso et al. 1995) in an open enclosure for all subjects to ensure the effectiveness of the contusion injury (for details, refer to Grau et al., 2004). Following baseline scores, the subjects were assigned to a treatment group (shock or unshock) in a manner that ensured that injury severity was balanced across groups prior to treatment. BBB scores were also collected daily from days 1–7 post-treatment for the 7 day subjects. Care was taken to ensure that the investigator scoring behavior was blind to the subject’s treatment group.
Sham controls
Eight rats underwent the surgical procedure described above, but did not receive a spinal cord injury. Four of these were sacrificed 25 hours following the surgical procedure with the other four sacrificed at 48 hours following. One centimeter of spinal cord section equivalent to and surrounding the lesion site (approximately L1-L3) was processed for RNA and total protein (as described below). These animals served as sham controls for the 1 hour and 24 hours following shock groups, respectively.
RNA extraction and RT-PCR
Animals were sacrificed at 1 hour, 24 hours or 7 days following shock or unshock treatment. All subjects were deeply anesthetized with pentobarbital (50mg/kg) and 1 centimeter of spinal cord around the lesion epicenter was rapidly removed. To determine the spatial (dorso-ventral) changes in the expression of genes of interest, the spinal cord tissue was further sectioned to yield dorsal and ventral portions in the 24 hours and 7 days after treatment groups. The cord was processed for the extraction of both total RNA (RNeasy Mini Kit; Qiagen, Valencia, CA) and total protein (see below). Total RNA (100 ng) was converted into cDNA using TaqMan EZ RT-PCR Core reagents (Applied Biosystems, Carlsbad, CA) and the mRNA levels of all targets were measured by TaqMan quantitative real-time (RT)-PCR using a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Carlsbad, CA.). β-actin served as a control gene. The sequences of probes, forward and reverse primers for β-actin, c fos and TrkB were obtained from Applied Biosystems, while the BDNF primer and probe sequences previously reported by Gómez-Pinilla et al. (2007) were obtained from Integrated DNA Technologies (Coralville, IA).
Subsequent to RNA extraction, total protein was extracted from the organic layer containing protein and DNA using the QIAzol™ lysis reagent protocol for isolation of genomic DNA and/or proteins from fatty tissue (Qiagen, Valencia, CA.). Following determination of the protein concentration, using the Bradford Assay (BioRad, Hercules, CA), protein samples were diluted in Laemmli sample buffer and stored at −80°C at known concentrations (usually 2-5μg/μl). Equal amounts (30μg; 40μg for BDNF) of total protein were subjected to SDS-PAGE using 12% Tris-HCl precast gels (Thermo Scientific, Rockford, IL) and then transferred to PVDF or nitrocellulose (for BDNF) membranes (Millipore, Bedford, MA). For quantification of non-phosphorylated proteins, the blots were blocked for one hour in 5% blotting grade milk (BioRad, Hercules, CA) in Tris-Buffered Saline Tween-20 (TBST), while blots for phosphorylated proteins were blocked in 5% bovine serum albumin in TBST. The blots were incubated in one of the following primary antibodies generated in rabbit for BDNF (1:100; #R-066-500 - Novus Biologicals, Littleton, CO), both the full-length (145kDa) and truncated (95kDa) forms of the TrkB receptor (1:500; #07-225 - Upstate Cell Signaling, Lake Placid, NY), ERK1/2 (1:2000; #06-182 - Millipore, Temecula, CA), pERK1/2 (1:500; #07-467 - Millipore, Temecula, CA) and pTrkB 145kDa (1:2000; #pY817 - Epitomic, Burlingame, CA); primary antibody generated in mouse for CAMKII alpha subunit (1:250; #05-532 - Upstate Cell Signaling, Lake Placid, NY) and β-tubulin (1:1000; #05-661 - Upstate Cell Signaling, Lake Placid, NY), overnight at 4° C. The following day, blots were washed in TBST (3 × 10 min) at room temperature, then incubated in HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies (1:5,000; #31460 or 31430, respectively; Pierce, Rockford, IL) for 1 hour at room temperature. Following another 3 × 10 min series of washes, the blots were developed with ECL (Pierce, Rockford, IL) and imaged with Fluorchem HD2 (Cell Biosciences, Santa Clara, CA). Ratios of the integrated densitometry of each protein of interest to β-tubulin (loading control) were calculated, normalized to sham controls and averaged for animals within each group.
Immunohistochemistry
Immunohistochemistry was performed to determine the effect of shock following contusion injury on the viability of neurons (NeuN expression) and also to determine the cellular expression pattern of c fos and TrkB. This was done at 24 hours after shock/unshock treatment, because at this time most changes in mRNA and protein levels were observed. An additional 9 subjects [contusion-unshock (n = 4) and contusion-shock (n = 5)] were anesthetized with pentobarbital (50mg/kg) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. The spinal cord was dissected and post-fixed in 4% PFA for 2 hours before being transferred to 30% sucrose for cryoprotection. Following at least 72 hours in sucrose, one centimeter of spinal cord around the lesion epicenter was removed and cut into 3 sections (rostral, lesion and caudal; approximately 3 mm each) and frozen into a mold with optimal cutting temperature (OCT) compound. Twenty microns (20μm) horizontal sections were cut with a cryostat (Leica Microsystems, Buffalo Grove, IL). The sections were subsequently used for immunohistochemistry as described below.
Light microscopic immunohistochemistry
Spinal sections were washed twice for 10 min each in Tris buffered saline (TBS), then quenched in 0.6% H2O2 in TBS for 30 min. Following a series of 3 × 5 min washes, the sections were incubated in blocking solution (3% normal goat serum, 0.1% Triton X-100 in TBS) for 30 min, then incubated in mouse anti-NeuN (1:400; #MAB377 - Millipore, Bedford, MA) in blocking solution, overnight at room temperature. The following day, the sections were first washed in cold TBS (2 × 10 min), and then incubated in biotinylated goat anti-mouse secondary antibody (1:250; Vector Laboratories, Burlingame, CA) for 2 hours at room temperature. Following a series of washes in TBS (3 × 5 min), immunoreactivity was detected with the avidin-biotin-peroxidase complex (ABC)-3,3-diaminobenzidinetetra-hydrochloride technique (Hsu et al., 1981).
Double fluorescent labeling
To determine the specific cell type expressing c fos and TrkB, double fluorescent labeling was conducted. Spinal sections were incubated in blocking solution for 1 hour. This was followed by incubation in a combination of rabbit anti- c Fos antibody (1:2000; #sc-52 - Santa Cruz Biotechnology, Santa Cruz, CA) or TrkB (1:250; # NB100-98826 - Novus Biologicals, Littleton, CO) with anti mouse markers for neurons (NeuN, 1:400), astrocytes (Glial fibrillary acidic protein; GFAP, 1:500; #610565 - BD Biosciences, San Diego, CA) or microglia (OX42/CD11b/c, 1:1000; #550299 - BD Biosciences, San Diego, CA) overnight at room temperature. The spinal sections were washed in cold TBS, and then incubated in the appropriate alexa fluor-conjugated secondary antibodies (goat anti-rabbit; #A11008 or goat anti-mouse; #A11030 - Invitrogen, Eugene, OR) for 1 hour at room temperature. Following another series of washes in TBS (3 × 5 min), the slides were mounted in Prolong Gold anti-fading mounting medium (Invitrogen, Eugene, OR). Serial images were taken with a confocal microscope (Olympus BX61 with a Plan Apo N 60x; N.A. = 1.42 oil immersion objective) using Olympus Fluoview version 5. Control slides for both light and fluorescent immunohistochemistry were exposed to diluted normal goat serum instead of the primary antibody.
Neuron count
The number of NeuN positive labeling in tissue obtained from subjects 24 hours following shock/unshock was estimated using the optical fractionator method (West et al., 1991). Unbiased estimation was performed on a modified light microscope (Olympus BX51) equipped with StereoInvestigator, version 9 (MBF Biosciences, Williston,VA). The region of interest was selected at low magnification (4x objective) and counting was performed at high magnification using a 40x; N.A = 0.9 Uplan SAPO objective (Olympus). Four randomly selected spinal cord sections of the lesion area were averaged per animal. The following parameters were used for the stereological analysis: height of optical dissector −20μm; base of optical dissector - 90 × 90μm; average grid size - 175 × 175μm; and average section thickness - 12μm.
Statistical Analyses
All data were analyzed using repeated measures analysis of variance (ANOVA) with an a priori alpha value of 0.05 or below considered significant (*, depicts significant effect of contusion injury, while #, depicts significant effect of noxious shock). Mean group differences were evaluated using Duncan’s New Multiple Range post hoc test. In both text and figures, all data are presented as Mean ± SEM.
Results
Previously, we showed that in adult rats with a complete T2 transection, uncontrollable intermittent shock inhibits adaptive plasticity (Grau et al., 1998) and causes a down-regulation in BDNF mRNA expression within the lumbar spinal cord (Gómez-Pinilla et al., 2007). The same intermittent stimulation also impairs recovery after a contusion injury (Grau et al., 2004). Here, we investigated the cellular pathways that may mediate the detrimental effects of intermittent noxious shock in spinal cord contusion subjects. Specifically, we assessed the mRNA and/or protein expression levels of BDNF, TrkB, pTrkB, the signaling proteins CAMKIIα, ERK1/2 and pERK1/2 at 1 hour, 24 hours and 7 days following noxious tailshock in spinal cord contused rats.
BBB Scores
Only subjects with a baseline BBB score less than 8 were used in this study. The mean baseline BBB score for shock and unshock subjects were 3.5± 0.5 and 3.8 ± 0.4, respectively, and did not differ prior to shock treatment (p>0.05).
We have previously shown that shock treatment impairs recovery and that this effect emerges over the first week of recovery (Grau et al., 2004). In the present study, a similar effect was observed in rats sacrificed 7 days after shock treatment (Fig. 1). An ANOVA confirmed that shock treatment had a significant effect (F(1, 10) = 7.25, p<0.05). While BBB scores generally increased across days (F(5, 50) = 41.9, p<.0001), the Day X Shock treatment interaction was not significant (F(5,50) < 1.0, p>.05). This suggests that the magnitude of the shock effect did not vary in a systematic way over days. We further assessed the magnitude of the shock effect over days using the Bonferroni t-test (one-tailed), which maintains the overall error rate at p<0.05. Significant differences were observed on days 2-6 (all t’s > 2.53, p<0.05). c fos
Fig. 1.
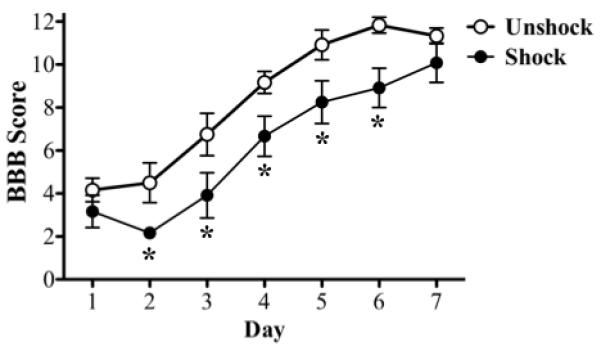
BBB score: Histogram shows the average locomotor performance of the rats. Shock subjects had significantly (p<0.05) lower BBB scores over the 7 days period than unshock subjects. Significant differences in shock and unshock subjects were observed on days 2-6 (*; p<0.05).
For additional confirmation of shock effects, we assessed the expression of c fos, a marker for neuronal activity. When compared to sham controls (1.0 ± 0.0), contusion-unshocks in the 1 hour group had elevated levels of c fos mRNA (fold change; 2.0 ± 0.2; p<0.05) (Fig. 2A). Intermittent tailshock induced an additional increase in c fos mRNA levels (3.6 ± 0.7). This change was significant compared to both unshock and sham controls (p<0.05). The increase in c fos waned over time and was not evident 24 hrs after shock treatment. c fos mRNA expression induced by shock was modestly elevated in both dorsal and ventral spinal cord. There was no difference in c fos mRNA levels in shock and unshock subjects at 7 days post treatment.
Fig. 2.
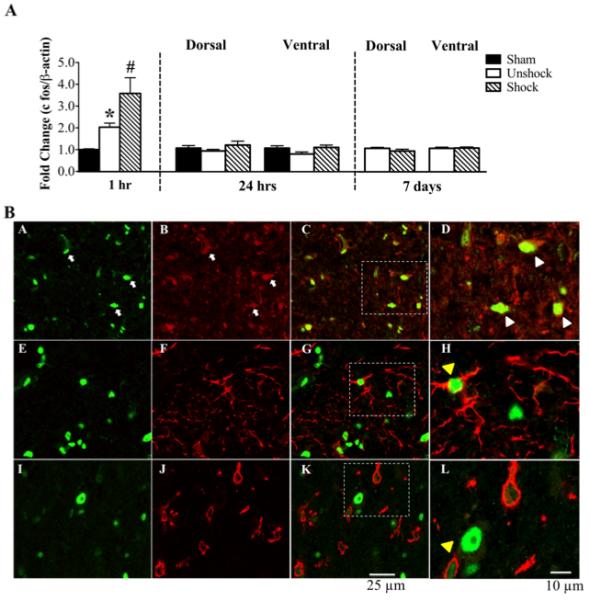
c fos mRNA and protein expression: A) qRT-PCR analyses show that both contusion injury alone (unshock) and shock significantly elevated c fos mRNA levels in the 1 hr group relative to sham controls. Shock significantly increased c fos levels above the increase induced by contusion injury alone. At 24 hr and 7 d post shock, there were no differences in c fos mRNA levels in either the dorsal or ventral spinal cords. [*, p<0.05; indicates significance relative to sham controls and #, p<0.05; indicates significance relative to unshock]. B) Double fluorescent labeling shows that c fos protein (green; A) is expressed primarily in neurons (red; B). Merged images in C and D indicate co-labeling of c fos with NeuN (yellow; white arrow heads). White arrows in A and B indicate c fos and NeuN positive labeling, respectively. There is no co-expression of c fos (green; E & I) with either GFAP (red; F & G) or OX42 (red; J & K). The enlarged images in H and L indicate that astrocytes and microglia are in close proximity to c fos positive labeling (yellow arrow heads), although not co-expressed.
Double fluorescent immunohistochemistry was performed at 24 hours post shock/unshock to assess the co-expression pattern of c fos with markers for neurons, astrocytes and microglia. Protein expression of c fos was increased in shock tissue compared to unshock tissue (not shown). Immunolabeling for c fos protein was observed throughout the dorso-ventral extent of the spinal cord, although labeling was most concentrated in the superficial and deep dorsal horn regions. As shown in Fig. 2B, c fos (green; A, E, I) was expressed primarily in neurons (red; B). Co-labeling with NeuN is shown as yellow in the merged images (C & D). There was no co-labeling of c fos with GFAP (red; F, G, H) or OX42 (red; J, K, L). These observations suggest that c fos is expressed in spinal cord neurons but not astrocytes or microglia.
BDNF
qRT-PCR and western blotting were conducted to determine the changes in the expression of BDNF mRNA and protein levels, respectively, at 1 hour, 24 hours and 7 days following shock.
As illustrated in Fig. 3A, neither contusion injury nor shock had any effect on BDNF mRNA levels compared to the sham controls in the 1 hour group. However at 24 hours, contusion injury significantly (p<0.05) decreased BDNF mRNA levels in both dorsal (fold change 0.6 ± 0.1) and ventral cords (0.6 ± 0.1) compared to sham controls. While shock had no effect on the BDNF mRNA levels at the 1 hour time point, it exerted differential effects on BDNF mRNA levels at 24 hours. In the dorsal spinal cord, shock produced an additional 42% decrease in BDNF mRNA levels relative to unshock (p<0.05), but exerted no effect ventrally. At 7 days, shock no longer had a significant effect on BDNF mRNA levels.
Fig. 3.
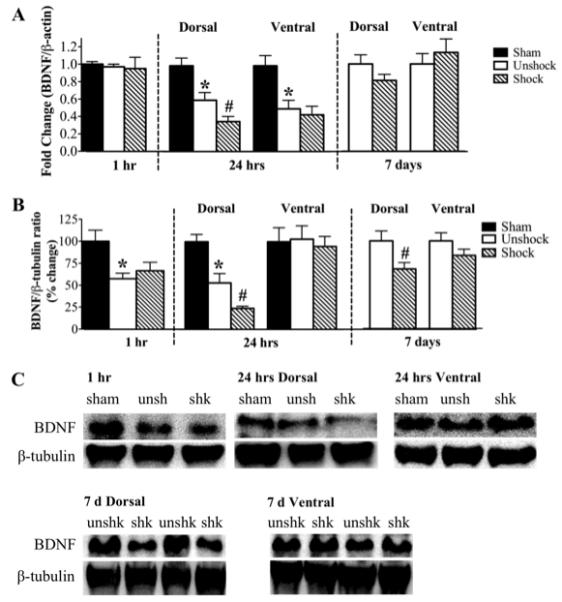
BDNF mRNA and protein expression: A) qRT-PCR analyses show that neither contusion injury alone (unshock) nor shock changed BDNF mRNA levels in the 1 hr group relative to sham controls. At 24 hr post treatment, BDNF mRNA levels in both dorsal and ventral spinal cords were significantly reduced in the unshock group. Shock additionally decreased BDNF mRNA levels in the dorsal spinal cord. At 7 d post treatment, BDNF mRNA levels returned to unshock levels. B) Western blotting shows that contusion injury alone significantly reduced BDNF protein levels, whereas shock had no additional effect relative to sham controls in the 1 hr group. At 24 hr and 7 d post treatment, shock significantly decreased BDNF levels in the dorsal spinal cord only. [*, p<0.05; indicates significance relative to sham controls and #, p<0.05; indicates significance relative to unshock]. C) Examples of representative western blot images for BDNF. β-tubulin served as a control gene.
The effect of intermittent shock on BDNF protein expression, illustrated in Figs. 3B & C, followed a similar trend as its effect on BDNF mRNA. In the 1 hour group, western blot analyses revealed a significant decrease in BDNF protein levels in both unshock and shock groups (57 ± 6% and 67 ± 10%, respectively; p<0.05) relative to sham controls. However, there was no difference in BDNF expression between these two groups. At 24 hours, contusion injury alone produced a significant decrease in BDNF protein levels in the dorsal cord compared to sham controls (53 ± 11 versus 100 ± 9; p<0.05). Shock induced additional reduction in BDNF protein levels to 24 ± 3% of sham controls. These changes in BDNF protein levels were only observed in the dorsal spinal cord, with neither contusion injury nor shock altering its expression in the ventral spinal cord. At 7 days, BDNF protein levels were still significantly decreased by shock in the dorsal spinal cord only (68 ± 7%; p<0.05) compared to unshock. Shock had no effect on BDNF levels ventrally.
TrkB
BDNF has high affinity for the TrkB receptor (Klein et al., 1991), through which BDNF mediates most of its cellular functions. To elucidate the interaction among shock, BDNF signaling and the shock-induced behavioral deficit, we also investigated the effect of intermittent shock on the mRNA and protein expression of the TrkB receptor at 1 hour, 24 hours and 7 days following shock.
Contusion injury alone induced a significant decrease in TrkB mRNA levels in the 1 hour group (0.45 ± .03; p<0.05) and in both dorsal (0.40 ± .02; p<0.05) and ventral spinal (0.34 ± .05; p<0.05) cords of the 24 hour group (Fig. 4Ai). Shock-induced change in TrkB mRNA levels was observed only in the dorsal spinal cord at 24 hours (p<0.05). In this group, TrkB mRNA levels were decreased by 30% compared to contusion unshock. As observed with BDNF mRNA levels, the effect of shock on TrkB mRNA levels was reversed by 7 days post shock.
Fig. 4.
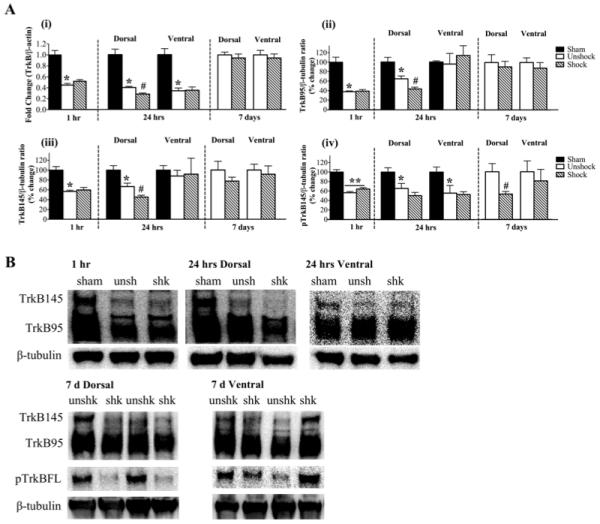
TrkB mRNA and protein expression: A) (i) Contusion injury alone significantly decreased TrkB mRNA at 1 hr post treatment compared to sham controls. Shock had no additional effect. At 24 hr, both dorsal and ventral levels were decreased in unshock subjects compared to sham controls. However, shock significantly reduced TrkB mRNA level in the dorsal spinal cord only. Shock had no effect on TrkB mRNA levels at 7 d. (ii) At 1 hr post treatment, TrkB95 protein levels were significantly reduced by contusion injury alone. At 24 hr, shock significantly reduced its levels in the dorsal spinal cord only. Shock had no effect on TrkB95 levels at 7 d. (iii) TrkB145 protein level was significantly decreased in the 1 hr group by contusion injury alone. At 24 hr, shock significantly reduced its levels in the dorsal spinal cord, but had no effect on the ventral levels. Shock had no effect on TrkB145 levels at 7 d. (iv) In the 1 hr group, pTrkB145 levels were significantly decreased by contusion injury alone. This decrease persisted at 24 hr in both dorsal and ventral spinal cords, although shock did not induce any additional effects. Dorsal levels of pTrkB145 were significantly decreased by shock in the 7 d group. [**, p<0.001, and *, p<0.05; indicate significance relative to sham controls and #, p<0.05; indicates significance relative to unshock]. B) Examples of representative western blot images for TrkB95, TrkB145 and pTrkB145. β-tubulin served as a control gene.
The TrkB receptor exists in truncated (95kD) and full-length (145kD) forms, but only the full-length receptor contains intracellular tyrosine kinase activity (Allendoerfer et al., 1994). Several studies have reported that both isoforms mediate cellular actions of BDNF and play a role in central plasticity (Seebach et al., 1999; Xu et al., 2000; Koponen et al., 2004; Gómez-Pinilla et al., 2007; Michaelsen et al., 2010). Western blotting was used to assess changes in the protein levels of the TrkB receptor, focusing on both the truncated and full-length isoforms of the receptor.
TrkB95
In the 1 hour group, contusion injury alone produced a significant decrease in the expression of the truncated TrkB (38 ± 2%; p<0.05) receptor compared to sham controls (Fig. 4Aii). Shock failed to induce any additional changes in the expression of the isoform in this group. However, at 24 hours, shock significantly decreased TrkB95 expression in the dorsal cord (p<0.05). This reduction was in addition to the decrease caused by the contusion injury (32%; p<0.05). The expression of TrkB95 was not altered by contusion injury or shock in the ventral spinal cord. In addition, the effect of shock seen in the dorsal cord was completely reversed at 7 days post treatment.
TrkB145
The effect of both injury and shock on the expression of the full-length TrkB receptor followed a very similar trend to that seen with the truncated TrkB receptor. In the 1 hour group, contusion injury alone significantly decreased TrkB145 expression (57 ± 3%; p<0.05) compared to sham controls (Fig. 4Aiii). Shock did not induce any additional effect on TrkB145 expression. At 24 hours, shock significantly decreased TrkB145 in the dorsal cord (33%; p<0.05), which was in addition to the reduction produced by contusion injury alone. Ventral cord expression of TrkB145 was not altered by contusion injury or shock at 24 hours. No differences were observed in TrkB145 expression at 7 days post shock. Examples of western blot images for TrkB expression at the various spatial and temporal points are shown in Fig. 4B.
TrkB immunohistochemistry
We used double fluorescent immunohistochemistry to further investigate the expression and cellular localization of TrkB receptors. Our results confirmed our previous observation in SC transected subjects that TrkB is expressed primarily in neurons (Huie, et al., unpublished results; also refer to Garraway et al., 2003). As shown in Fig. 5, there is extensive co-labeling of TrkB (green; A & E) with NeuN (red) in both the dorsal (B) and ventral (F) spinal cord (merged images, yellow; C & G, respectively). Interestingly, although not all TrkB positive labeling was neuronal in nature (white arrows; C & G); there was no obvious expression of TrkB (green; I & M) in astrocytes (GFAP, red; J & K) or in microglia (OX42, red; M & O). To further illustrate TrkB and NeuN, GFAP or OX42 co-labeling, the areas within the white boxes in C, G, K and O are enlarged and shown as D, H, L and P, respectively. These patterns of immunolabeling were observed in both shock and unshock tissue at 24 hours post treatment.
Fig. 5.
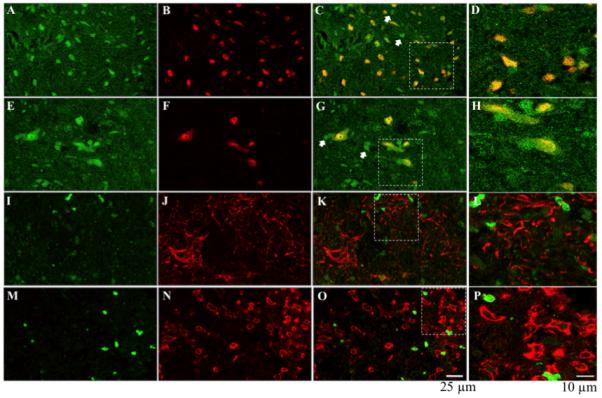
TrkB immunolabeling: TrkB (green; A, E, I & M) is expressed in both dorsal (NeuN: red; B & C) and ventral (red; F & G) neurons, but is not expressed in astrocytes (GFAP: red; J & K) or microglia (OX42: red N & O). Not all TrkB positive labeling is neuronal (white arrows, C & G). To further illustrate TrkB co-labeling with NeuN in the dorsal and ventral spinal cord, GFAP and OX42, the areas indicated by the white rectangles are enlarged in D, H, L & P, respectively.
Phospho-TrkB protein
Western blotting was used to assess the effect of shock on the expression of the activated (phosphorylated) full-length TrkB receptor (pTrkB145) (Fig. 4Aiv). One hour post treatment, the expression of pTrkB145 was significantly decreased in both unshock (56 ± 3%) and shock (64 ± 3%) groups compared to sham controls (**; p<0.001). There was no additional effect of shock. At 24 hours, contusion injury alone significantly decreased pTrkB145 levels in both the dorsal (66 ± 11%) and ventral spinal cords (56 ± 16%) (p<0.05). However, shock did not induce any additional alteration in pTrkB145 expression. At 7 days post shock, pTrkB145 expression was significantly decreased by shock in the dorsal spinal cord (53 ± 6%) compared to unshock (100 ± 17%) (p<0.05), but was unchanged in the ventral spinal cord. In addition to assessing pTrkB145 to β-tubulin ratios, we also assessed the ratio of pTrkB145 to TrkB145 levels. The ratio of pTrkB145/TrkB145 was significantly reduced only at 7 days in the spinal dorsal cord compared to the unshock group (71 ± 10 versus 100 ± 8; not shown). No other differences were observed in this ratio. Examples of western blot images for pTrkB expression at 7 days are shown in Fig. 4B.
Protein kinase signaling
BDNF signaling has been shown to involve recruitment and activation of several signaling pathways and kinases), including MAPK/ERK (e.g. Patapoutian and Reichardt, 2001; Slack et al., 2004). Previously, we showed that controllable shock-induced spinal learning might also depend on increases in the expression and activation of CAMKII (Gómez-Pinilla et al., 2007). Therefore, in this study, we used western blotting to assess the effect of intermittent shock on kinase-mediated signaling pathways, focusing on the protein expression levels of CAMKII alpha subunit (CAMKIIα), ERK1/2 and pERK1/2.
CAMKIIα
Consistent with the pattern of gene regulation seen with BDNF and TrkB, CAMKIIα protein levels were significantly decreased in both contusion unshock (63 ± 7%) and contusion shock (74 ± 4%) groups in the 1 hour group compared to sham controls (p<0.05). However, there was no effect of shock compared to unshock. At 24 hours following shock, both injury and shock affected CAMKIIα protein levels, although these effects were only observed dorsally. Specifically, there was a significant decrease in CAMKIIα protein levels in the unshock group (74 ± 8%; p<0.05) compared to sham controls. Shock added to the decrease (48 ± 6%; p<0.05) compared to sham controls. This additional decrease in CAMKIIα protein in the shock group was significant compared to unshock subjects (p<0.05). Unlike the effects observed dorsally, neither contusion injury alone nor shock altered CAMKIIα protein levels in the ventral cord. However, the additive effect of both contusion injury and shock caused a significant decrease in CAMKIIα expression level in the ventral spinal cord of shock subjects (69 ± 6%; p<0.05) compared to just sham controls. At 7 days the effect of shock on CAMKIIα expression persisted. Furthermore, not only were the dorsal levels of CAMKIIα significantly decreased by shock (67 ± 5%; p<0.05), ventral levels were also significantly reduced (65 ± 10%; p<0.05) relative to unshock. CAMKIIα expression is depicted in Fig. 6Ai.
Fig. 6.
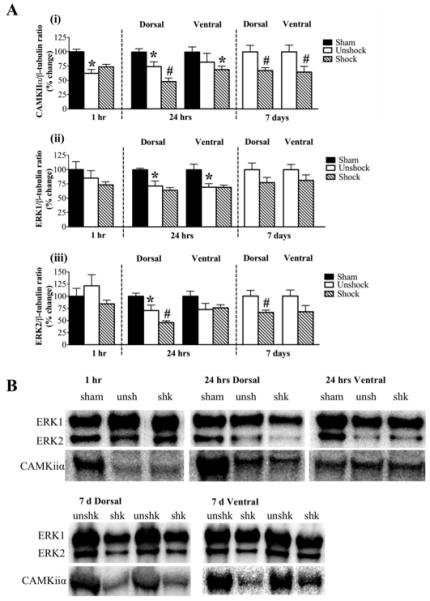
Protein kinase expression: A) (i) CAMKIIα protein level is significantly reduced by contusion injury only at 1 hr and in the dorsal cord at 24 hr. At 7 d, shock significantly reduced both dorsal and ventral levels of CAMKIIα. (ii) ERK1 levels are decreased by contusion injury alone at 24 hr. (iii) Contusion injury alone decreased the dorsal levels of ERK2 at 24 hr. In addition, shock significantly decreased ERK2 levels in the dorsal cord at both 24 hr and 7 d post treatment. [*, p<0.05; indicates significance relative to sham controls and #, p<0.05; indicates significance relative to unshock]. B) Examples of western blot images for ERK1, ERK2 and CAMKIIα for each time point are shown in to the right of each histogram. β-tubulin (not shown) served as a control gene.
ERK1
As illustrated in Fig. 6Aii, neither contusion injury nor shock had any effect on ERK1 protein expression level in the 1 hour group. At 24 hour after shock, ERK1 expression in both dorsal and ventral spinal cords was significantly decreased in both contusion groups compared to sham controls. However, shock had no additional effect on ERK1 expression at this time point or at 7 days.
ERK2
As seen with ERK1 protein expression, neither contusion injury nor shock had any effect on ERK2 protein expression level in the 1 hour group. However at 24 hours, shock produced a significant decrease (46 ± 4%; p<0.05) in ERK2 expression in the dorsal cord. This effect was in addition to the significant decrease produced by the contusion injury (unshock; 71 ± 11% compared to sham; 100 ± 6%; p<0.05). ERK2 levels were modestly decreased by contusion injury in the ventral spinal cord at 24 hours following shock treatment. This decrease was not significant nor did shock exert any additional effect on ERK2 levels. Shock-induced decrease in ERK2 levels in the dorsal spinal cord persisted for up to 7 days post shock (66 ± 5%) compared to unshock (100 ± 12%; p<0.05). Changes in ERK2 expression is represented in Fig. 6Aiii. Examples of western blot images for CAMKIIα, ERK1 and ERK2 expression are shown in Fig. 6B.
PhosphoERKs expression
We also investigated the effect of shock on the expression of activated ERK1 (pERK1) and ERK2 (pERK2) levels (Fig. 7). Neither contusion injury nor shock exerted any effect on the expression of pERK1 at 1 or 24 hours compared to sham controls. However at 7 days, only in the dorsal spinal cord was pERK1 level significantly decreased by shock (65 ± 13%; p<0.05) (Fig. 7A). Neither contusion injury nor shock induced any spatial or temporal changes in pERK2 levels (Fig. 7B). Although dorsal pERK2, ventral pERK1 and pERK2 levels were modestly decreased by shock at 7 days, none of these changes were significant. We also assessed the ratios of phosphorylated ERKs to total ERKs (pERK1/ERK1 and pERK2/ERK2). There were no differences in these ratios in any of the spatial or temporal groups.
Fig. 7.
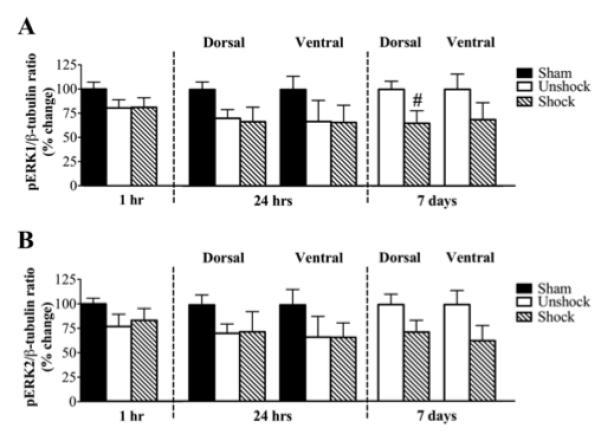
Expression of pERK1 and pERK2: A) Neither contusion injury nor shock altered the expression of pERK1 at 1hr or 24 hr. At 7 d post shock, pERK1 levels were significantly (#, p<0.05) reduced by shock in the dorsal spinal cord only. B) pERK2 levels were not affected by contusion injury or shock at any time point.
Neuron positive labeling and count
The total number of NeuN positive labeling in the lesion area of the spinal cord was estimated with optical fractionator to determine the overall effect of intermittent shock on neuronal survival. Because most of the shock effects were observed at 24 hours post treatment, immunolabeling was conducted on spinal sections obtained from contusion unshock and contusion shock subjects, 24 hours post treatment. The results of the unbiased stereological analysis are summarized in Table 1. As shown in Fig. 8A, there was no difference in the average estimated number of neurons in the shock (1302 ± 179; n = 5) and unshock (1162 ± 129; n = 4) groups (p<0.05). Examples of spinal cord sections and NeuN labeling from unshock (i, iii and v) and shock (ii, iv and vi) subjects respectively, are shown in Fig. 8B. For comparison, NeuN labeling immediately rostral (vii) and caudal (viii) are also shown. These sections were taken from a shock subject.
Table 1.
Summary of estimated neuronal count
| Subject | Average # of sections |
Average # of neurons |
SEM | CE |
|---|---|---|---|---|
| Unshock #1 | 4 | 1518 | 143 | 0.05 |
| Unshock #2 | 4 | 934 | 61 | 0.07 |
| Unshock #3 | 4 | 1178 | 254 | 0.07 |
| Unshock #4 | 4 | 1020 | 51 | 0.07 |
| Shock #1 | 4 | 1364 | 129 | 0.06 |
| Shock #2 | 4 | 1555 | 442 | 0.06 |
| Shock #3 | 4 | 1795 | 342 | 0.05 |
| Shock #4 | 4 | 867 | 79 | 0.07 |
| Shock #5 | 4 | 928 | 99 | 0.07 |
Fig. 8.
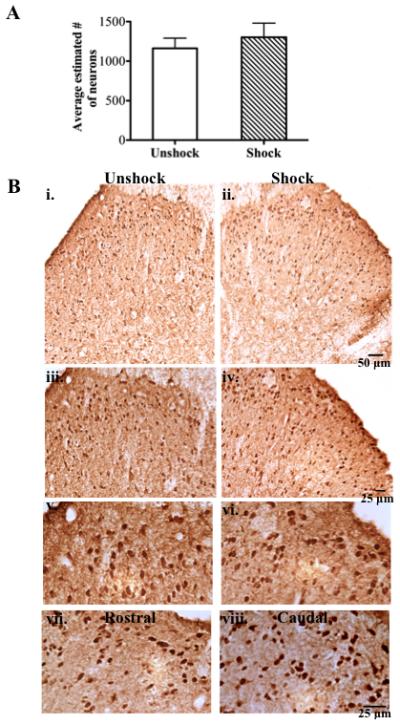
NeuN immunoreactivity: A) Average estimated number of NeuN positive labeling (viable neurons) at 24 hr post shock is unchanged relative to unshock. B) Low magnification (10x) images showing the overall integrity and NeuN labeling in the dorsal horn of an unshock (i) and shock (ii) subject. NeuN labeling from the superficial dorsal is shown at higher magnifications for unshock (iii; 20x & v; 40x) and shock (iv; 20x & vi; 40x) subjects. NeuN labeling from spinal sections of a shock subject immediately rostral (vii) and caudal (viii) to the lesion are shown for comparison.
Correlations with Behavioral Recovery
Additional analyses were conducted to examine whether the behavioral recovery observed in subjects sacrificed at day 7 was correlated with expression of BDNF, TrkB, and downstream signaling proteins. For these analyses, the average BBB score observed after treatment (Days 2-7) was correlated with cellular expression. Given our moderate sample size (six per condition), a robust correlation (r>0.81) was required for statistical significance. Interestingly, the two strongest correlations emerged from our measures of BDNF and TrkB mRNA expression (Fig. 9). In unshock rats, locomotor recovery was positively correlated with an increase in BDNF and TrkB expression (r = 0.78 and r = 0.93 respectively; Figs. 9A and C). No such correlation was observed in shock subjects (Figs. 9B and D). Further analysis showed that, for TrkB mRNA expression, the correlations observed for shock and unshock subjects were significantly different (z = 2.48, p<0.05). No significant correlations were observed between BBB performance and protein expression.
Fig. 9.
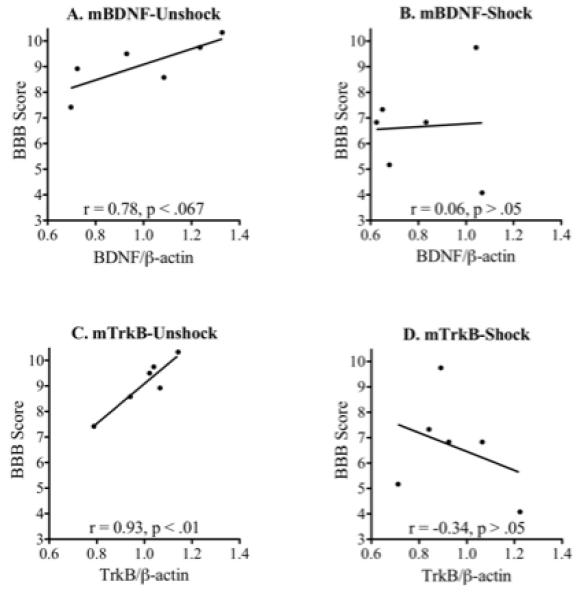
Behavioral correlation: BDNF mRNA expression was moderately correlated with locomotor recovery in unshock (A) but not shock (B), subjects. A strong correlation was observed between TrkB expression and locomotor performance in unshock (C), but not shock (D), contused rats.
Discussion
In several previous studies, we have identified conditions that undermine plasticity or recovery of locomotor functions following SCI. For example, administration of intermittent stimulation to the leg or tail disrupts adaptive plasticity and induces a learning deficit, which lasts up to 48 hours in completely transected rats (Crown et al., 2002a). This stimulation paradigm undermines the recovery of locomotor functions in spinal contused rats (Grau et al., 2004). In addition to electrical stimulation, peripheral inflammation has also been shown to undermine spinal adaptive plasticity (Hook et al., 2008). These early studies generally focused on the behavioral consequences of noxious shock rather than the underlying cellular mechanisms. Further, no prior work had explored the mechanisms that underlie the long-term effects of uncontrollable stimulation on recovery after a contusion injury. In the present study, we examined the molecular modifications produced by noxious tailshock in adult rats following a spinal cord contusion injury to determine the cellular mechanisms that may account for these behavioral deficits. In addition, by utilizing the contusion injury model, we were able to evaluate the recovery of locomotor functions during the first week post injury, a critical period in long-term recovery following SCI. We observed that noxious shock significantly altered BDNF-TrkB signaling following contusion injury and that these effects were both temporally and spatially regulated. Specifically, shock consistently decreased BDNF, TrkB and several downstream signaling kinases at 24 hours in the dorsal spinal cord. Although the predominant effects of shock on mRNA and protein levels were observed at 24 hours, its effect on BDNF, CAMKIIα, ERK2 and pERK1 protein levels persisted up to 7 days. In addition, we also replicated previous observations that shock significantly undermines recovery of locomotor functions following contusion SCI (Grau et al., 2004). Furthermore, a positive trend existed between BDNF/TrkB mRNA expression and behavioral recovery in unshock, but not shock, subjects.
At the molecular level, the current findings are consistent with our prior work in spinally transected rats demonstrating that uncontrollable shock decreases BDNF mRNA levels relative to both unshock and controllably shocked subjects, with the latter exhibiting elevated levels of BDNF (Gómez-Pinilla et al., 2007). Similarly, at the behavioral level controllable shock exerts actions that are opposite to that of uncontrollable shock. For instance, unlike the detrimental effect of uncontrollable shock reported both here and previously, controllable shock protects the recovery of locomotor functions in rats with a moderate contusion injury (Grau et al., 2004). These observations create a potential link between spinal levels of BDNF and functional recovery after SCI; treatments that decrease BDNF spinal levels exert additional detrimental effects following SCI. In addition, they corroborate several previous studies showing the restorative capabilities of neurotrophic factors, BDNF and neurotrophin 3 following SCI, at both cellular (Jin et al., 2002; Tobias et al., 2003) and behavioral (Coumans et al., 2001; Boyce et al., 2007) levels.
It is not surprising that SCI alone significantly decreased several genes. Previous studies have reported similar effects of SCI on BDNF and/or TrkB expression (King et al., 2000; Liebl et al., 2001; Hajebrahimi et al., 2008). Although SCI alone decreased gene expression, the effect of shock was clearly distinct from the injury-induced effects. Most genes were both spatially and temporally regulated by intermittent shock. In general, the changes occurred at 24 hours and were localized to the dorsal spinal cord. This is unlike the less selective effects of SCI, which were observed at both 1 hour and 24 hours in the dorsal and ventral spinal cord (approximately 25, and 48 hrs after injury, respectively). It is conceivable that the reduction in gene expression caused by the SCI may be related to injury-induced neuronal cell death. Several studies have noted that within the first 24 hours following SCI, there is a tremendous amount of apoptotic and necrotic neuronal and glial cell death (Liu et al., 1997; reviewed by Beattie et al., 2000, 2002). While a decrease in viable cells, specifically neurons, can cause a dramatic decrease in gene expression, it is evident here that increased neuronal loss is not responsible for the behavioral deficit caused by intermittent shock for the following reasons. First, shock does not induce any additional decrease in NeuN immunoreactivity, which would be indicative of neuronal loss, at the time when the shock effects are most prevalent. Previously, we reported that shock significantly increases tissue loss in the region of the injury, when examined 42 days post shock (Grau et al., 2004). Our current observation suggests that the short-term behavioral and cellular changes induced by shock are not related to concomitant neuronal death. In addition, our immunohistochemical analyses reveal that shock increases c fos protein expression in spinal neurons, suggesting that shock is engaging a neuronal circuitry. Second, the behavioral deficit seen in transected subjects (Grau et al., 1998; Crown et al., 2002a) and in contusion subjects both here and as reported in Grau et al. (2004) is induced only by uncontrollable or intermittent shock and not merely by SCI or shock per se. Third, while it is clear that SCI can induce neuronal and glial cell death soon after injury (Liu et al., 1997), what is critical in the present study is whether the short-term effect of shock treatment in injured rats (relative to unshocked injured rats) is due to enhanced cell loss. In the long-term (6 weeks after injury), shock treatment can increase tissue loss (Grau et al., 2004). However, within the time frame studied here (at least at 48 hours after SCI, when cellular changes are most robust), there was no evidence of a shock-induced cell loss. During this period of secondary injury, alterations in function may be tied to changes at a cellular level (e.g., decreased BDNF-TrkB levels) that set the stage for impaired recovery and further cell loss.
Together with these previous reports, our current study shows that intermittent shock to dermatomes distal to a contusion injury, engages a very selective cellular pathway that is maladaptive in nature and capable of producing short-term and long-lasting detrimental effects. This pathway is opposite to that induced by controllable shock, which results in corrective and beneficial effects (Grau et al., 2004).
The present study is consistent with past work reporting the beneficial effects of BDNF-TrkB signaling following SCI. For example, administrations of exogenous BDNF (Jin et al., 2002), or training paradigms that increase its endogenous levels (Gómez-Pinilla et al., 2002; Ying et al., 2005) provide positive outcomes following SCI. Thus, measures that are taken to compensate for BDNF loss following injury are expected to provide some potential therapeutic benefits, while those that decrease BDNF may have devastating consequences. Here we show that a nociceptive stimulation paradigm that overlaps with that used to induce wind-up (0.5Hz) (Mendell and Wall, 1965; Mendell, 1966) not only produces detrimental effects such as a learning deficit and impairment of locomotor recovery, but also dramatically decreases the expression of BDNF, TrkB and several signaling kinases in the dorsal spinal cord. Hence, we can deduce that adverse nociceptive stimulation following SCI is potentially detrimental to future recovery or adaptive plasticity. Interestingly, this relation is not unique to the spinal cord, but has been reported elsewhere. Rasmusson et al. (2002) showed that footshock-induced stress caused a down-regulation of BDNF mRNA level in the hippocampus, while more recently; Barichello et al. (2010) showed that meningitis-induced memory impairment is associated with a decrease in hippocampal BDNF levels. Throughout the CNS, it appears that down-regulation of BDNF has a detrimental effect.
While numerous reports have shown that BDNF can promote adaptive plasticity, it must also be recognized that an increase in BDNF could adversely affect some spinal processes. These paradoxical effects of BDNF can ultimately limit its therapeutic benefits. For instance, BDNF has been implicated in central sensitization in the spinal cord (Mannion et al., 1999), the neural mechanism proposed to underlie inflammatory and chronic pain. Also, BDNF induces synaptic facilitation of pain fiber evoked responses in neonatal rat spinal cord (Garraway et al., 2003), increases phosphorylation of the NMDAR1 subunit (Slack et al., 2004) and treatments that increase endogenous BDNF have contributed to pain hypersensitivity (Kerr et al., 1999). These studies, although not conducted in SCI subjects, suggest that BDNF is pro-nociceptive, at least in the naïve spinal cord. While several studies suggest that elevated levels of BDNF in the naïve spinal cord can have a pro-nociceptive effect, relatively few studies have examined the relation between BDNF and nociception following SCI. Hutchinson et al. (2004) addressed this issue in contused subjects undergoing treadmill training. They found that this training restored BDNF mRNA to control levels and attenuated allodynia. However, these effects were observed at a much later time point (three weeks or more following injury), suggesting again that impact of BDNF on recovery and function may vary over time after injury. Consequently, given the prevalence of pain following SCI; nearly 70% of patients experience pain, (e.g. Siddall & Loeser, 2001; Felix et al., 2007), there is a need to carefully evaluate BDNF functions; given the likelihood that it too can produce pain hypersensitivity.
Our current work suggests that the cellular mechanisms that underlie the adverse effect of nociceptive stimulation are neuronal and dorsally mediated. The first observation in support of this is the fact that c fos labeling was observed only in neurons. In addition to being an immediate early gene, c fos has been linked to nociceptive activity and the expression of pain for over 2 decades (e.g. Bullitt, 1989). Similarly, TrkB labeling was observed primarily in neurons, with no evidence of glial expression. While these observations suggest that activation of TrkB by BDNF and subsequent engagement of downstream signaling protein will most likely recruit neuronal mechanisms, they do not rule out the involvement of glia cells. Furthermore, this study does not identify the source of spinal BDNF, which can be released from both primary afferents (Thompson et al., 1999) and activated microglia (Coull et al., 2005). Given that we previously reported that intrathecal administration of the glial blocker fluorocitrate blocks the induction of shock-induced learning deficit (Vichaya et al., 2009), we infer that two (or more) distinct pathways are involved in mediating the deleterious effects of intermittent shock: one that is glial-mediated and another that may involve a decrease in neuronal BDNF-TrkB signaling.
It is not surprising that shock exerted very similar effects on the expression of CAMKII and ERK1/2. These signaling proteins are also implicated in a variety of neural processes such as hippocampal LTP, learning and memory (Silva et al., 1992; Lisman et al., 2002; Selcher et al., 2003), spinal LTP (Yang et al., 2004; Pedersen et al., 2005; Xin et al., 2006; Zhou et al., 2008), and central sensitization and pain (Fang et al., 2002; Ji et al., 1999, 2002; Xu et al., 2008). Furthermore, because activation of TrkB can turn on these protein kinases, both post-translational and transcriptional effects, critical steps in adaptive plasticity, can be induced secondary to TrkB receptor activation. A decrease in BDNF and TrkB expression will unequivocally reduce the expression of the signaling kinases and ultimately diminish these processes. Therefore, these observations substantiate the idea that intermittent shock engages a very specific dorsal spinal cord neuronal pathway; one that prevents two processes (post-translation and transcription) that are necessary for beneficial plasticity, such as functional recovery and learning.
Conclusion
This study identifies a potential mechanism for stimulation-induced maladaptive plasticity following SCI. Stimulation that produces a learning deficit significantly decreased components of the BDNF pathway at the lesion site. While excess BDNF, indicative of peripheral injury or inflammation, can increase BDNF levels beyond normal and consequently lead to central sensitization or pain, following SCI, an increase in spinal BDNF levels can be restorative. On the other end of the spectrum, significant decreases in BDNF can result in a behavioral deficit and compromised locomotor recovery. We therefore propose that carefully titrated levels of BDNF are essential to maintaining adaptive spinal plasticity following SCI.
Highlights.
Noxious stimulation after SCI impairs the recovery of locomotor functions
Spinal BDNF and TrkB levels are decreased by noxious stimulation following SCI
Shock engages a neuronal circuitry in the dorsal spinal cord
Impaired locomotor recovery is associated with decreased BDNF-TrkB levels
Acknowledgement
This work was funded by National Institute of Neurological Disorders and Stroke (NS41548) and National Institute of Child Health and Human Development (HD058412). The authors wish to thank John Hartman for technical assistance and Dr Kevin Hoy for reviewing an earlier version of this manuscript.
List of Abbreviations
- ANOVA
analysis of variance
- BBB
Basso, Beattie and Bresnahan
- BDNF
brain-derived neurotrophic factor
- CAMKIIα
calcium-calmodulin kinase II, alpha subunit
- ERK
extracellular related kinase
- mRNA
messenger ribonucleic acid
- PBS
phosphate buffered saline
- PFA
paraformaldehyde
- qRT-PCR
quantitative real time-polymerase chain reaction
- SCI
spinal cord injury
- TBS
tris-buffered saline
- TBST
tris-buffered saline -Tween 20
- TrkB
tropomyosin-receptor-kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allendoerfer KL, Cabelli RJ, Escandón E, Kaplan DR, Nikolics K, Shatz CJ. Regulation of neurotrophin receptors during the maturation of the mammalian visual system. J Neurosci. 1994;14:1795–1811. doi: 10.1523/JNEUROSCI.14-03-01795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barichello T, Belarmino E, Jr, Comim CM, Cipriano AL, Generoso JS, Savi GD, Stertz L, Kapczinski F, Quevedo J. Correlation between behavioral deficits and decreased brain-derived neurotrophic [correction of neurotrofic] factor in neonatal meningitis. J Neuroimmunol. 2010;223:73–76. doi: 10.1016/j.jneuroim.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Li Q, Bresnahan JC. Cell death and plasticity after experimental spinal cord injury. Prog Brain Res. 2000;128:9–21. doi: 10.1016/S0079-6123(00)28003-5. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res. 2002;137:37–47. doi: 10.1016/s0079-6123(02)37006-7. [DOI] [PubMed] [Google Scholar]
- Boyce VS, Tumolo M, Fischer I, Murray M, Lemay MA. Neurotrophic factors promote and enhance locomotor recovery in untrained spinalized cats. J Neurophysiol. 2007;98:1988–1996. doi: 10.1152/jn.00391.2007. [DOI] [PubMed] [Google Scholar]
- Bullitt E. Induction of c-fos-like protein within the lumbar spinal cord and thalamus of the rat following peripheral stimulation. Brain Res. 1989;493:391–397. doi: 10.1016/0006-8993(89)91177-3. [DOI] [PubMed] [Google Scholar]
- Constantini S, Young W. The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg. 1994;80:97–111. doi: 10.3171/jns.1994.80.1.0097. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Coumans JV, Lin TT, Dai HN, MacArthur L, McAtee M, Nash C, Bregman BS. Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci. 2001;21:9334–9344. doi: 10.1523/JNEUROSCI.21-23-09334.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord: IV. Induction and retention of the behavioral deficit observed after noncontingent shock. Behav Neurosci. 2002a;116:1032–1051. doi: 10.1037//0735-7044.116.6.1032. [DOI] [PubMed] [Google Scholar]
- Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord: II. Evidence for central mediation. Physiol. Behav. 2002b;77:259–267. doi: 10.1016/s0031-9384(02)00859-4. [DOI] [PubMed] [Google Scholar]
- Crown ED, Grau JW. Preserving and restoring behavioral potential within the spinal cord using an instrumental training paradigm. J Neurophysiol. 2001;86:845–855. doi: 10.1152/jn.2001.86.2.845. [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix ER, Cruz-Almeida Y, Widerström-Noga EG. Chronic pain after spinal cord injury: what characteristics make some pains more disturbing than others? J Rehabil Res Dev. 2007;44:703–715. doi: 10.1682/jrrd.2006.12.0162. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Crown ED, Grau JW. Nociceptive plasticity inhibits adaptive learning in the spinal cord. Neuroscience. 2006;141:421–431. doi: 10.1016/j.neuroscience.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Frei E, Klusman I, Schnell L, Schwab ME. Reactions of oligodendrocytes to spinal cord injury: cell survival and myelin repair. Exp Neurol. 2000;163:373–380. doi: 10.1006/exnr.2000.7379. [DOI] [PubMed] [Google Scholar]
- Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci. 2003;18:2467–2476. doi: 10.1046/j.1460-9568.2003.02982.x. [DOI] [PubMed] [Google Scholar]
- Grau JW, Barstow DG, Joynes RL. Instrumental learning within the spinal cord: I. Behavioral properties. Behav Neurosci. 1998;112:1366–1386. doi: 10.1037//0735-7044.112.6.1366. [DOI] [PubMed] [Google Scholar]
- Grau JW, Washburn SN, Hook MA, Ferguson AR, Crown ED, Garcia G, Bolding KA, Miranda RC. Uncontrollable stimulation undermines recovery after spinal cord injury. J Neurotrauma. 2004;21:1795–1817. doi: 10.1089/neu.2004.21.1795. [DOI] [PubMed] [Google Scholar]
- Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992 Summer;9:123–126. doi: 10.1089/neu.1992.9.123. discussion 126-128. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Huie JR, Ying Z, Ferguson AR, Crown ED, Baumbauer KM, Edgerton VR, Grau JW. BDNF and learning: Evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience. 2007;148:893–906. doi: 10.1016/j.neuroscience.2007.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 2002;88:2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajebrahimi Z, Mowla SJ, Movahedin M, Tavallaei M. Gene expression alterations of neurotrophins, their receptors and prohormone convertases in a rat model of spinal cord contusion. Neurosci Lett. 2008;441:261–266. doi: 10.1016/j.neulet.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Hathway GJ, Vega-Avelaira D, Moss A, Ingram R, Fitzgerald M. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain. 2009;144:110–118. doi: 10.1016/j.pain.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook MA, Ferguson AR, Garcia G, Washburn SN, Koehly LM, Grau JW. Monitoring recovery after injury: procedures for deriving the optimal test window. J Neurotrauma. 2004;21:109–118. doi: 10.1089/089771504772695995. [DOI] [PubMed] [Google Scholar]
- Hook MA, Huie JR, Grau JW. Peripheral inflammation undermines the plasticity of the isolated spinal cord. Behav Neurosci. 2008;122:233–249. doi: 10.1037/0735-7044.122.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- Hutchinson KJ, Gómez-Pinilla F, Crowe MJ, Ying Z, Basso DM. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain. 2004;127:1403–1414. doi: 10.1093/brain/awh160. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Fischer I, Tessler A, Houle JD. Transplants of fibroblasts genetically modified to express BDNF promote axonal regeneration from supraspinal neurons following chronic spinal cord injury. Exp Neurol. 2002;177:265–275. doi: 10.1006/exnr.2002.7980. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King VR, Bradbury EJ, McMahon SB, Priestley JV. Changes in truncated trkB and p75 receptor expression in the rat spinal cord following spinal cord hemisection and spinal cord hemisection plus neurotrophin treatment. Exp Neurol. 2000;165:327–341. doi: 10.1006/exnr.2000.7480. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen E, Lakso M, Castrén E. Overexpression of the full-length neurotrophin receptor trkB regulates the expression of plasticity-related genes in mouse brain. Brain Res Mol Brain Res. 2004;130:81–94. doi: 10.1016/j.molbrainres.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Kuzhandaivel A, Nistri A, Mazzone GL, Mladinic M. Molecular mechanisms underlying cell death in spinal networks in relation to locomotor activity after acute injury in vitro. Front Cell Neurosci. 2011;5:9. doi: 10.3389/fncel.2011.00009. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert A, Hains BC, Waxman SG. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp Brain Res. 2006;174:660–666. doi: 10.1007/s00221-006-0511-x. [DOI] [PubMed] [Google Scholar]
- Liebl DJ, Huang W, Young W, Parada LF. Regulation of Trk receptors following contusion of the rat spinal cord. Exp Neurol. 2001;167:15–26. doi: 10.1006/exnr.2000.7548. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu D, Xu GY, Pan E, McAdoo DJ. Neurotoxicity of glutamate at the concentration released upon spinal cord injury. Neuroscience. 1999;93:1383–1389. doi: 10.1016/s0306-4522(99)00278-x. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Horner PJ, Stokes BT, Gage FH. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci. 1998;18:5354–5365. doi: 10.1523/JNEUROSCI.18-14-05354.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell LM. Physiological properties of unmyelinated fiber projection to the spinal cord. Exp Neurol. 1966;16:316–332. doi: 10.1016/0014-4886(66)90068-9. [DOI] [PubMed] [Google Scholar]
- Mendell LM, Wall PD. Responses of single dorsal cord cells to peripheral cutaneous unmyelinated fibres. Nature. 1965;206:97–99. doi: 10.1038/206097a0. [DOI] [PubMed] [Google Scholar]
- Michaelsen K, Zagrebelsky M, Berndt-Huch J, Polack M, Buschler A, Sendtner M, Korte M. Neurotrophin receptors TrkB.T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur J Neurosci. 2010;32:1854–1865. doi: 10.1111/j.1460-9568.2010.07460.x. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pedersen LM, Lien GF, Bollerud I, Gjerstad J. Induction of long-term potentiation in single nociceptive dorsal horn neurons is blocked by the CaMKII inhibitor AIP. Brain Res. 2005;1041:66–71. doi: 10.1016/j.brainres.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Rasmusson AM, Shi L, Duman R. Downregulation of BDNF mRNA in the hippocampal dentate gyrus after re-exposure to cues previously associated with footshock. Neuropsychopharmacology. 2002;27:133–142. doi: 10.1016/S0893-133X(02)00286-5. [DOI] [PubMed] [Google Scholar]
- Rossignol S, Schwab M, Schwartz M, Fehlings MG. Spinal cord injury: time to move? J Neurosci. 2007;27:11782–11792. doi: 10.1523/JNEUROSCI.3444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach BS, Arvanov V, Mendell LM. Effects of BDNF and NT-3 on development of Ia/motoneuron functional connectivity in neonatal rats. J Neurophysiol. 1999;81:2398–2405. doi: 10.1152/jn.1999.81.5.2398. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall PJ, Loeser JD. Pain following spinal cord injury. Spinal Cord. 2001;39:63–73. doi: 10.1038/sj.sc.3101116. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1178. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- Thompson SW, Bennett DL, Kerr BJ, Bradbury EJ, McMahon SB. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci U S A. 1999;96:7714–7718. doi: 10.1073/pnas.96.14.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SW, King AE, Woolf CJ. Activity-dependent changes in rat ventral horn neurons in vitro; summation of prolonged afferent evoked postsynaptic depolarizations produce a d-2-amino-5-phosphonovaleric acid sensitive windup. Eur J Neurosci. 1990;2:638–649. doi: 10.1111/j.1460-9568.1990.tb00453.x. [DOI] [PubMed] [Google Scholar]
- Tobias CA, Shumsky JS, Shibata M, Tuszynski MH, Fischer I, Tessler A, Murray M. Delayed grafting of BDNF and NT-3 producing fibroblasts into the injured spinal cord stimulates sprouting, partially rescues axotomized red nucleus neurons from loss and atrophy, and provides limited regeneration. Exp Neurol. 2003;184:97–113. doi: 10.1016/s0014-4886(03)00394-7. [DOI] [PubMed] [Google Scholar]
- Vichaya EG, Baumbauer KM, Carcoba LM, Grau JW, Meagher MW. Spinal glia modulate both adaptive and pathological processes. Brain Behav Immun. 2009;23:969–976. doi: 10.1016/j.bbi.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn SN, Patton BC, Ferguson AR, Hudson KL, Grau JW. Exposure to intermittent nociceptive stimulation under pentobarbital anesthesia disrupts spinal cord function in rats. Psychopharmacology (Berl) 2007;192:243–252. doi: 10.1007/s00213-007-0707-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. J Neurosci Res. 2003;74:227–239. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin WJ, Gong QJ, Xu JT, Yang HW, Zang Y, Zhang T, Li YY, Liu XG. Role of phosphorylation of ERK in induction and maintenance of LTP of the C-fiber evoked field potentials in spinal dorsal horn. J Neurosci Res. 2006;84:934–943. doi: 10.1002/jnr.21013. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Hughes MG, Ye Z, Hulsebosch CE, McAdoo DJ. Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Exp Neurol. 2004;187:329–336. doi: 10.1016/j.expneurol.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund’s adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–14096. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Guénard V, Kleitman N, Aebischer P, Bunge MB. A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol. 1995;134:261–72. doi: 10.1006/exnr.1995.1056. [DOI] [PubMed] [Google Scholar]
- Yang HW, Hu XD, Zhang HM, Xin WJ, Li MT, Zhang T, Zhou LJ, Liu XG. Roles of CaMKII, PKA, and PKC in the induction and maintenance of LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2004;91:1122–1133. doi: 10.1152/jn.00735.2003. [DOI] [PubMed] [Google Scholar]
- Ying Z, Roy RR, Edgerton VR, Gómez-Pinilla F. Exercise restores levels of neurotrophins and synaptic plasticity following spinal cord injury. Exp Neurol. 2005;193:411–419. doi: 10.1016/j.expneurol.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Zhou LJ, Zhong Y, Ren WJ, Li YY, Zhang T, Liu XG. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp Neurol. 2008;212:507–514. doi: 10.1016/j.expneurol.2008.04.034. [DOI] [PubMed] [Google Scholar]