

Abstract
Exposure to glucocorticoids (GCs) in early development can lead to long-term changes in brain function and behavior although little is known about the underlying neural mechanisms. Perinatal exposure to GCs alters adult anxiety and neuroendocrine responses to stress. Therefore, we investigated the effects of either late gestational or neonatal exposure to the GC receptor agonist dexamethasone (DEX), on apoptosis within the amygdala, a region critical for emotional regulation. DEX was administered to timed-pregnant rat dams from gestational day 18 until parturition, or postnatal day 4-6. Offspring were sacrificed the day following the last DEX treatment and tissue was processed for immunohistochemical detection of cleaved caspase-3, a marker for apoptotic cells. Prenatal DEX treatment significantly increased the number of cleaved caspase-3 positive cells in the amygdala of both sexes, largely due to increases within the medial and basomedial sub-regions. Postnatal DEX treatment also increased cleaved caspase-3 immunoreactivity within the amygdala, although effects reached significance only in the central nucleus of females. Overall, DEX induction of cleaved caspase-3 in the amygdala was greater following prenatal compared to postnatal treatment, yet in both instances elevations in cleaved caspase-3 correlated with an increase in pro-apoptotic Bax mRNA expression. Dual-label immunohistochemistry of cleaved caspase-3 and the neuronal marker NeuN confirmed that virtually all cleaved caspase-3 positive cells in the amygdala were neurons and a subset of these cells (primarily following postnatal treatment) expressed a GABAergic calcium binding protein phenotype (calbindin or calretinin). Together these results indicate that early developmental GC exposure induces neuronal apoptosis within the amygdala in an age, sex, and region dependent manner.
Keywords: dexamethasone, glucocorticoid, apoptosis, prenatal, neonatal, sex difference
Pregnant women commonly undergo synthetic glucocorticoid (GC) therapy, such as dexamethasone (DEX), to improve the survival rate of potentially preterm infants or prevent virilization of females at risk for congenital adrenal hyperplasia (CAH; Liggins and Howie, 1972; Ritzen, 2001). In the case of potentially preterm and preterm infants, DEX is administered to induce production of surfactant in the fetal lungs which increases their ability to function without the use of a ventilator (Liggins and Howie, 1972). As a result, the risk of chronic lung disease is reduced. Less commonly, women who are at risk for carrying a CAH fetus are given DEX to suppress fetal adrenal androgen production, thereby preventing fetal genital virilization (Vos and Bruinse, 2010). These treatments are performed despite evidence that they retard growth and dramatically reduce body weight (Seckl, 2001). Infants treated with DEX, both preterm or following preterm delivery, show detriments in cognitive and motor development as well as disrupted hypothalamic-pituitary-adrenal (HPA) axis function (Barrington, 2001; Jones, 2005; Karemaker et al., 2008; Guilherme et al., 2009).
In rodent models, perinatal GC exposure causes increases in anxiety-related behaviors, disruptions in sexual behaviors and HPA axis function, and memory deficits in adulthood (Hossain et al., 2008; Nagano et al., 2008; Holson et al., 1995; Brabham et al., 2000). These behavioral abnormalities correspond to alterations in brain development that have been described in both rodent and non-human primate models. For example, in rodents, GC exposure in early development results in smaller brain size/weight (Holson et al., 1995). Furthermore, within brain regions such as the hippocampus and cortex, developmental GC exposure can retard neuronal migration (Fukumoto et al., 2009), decrease cell proliferation (Noorlander et al., 2008), induce apoptosis (Yu et al., 2010), and reduce cell size (Kreider et al., 2006). It is thought that synthetic GCs such as DEX can exert effects early in development because of the immaturity of the blood-brain barrier (BBB), which, unlike the adult BBB, allows access of these substances into the brain (Arya et al., 2006).
Exposure to high levels of GCs, either following a stressor, or by administration, induces apoptosis in regions of the rodent brain including the hippocampus, hypothalamus, and cortex (Yu et al., 2010; Zhang et al., 2002; Tobe et al., 2005). The hippocampus has high concentrations of GR and in adulthood it has been shown to be particularly sensitive to death promoting effects of GCs. Aging mice and rats as well as juvenile rats show increased hippocampal apoptosis following DEX administration (Hassan et al., 1996; Li et al., 2010). Recent evidence also indicates that neonatal GC treatments increase programmed cell death within the hippocampus and cerebellum (Duksal et al., 2009; Noguchi et al., 2008). GCs may influence apoptosis by altering expression of apoptosis-related genes such as pro-apoptotic Bax and anti-apoptotic Bcl2. GCs induce expression of pro-apoptotic Bax and this increase corresponds with increased hippocampal apoptosis in adult rodents (Almeida et al., 2000). Furthermore, Bax deficient mice show decreased levels of apoptosis in response to DEX treatment compared to wild type mice (Almeida et al., 2000), suggesting that these GC induced increases in apoptosis are dependent on Bax gene regulation.
The amygdala regulates a variety of emotional behaviors (including fear, depressive and anxiety behaviors), emotional memory processing, and reactivity of the HPA axis to stressors (Whalen and Phelps, 2009). The rodent amygdala is divided into several subdivisions including the medial, central, and basolateral complex (comprises the lateral, basolateral, and basomedial portions) each of which control distinct emotion-related behaviors (Whalen and Phelps, 2009). Developmental alterations in amygdala morphology and function correlate with later disturbances in emotional behaviors in both humans and rodents (Yilmazer-Hanke et al., 2004; Sheline et al., 2001; Zhou et al., 2010). Early life stress, which may also include increased GC levels, alters the development of the amygdala resulting in compromised adult mental function in humans and rodents. For example, in humans, orphanage rearing is associated with an atypically large amygdala volume and an increased incidence of anxiety disorders (Tottenham et al., 2010). In rats, 7 and 25 day old offspring of dams exposed to late gestational stress have smaller amygdala volumes and a decreased number of both neurons and glia compared to unstressed controls (Kawamura et al., 2006). The fact that all amygdala areas express high levels of GRs (McGimsey et al., 1991; McEwen et al., 1975) supports the possibility that GCs can act directly within the amygdala and subsequently modulate behavioral outcomes.
Within the amygdala, subtypes of GABAergic neurons are critical for the regulation of amygdala functions, including anxiety/fear behaviors, which are altered in both rodents and children who were treated with DEX in early development (Wiltgen et al., 2009; Sanders and Shekhar, 1995a; Lajic et al., 2011). GABAergic neurons are found in high concentrations throughout amygdala sub-regions and are critical for normal amygdala activity (Paré and Smith, 1993). GABA expressing neurons can be classified according to their expression of calcium-binding proteins (CBPs) including calbindin, calretinin, and parvalbumin. These CBPs identify subpopulations of GABA-ergic neurons that have distinct cell morphology, projections, and firing patterns (Conde et al., 1994; Zaitsev et al., 2005). CBPs are highly expressed in the amygdala although the distributions of CBP expressing neurons differ by amygdala sub-region and age (Berdel and Moryś, 2000; Legaz et al., 2005). Nonetheless, nearly all sub-regions contain cells that express each of these phenotypes (Kemppainen and Pitkänen, 2000). Alterations in CBPs have also been linked to anxiety/fear related behaviors and depression in humans and rodents (Maciag et al., 2010; Helmeke et al., 2008; Yilmazer-Hanke et al., 2002). Furthermore, neonatal stress has been shown to cause region specific increases/decreases in the expression of CBPs in adult rodents and these alterations correspond with disturbances in emotional behavior (Seidel et al., 2008; Giachino et al., 2007).
In the present study we investigated the effects of both late gestational and postnatal GC exposure on apoptosis within the amygdala. Since humans and rodents undergo different developmental trajectories prior to and after birth, our late gestational treatment period of rats relates to a late second early third trimester exposure in humans and is therefore analogous to a prenatal GC therapy regimen administered to pregnant women at risk for premature delivery or for treatment of very preterm infants (Seckl, 2001). The postnatal treatment regimen in this study (postnatal day 4-6) corresponds to a late gestational period in humans and can therefore mimic a treatment regimen administered to late-term premature infants. The results of these studies show increased neuronal apoptosis in the amygdala following DEX treatment at both time points, with greater GC-induced apoptosis in the prenatal treated cohort. These effects occurred in an amygdala sub-region and sex-specific manner, and a subset of these apoptotic cells (primarily following postnatal treatment) expressed a CBP phenotype (Calbindin or Calretinin- immunoreactivity).
Method
Animals
Timed-pregnant Sprague-Dawley dams were obtained from Charles River Laboratories (Wilmington, MA) at 7 days gestation and were handled for 4 days (GD14-17) prior to treatment. Dams were singly housed in a vivarium with a 14/10 L/D cycle, lights on at 0600, with food and water available ad libitum. For prenatal treatment, dams were subcutaneously injected with DEX dissolved in ethanol (2%) and diluted in safflower oil (0.4 mg/kg) or ethanol/safflower oil alone once daily at 1000 on gestation days 18-21. Another cohort of rats was administered subcutaneous injections of DEX (0.4 mg/kg) or oil vehicle daily on postnatal days 4-6. This dosage was designed to mimic that which pregnant women and premature infants would receive during GC therapy (Crane et al., 2003) and is within the normal range of doses administered experimentally to rats (Duksal et al., 2009; Kreider et al., 2006). All procedures were approved by the Arizona State University Institutional Animals Care and Use Committee, under subcontract from the University of Arizona College of Medicine - Phoenix and were in accord with National Institutes of Health guidelines.
Perfusion and Tissue Processing
On the day following the final DEX injection, pups were cryoanaesthetized on ice and intracardially perfused with 4% buffered paraformaldehyde. Brains were removed from the skull and placed in the same fixative at 4°C overnight. The following morning brains were transferred into a 30% sucrose cryoprotection solution, where they remained at 4° C until sectioning. Fixed brain weights were obtained after removal of the olfactory bulb and brain stem. Subsequently, brain tissue was sectioned at 35μm into 2 (prenatal tx (P0)) or 3 (postnatal tx (P7)) alternate series at -20°C using a Leica CM3050S cryostat (Leica, Buffalo Grove, IL). Tissue was placed in cryopreservative at 4° C until immunohistochemistry was performed.
Immunohistochemistry
For cleaved caspase-3 immunohistochemistry, sections were rinsed in tris-buffered saline (TBS), incubated in 1% hydrogen peroxide and 0.3% Triton-X in TBS (TBS-TX) for 10 minutes, again rinsed in TBS, then incubated in 4% normal goat serum (NGS) in TBS-TX for 1 hour. After rinsing in TBS, tissue was incubated in primary antisera (cleaved caspase-3 (1:500, rabbit, Cell Signaling, Danvers, MA) in 4% NGS and TBS-TX overnight at room temperature. Tissue was then rinsed in TBS and incubated for 1 hour in biotinylated goat-anti rabbit antibody in TBS-TX (1:1000; Vector Laboratories, Burlingame, CA) followed by rinses in TBS and a 1 hour incubation in avidin-biotin complex (ABC Elite kit, Vector Laboratories, Burlingame, CA). Following rinses in TBS, tissue was then developed for visualization of cleaved caspase-3 positive cells using diaminobenzidine as the chromagen. Subsequently, sections were rinsed in TBS and mounted on gelatin-coated slides. Sections were counterstained with cresyl violet for visualization of amygdala sub-regions, and coverslipped using permount (Sigma Chemical Co., St. Louis MO).
Dual Label Immunohistochemistry
To determine the co-localization of cleaved caspase-3 and NeuN or CBPs in the neonatal rat brain we performed dual label immunohistochemistry. To visualize CBP positive cells we examined calbindin and calretinin immunoreactivity (-ir), but not parvalbumin-ir because it is not expressed in the rat amygdala until 17 days postnatal (Berdel and Morys, 2000). For cleaved Caspase-3-ir and NeuN-, Calbindin-, or Calretinin-ir double-labeling, free-floating sections were rinsed with TBS 3 times, then blocked with 4% normal goat serum in TBS-TX for 60 min. Sections were then incubated in cocktail of the anti-NeuN (1:1000, mouse, Millipore, Billerica, MA), Calbindin (1:10,000, mouse, Millipore), or Calretinin (1:1000, mouse, Millipore) and anti-cleaved caspase-3 (1:500, rabbit, Cell Signaling) overnight. Sections were then incubated in 1:500 goat anti-mouse Alexa 488 (NeuN, Calbindin, or calretinin) and goat anti-rabbit Alexa 546 (for cleaved-caspase-3) for 1 hour at room temperature. Tissue is then mounted and coverslipped with antifade reagent to preserve fluorescent signal (Vectashield with DAPI, Vector), light protected, and stored at 4°C.
Microscopy
Analysis of caspase-3 positive cells was conducted on a Zeiss Axioskop microscope equipped with Neurolucida v.7 software (MicroBrightField Inc, Williston, VT). Sub-regions of the amygdala (medial, basomedial, central, and basolateral) were identified by cresyl violet counterstain using a rat brain atlas (Paxinos and Watson, 2007), and cleaved caspase-3-ir cells were counted within these regions. Cleaved-caspase-3 ir cells were counted bilaterally within 3 consecutive sections and the mean of these counts was used as a single value for each amygdala sub-region for each animal. For quantification of caspase-3/NeuN, caspase-3/Calbindin, and caspase-3/Calretinin double labeling, 3 images of the amygdala containing either the medial, basomedial, or central portions of the amygdala, within 3 consecutive sections were captured unilaterally at 40× using an Olympus IX71 microscope equipped with Olympus DP Controller software (v. 2.2; Olympus, Tokyo, Japan). All cleaved caspase-3-ir cells within the 40× field were analyzed for co-localization. Cells were considered co-localized when they expressed both red (cleaved caspase-3-ir) and green (NeuN, Calbindin or Calretinin-ir) fluorescence. Between 10-16 (Postnatal Tx (P7)) and 18-25 (Prenatal Tx (P0)) caspase-3 positive cells were assessed per animal for NeuN dual labeling. For analysis of caspase-3/calbindin and caspase-3/calretinin co-localization between 10-15 (Postnatal Tx (P7)) and 10-22 (Prenatal Tx (P0)) caspase-3 positive cells were assessed per animal for calbindin and calretinin dual labeling. Numbers were generally higher in P0 rats given the greater number of cleaved caspase-3 positive cells found at this time point.
Brain microdissection, RNA isolation, and Quantitative Real Time Polymerase Chain Reaction (Q-RT-PCR)
Frozen brains from prenatal treated rats at postnatal day 0 and postnatal treated rats at postnatal day 7 were sectioned at 80μm using a Leica CM3050S cryostat (Leica, Buffalo Grove, IL). The amygdala was identified using a brain atlas (Paxinos and Watson, 2007) and areas were punched bilaterally using a 1 mm diameter stainless steel cannula. Tissue was homogenized in guanidium isothiocyanate supplemented with beta-mercaptoethanol and RNA was extracted using a standard phenol/chloroform/isoamyl procedure (Ribaudo et al., 2001). Purity and concentration of RNA was confirmed spectrophotometrically using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE) and 1 μg total RNA per sample was reverse-transcribed using iScript cDNA synthesis kit (BioRad, Hercules, CA). The resulting cDNA was quantified using a fluorescent detection reagent (Molecular Probes, Eugene, OR). The quantity of cDNA in each PCR reaction was normalized based on the fluorescent quantification, and real-time RT-PCR was performed using a Roche LightCycler 480 employing SYBR green detection chemistry. The initial template for each gene was quantified by comparison to a standard curve generated with product formed by the respective primers. Primers used were: Bax (sense: 5′-GACACCTGACTGACCTTGGAGCA-3′) (antisense: 5′-AAACATGTCAGCTGCCACCCGG-3′), and Bcl2 (sense: 5′-GCAAGCCGGGAGAACAGGGTATGA-3′) (antisense: 5′-ATCTCTGCAAAGTCGCGACGGTAG-3′). The real-time RT-PCR conditions were 95 °C for 3 min, followed by 50 cycles of 95 °C for 15 s and 60 °C for 1 min. After the last PCR cycle, each sample was subject to thermal melting curve analysis according to the Roche LightCycler 480 software protocol (Roche, Indianapolis, IN). For each RNA sample a no-reverse transcriptase reaction was run in parallel to cDNA synthesis, and measured by RT-PCR to control for genomic DNA contamination. Each RT-PCR reaction was verified for a single PCR product of expected size using thermal melt curve disassociation. Furthermore, PCR products of each primer were checked for correct size using 2% agarose gel electrophoresis. Absolute values of product were generated by plotting the crossing point against a standard curve.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad, La Jolla, CA). The number of cleaved caspase-3 positive cells was quantified within the amygdala as a whole or by sub-region using 2-way ANOVA with treatment (DEX or vehicle) and sex (male or female) as factors. Apoptosis associated mRNA expression and co-localization of cleaved caspase-3 positive cells with NeuN-, calbindin-, or calretinin-ir neurons were also analyzed using 2-way ANOVA with treatment (DEX or vehicle) and sex (male or female) as factors. All significant main effects or interactions were further analyzed using Students T-tests. Differences were considered significant when p<0.05 and all data are reported as means ± standard error of the mean (SEM).
Results
Brain and Body Weights
Separate two way ANOVAs revealed that both pre and postnatal DEX treatments decreased brain [Prenatal tx: F(1,20)= 34.35, p<.001) Postnatal tx: (F(1,20)= 21.19, p<.001] and body weights [Prenatal tx: F(1,20)= 65.57, p<.001; Postnatal tx: (F(1,20)= 13.9, p<.01]. No significant sex or interaction effects were found for either treatment period. Post hoc comparisons revealed that body weights taken at the time of euthanasia were significantly decreased in both males and females following both DEX treatment times (p<.001, Table 1). Brain weight was significantly decreased in prenatally treated males (p<.05, Table 1) and females (p<.01, Table 1). Although there was no significant interaction between sex and treatment for postnatal treatment brain weight, post hoc tests revealed a significant reduction by DEX only in females (p<.01, Table 1). Given the near significant decrease in DEX male brain weight we performed a power analysis which indicated that an approximate doubling in sample size could potentially result in a significant effect.
Table 1. Effect of pre and postnatal dexamethasone treatment on body and brain weight.
Body and brain weights were measured within 12 hours of birth following prenatal treatment [prenatal tx (P0)] with DEX or oil vehicle from GD18-22 or following administration of DEX or vehicle from P4-6 [postnatal tx (P7)]. All values are in grams and are shown as mean +/- SEM.
| Prenatal tx (P0) | Postnatal tx (P7) | |
|---|---|---|
| Body Weight | ||
| Veh Male | 6.95 ± .07 | 18.5 ± .19 |
| Dex Male | 5.19 ± .06* | 14.4 ± .19* |
| Veh Female | 6.61 ± .07 | 17.9 ± .26 |
| Dex Female | 4.96 ± .06* | 14.1 ± .18* |
| Brain Weight | ||
| Veh Male | .297 ± .008 | .637 ± .018 |
| Dex Male | .243 ± .007* | .580 ± .023 |
| Veh Female | .276 ± .007 | .688 ± .015 |
| Dex Female | .244 ± .007* | .564 ± .022* |
Post hoc comparisons indicate p<.01 compared to same sex DEX treatment.
Cleaved caspase-3 immunoreactivity in the amygdala
Two way analysis of variance revealed that there was a significant effect of DEX treatment on the number of cleaved capase-3 positive cells in the amygdala following both prenatal (F(1,20)= 13.9, p<.01; Figure 1a) and postnatal treatments (F(1,20)= 5.46, p<.05; Figure 1b). No significant sex or interaction of sex and treatment effects were found. However, post hoc comparisons revealed that cleaved caspase-3 ir was significantly elevated in both sexes following prenatal DEX treatment (p<.05) but following postnatal treatment this effect reached significance only in females (p<.05). Regardless of treatment, the total number of cleaved caspase-3 positive cells counted was greater in the P0 compared to the P7 amygdala (F(1,38)= 80.66, p<.001).
Figure 1. Cleaved caspase-3 immunoreactive cells in the amygdala following prenatal and postnatal dexamethasone treatment.

Pregnant dams were treated with DEX or oil from gestation day 18-21 (prenatal tx) and pups euthanized on the day of birth (P0), or neonates were treated from postnatal day 4-6 (postnatal tx) and euthanized on postnatal day 7 (P7). The number of cleaved caspase-3 immunoreactive cells was counted in the amygdala and are expressed as number of cells per tissue section examined. At least 3 sections were examined per individual. Each bar represents the mean +/- SEM. * Post hoc comparisons indicate p<.05 compared to same sex Oil vehicle treatment. N=5-6 per group.
A more detailed analysis of the location of cleaved-caspase-3-ir neurons revealed that prenatal DEX altered cleaved caspase-3-ir in a region specific manner. Specifically, prenatal DEX treatment increased cleaved caspase-3-ir within the medial (F(1,20)= 12.09, p<.01; Figure 2a,e,f) and basomedial amygdala (F(1,20)= 11.25, p<.01; Figure 2c,g,h). There was no sex or interaction effect. Post hoc comparisons revealed that DEX significantly increased cleaved caspase-3 ir in these regions in both males (p<.05) and females (p<.05). No significant treatment effects were found in the central or basolateral amygdala (Figure 2b,d). There was also no significant effect of sex or interaction, although there was a trend towards an increase in the basolateral amygdala of females (F(1,20)= 4.03, p=.059; Figure 2d).
Figure 2. Cleaved caspase-3 immunoreactive cells within amygdala sub-regions following prenatal dexamethasone treatment.
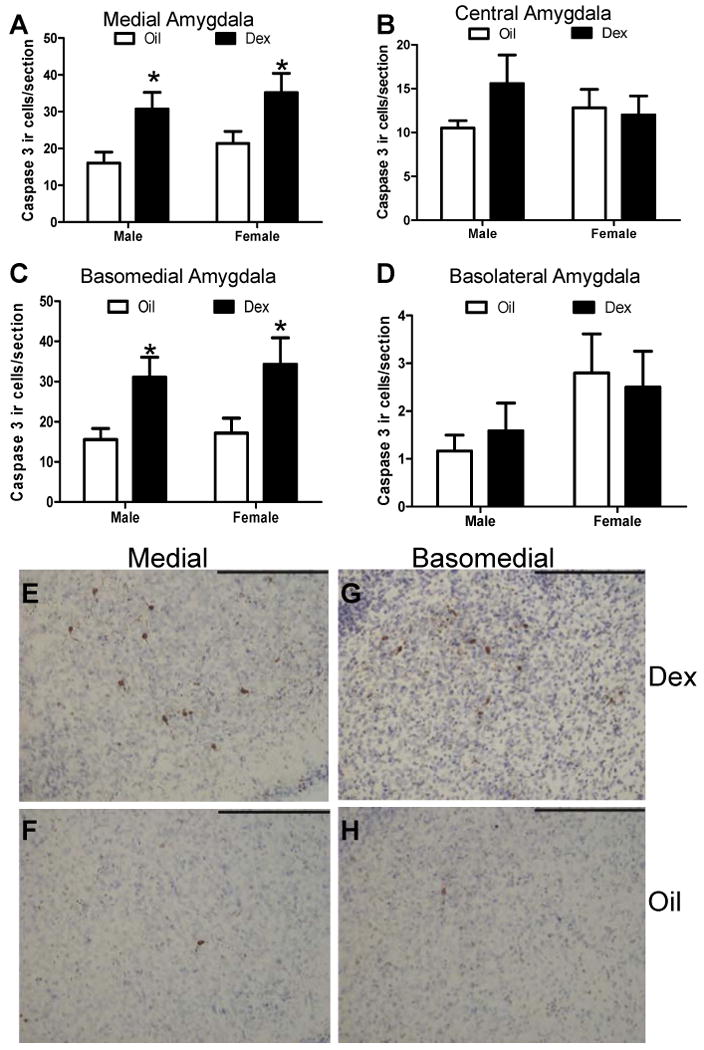
The number of cleaved caspase-3 immunoreactive cells was counted in the medial (A), central (B), basomedial (C), or basolateral (D) amygdala following prenatal treatment with DEX. Representative images of cleaved caspase-3 immunoreactive cells are shown in the medial and basomedial amygdala in P0 rats following treatment with DEX (E,G) or oil vehicle (F,H) on GD18-22. Images were captured using a 20× objective in cresyl violet counterstained sections. * Post hoc comparisons indicate p<.05 compared to same sex Oil treatment. N=5 per group. Scale bar= 200μm.
Postnatal DEX treatment also altered cleaved caspase-3-ir in a region specific manner. Following postnatal DEX treatment a significant increase in cleaved caspase-3-ir was found within the central amygdala (F(1,20)= 6.09, p<.05; Figure 3). Although there was no significant effect of sex or interaction between sex and treatment, post hoc comparisons revealed that DEX significantly increased cleaved caspase-3-ir only in females (p<.05; Figure 3b). No significant effects of treatment, sex, or interactions were found within other sub-regions of the amygdala (Figure 3a,c,d).
Figure 3. Cleaved caspase-3 immunoreactive cells within amygdala sub-regions following postnatal dexamethasone treatment.

The number of caspase-3 immunoreactive cells was quantitated in the medial (A), central (B), basomedial (C), and basolateral (D) amygdala regions at P7 following postnatal treatment (P4-6) with DEX. Each bar represents the mean +/- SEM. * Post hoc comparisons indicate p<.05 compared to same sex Oil vehicle treatment. N=5-6 per group.
Expression of Bax and Bcl2 mRNA in the amgdala
Two-way ANOVA of amygdala Bax mRNA revealed a significant increase in expression in DEX treated compared to vehicle treated rats following both prenatal (P0) (F(1,15)= 6.45, p<.05; Figure 4a) and postnatal (P7) (F(1,22)= 5.49, p<.05; Figure 4b) treatments. Post hoc analyses did not reveal any further significant effects in pre- or postnatal treated rats. No significant differences were found for Bcl2 expression in the amygdala following either prenatal (Figure 4c) or postnatal (Figure 4d) treatments. No significant effects of sex or interactions were found for either Bax or Bcl2.
Figure 4. Bax and Bcl2 mRNA in the amygdala following pre- and postnatal dexamethasone treatment.

Bax (A,B) and Bcl2 (C,D) mRNA expression in microdissected amygdala excised from prenatal (P0) and postnatal (P7) DEX and vehicle treated rats. Bax mRNA expression was significantly increased in DEX compared to vehicle treatment groups; * p<.05. N=3-7 per group for prenatal treatment and N=5-8 per group for postnatal treatment. MOIL, male oil vehicle treated; FOIL, female oil vehicle treated; MDEX, male DEX treated; FDEX, female DEX treated.
Co-localization of cleaved caspase-3 and NeuN
Analysis of amygdala sections dual labeled for cleaved caspase-3-ir and NeuN-ir revealed a 92% co-localization in prenatal treated rats and 87% in postnatal treated rats (Figure 5a). The percentage of cells dual labeled for cleaved caspase-3 and NeuN did not differ between DEX and oil vehicle treated rats, nor were there significant sex or interaction effects. Cells co-expressing cleaved caspase-3 and NeuN often displayed a faint NeuN label (Figure 6a-c), likely because the cell is undergoing apoptosis and may therefore be decreasing its expression of NeuN. Using confocal microscopy, we analyzed a total of 20 caspase-3 positive cells within 4 randomly selected brains and found that every cell exhibited characteristics of cell death including condensed/fragmented nuclei and membrane blebbing, as visualized by dual labeling with the nuclear marker DAPI (See figure 6j-l for a representative apoptotic cell). This indicates that these cleaved caspase-3 protein expressing cells represent at least a subset of the cells undergoing apoptosis.
Figure 5. Co-localization of cleaved caspase-3 with markers for neurons and calcium binding proteins in dexamethasone and vehicle treated rats.

(A) The percentage of cleaved caspase-3 immunoreactive cells co-expressing the neuronal marker NeuN. (B) The percentage of cleaved caspase-3 immunoreactive cells co-expressing calbindin and calretinin at P7 (postnatal tx). * Post hoc comparisons indicate p<.05 compared to same sex Oil treatment. N=3 per group for NeuN/caspase-3. N=5 per group for calbindin/caspase-3 and calretinin/caspase-3. MOIL, male oil vehicle treated; FOIL, female oil vehicle treated; MDEX, male DEX treated; FDEX, female DEX treated.
Figure 6. Photomicrographs showing immunofluorescent co-localization of caspase-3 and NeuN, calbindin, and calretinin.
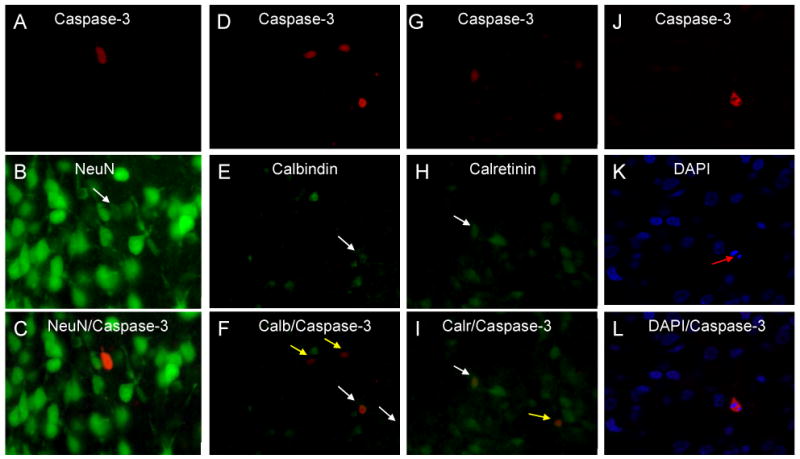
(A,D,G,J) Photomicrographs showing representative cells which express cleaved caspase-3 (red). (B) NeuN, (E) calbindin, and (H) calretinin positive cells (green). DAPI nuclear label is shown in blue (K). Merged photographs which reveal co-localization of caspase-3 with NeuN, calbindin, calretinin, and DAPI (C,F,I,L). Note the relatively weak NeuN labeling of the caspase-3 positive cell (B) indicating a decreased NeuN phenotype in this apoptotic neuron. Images are captured using a 40× objective. White arrow indicates co-localized cells; yellow arrow indicates a caspase-3 positive cell that is not co-localized; red arrow indicates a fragmented nucleus with condensed chromatin.
Co-localization of CBPs in cleaved caspase-3-ir neurons
At P0 calbindin immunoreactivity was low in the amygdala with scattered cells found within the basolateral and basomedial regions. Calretinin-ir was sparse in the P0 amygdala with a few scattered cells found in the medial and basomedial regions. Immunoreactivity for both CBP proteins was much greater in the P7 brain, where calbindin-ir cells populated the basolateral and basomedial divisions with many cells also found in the medial and central divisions. At this age, calretinin positive cells were most dense within the basomedial and medial divisions with cells also localized within the basolateral and central regions.
Analysis of cleaved caspase-3 positive cells co-labeled for calbindin revealed 2.3% of cleaved caspase-3-ir cells expressed calbindin in prenatal treated rats and 13.3% in postnatal treated rats. For both pre- and postnatal treated rats the percentage of cleaved caspase-3 cells expressing calbindin did not differ significantly between DEX and oil vehicle treatments, nor were there significant effects of sex, or a sex × treatment interaction. No co-localization of cleaved caspase-3 and calretinin was found following prenatal treatment but 11% of cleaved caspase-3-ir neurons were calretinin positive in the amygdala of postnatal treated rats. No significant differences in the percentage of cleaved caspase-3-ir cells that co-expressed calretinin were found between DEX and vehicle-treated rats, nor were there significant sex differences, or a sex × treatment interaction. Figure 6 shows photomicrographs with representative immunostaining for caspase-3 and calbindin/calretinin positive cells.
Since calbindin and calretinin show virtually no co-localization within the amygdala (McDonald and Mascagni, 2001), the percent of cleaved caspase-3-ir cells that express calbindin and calretinin were combined as an indication of the percent of cleaved caspase-3-ir cells that are CBP-ir for postnatal treated rats. This analysis was not performed for prenatal treated rats because no co-localization of caspase-3/calretinin was found. Combined analysis for postnatal treated rats revealed a significant main effect of treatment in which DEX treated rats showed a greater percentage of cleaved caspase-3-ir /CBP-ir cells than did vehicle treated rats (Figure 5b). Although there were no significant effects of sex or a sex × treatment interaction, post hoc analyses revealed that DEX treated females had a significant increase in cleaved-caspase-3-/CBP-ir cells compared to vehicle females (p<.05, Figure 5b) while male groups did not significantly differ. A subset of P7 cleaved caspase-3/CBP dual labeled tissue sections were reanalyzed using confocal microscopy to confirm co-localization within cells. The percentage of cells identified as co-labeled for caspase-3/CBP was similar to those revealed by epifluorescence microscopy.
Discussion
In this series of studies, we examined the effects of both late gestational and postnatal exposure to DEX on neural cell death. The results of these studies show that brain and body weight were decreased in male and female offspring of DEX treated dams. Postnatal treatment of rats also caused overall decreases in brain and body weight. Of importance, DEX treatment had significant effects on the degree of cleaved caspase-3 positive cells in the amygdala, and these effects occurred in an age, sex, and subregion specific fashion. Prenatal DEX treatment increased cleaved caspase-3-ir in the amygdala specifically within the medial and basomedial subregions, while postnatal DEX induced increases in cleaved caspase-3-ir reached significance only in the female central nucleus. In both instances DEX induced elevations in cleaved caspase-3-ir within the amygdala correlated with an increase in pro-apoptotic Bax mRNA expression. Dual-label immunohistochemistry of cleaved caspase-3-ir and the neuronal marker NeuN confirmed that nearly all cleaved caspase-3-ir cells in the amygdala were neurons. Furthermore, a subset of these cells (primarily following postnatal treatment) expressed a GABAergic calcium binding protein phenotype.
The reduction in brain weight following prenatal DEX is in line with previous studies that have also indicated dramatic decreases following gestational treatment with GCs such as DEX (Holson et al., 1995; Duksal et al., 2009). We also demonstrate that postnatal DEX exposure reduced brain weight, confirming previous findings (Duksal et al., 2009; Ferguson et al., 2001). Furthermore, our results indicate that the female brain may be somewhat more vulnerable to GC effects during the postnatal period since only females brains weights were significantly reduced by DEX following postnatal treatment although there was also a trend toward a decrease in males.
Naturally occurring cell death is prevalent throughout the developing brain. It occurs through an intrinsic mechanism within the cell and is essential for the removal of damaged and superfluous cells (Pettmann and Henderson, 1998). Naturally occurring cell death occurs during the postnatal period within many brain regions including the amygdala, and inhibition or exacerbation of cell death can lead to later impairments in behavior (Jyotika et al., 2007; Broad et al., 2009; Rondi-Reig and Mariani, 2002). In the present study, the administration of DEX during both the late gestational and neonatal periods significantly increased apoptosis (as measured by cleaved caspase-3) within the amygdala. Similar inductions in programmed cell death have been reported in select brain regions following GC administration both in adulthood (Hassan et al., 1996; Li et al., 2010) and in early development (Duksal et al., 2009; Noguchi et al., 2008) but to our knowledge this is the first evidence indicating that the amygdala is similarly affected directly by GCs. Our results also demonstrate that DEX treatment can increase cleaved caspase-3 positive cells within the amygdala when it is delivered during multiple developmental periods, and the late prenatal period of rat gestation may represent a highly sensitive period for GCs to induce apoptosis.
Prenatal DEX exposure specifically increased cleaved caspase-3 ir within the medial and basomedial amgdala but not within the central and basolateral regions indicating an increased sensitivity to GCs in cells of the medial and basomedial regions. Neurons in the amygdala have been detected as early as GD12 (Manolova and Manolova, 1991) and these cells begin to express GRs during late gestation beginning at GD 17 or 18 (Kitraki et al., 1996; Yi et al., 1994; Cintra, 1993), with maximal levels of amygdala GR reached at P10 (Yi et al., 1994). This indicates a potential direct mechanism through which GCs can induce apoptosis. However, why the medial and basomedial regions specifically show the greatest levels of GC induced cleaved caspase-3 remains elusive, as GR levels in these regions are not greater than levels in other amygdala regions (Yi et al., 1994). It may be that there are cell types specific to the medial and basomedial region that are vulnerable to GC activation. Alternatively GC activation of cells outside of the amygdala which project to the medial and basomedial regions may induce cleaved caspase-3 activation within these regions. The medial and basomedial regions of the amygdala share homogeneous projection patterns from the GC rich hippocampus which are distinct from projection patterns of the central and basolateral amygdala (Kishi et al., 2006). Damage to neurons in several brain regions, including the hippocampus, have been shown to induce apoptosis in remote but functionally connected brain regions (Khaing et al., 2000; Zin et al., 2000), suggesting one mechanism through which these regions may be specifically affected by prenatal DEX.
In contrast, postnatal DEX treatment significantly increased cleaved caspase-3 ir only in the central amygdala. The central amygdala, particularly during the postnatal period, expresses very high levels of GRs even when compared to other GR-rich amygdala sub-regions (Yi et al. 1994; Morimoto et al., 1996). In this instance, if levels of GR were the only driving force behind the incidence of apoptosis, then the postnatal central amygdala should be most vulnerable to effects of GCs.
Since amygdala GRs are more abundant during the postnatal period it may be expected that apoptosis would be greater following postnatal DEX treatment, but this was not the case. Alternatively the amygdala cells expressing GRs may possess other characteristics that make them more prone to apoptosis during prenatal development. The P0 amygdala showed much higher levels of cleaved caspase-3 positive cells in vehicle treated rats compared to the P7 amygdala indicating that the P0 time point represents a period of abundant naturally occurring cell death. This may suggest that these cells are particularly vulnerable at P0 and are therefore more prone to apoptosis following a physiological challenge. Thus, naturally occurring cell death may be enhanced by GC exposure.
Interestingly, postnatal GC administration significantly increased cleaved caspase-3 ir only in the central nucleus of females, indicating a possible role of perinatal gonadal hormones in this effect. In adults, both androgens and estrogens have been shown to be neuroprotective in the hippocampus following adrenalectomy or kainic acid administration (Frye 2001; Frye and McCormick, 2000a,b; Veiga et al., 2003). Testicular surges of testosterone occur during the prenatal and early neonatal periods and these may inhibit the effects of DEX on apoptosis. Testosterone has been shown to decrease naturally occurring cell loss within several regions of the postnatal central nervous system (Tsukahara et al., 2008; Breedlove and Arnold, 1980). Therefore, postnatal testosterone may play a role in reducing GC-induced cell death in the amygdala, although further studies involving postnatal androgen manipulation are needed to test this hypothesis.
Similar to the differential effects of pre- and postnatal GCs on amygdala apoptosis reported here, developmental timing of GC treatment can also differentially alter the display of amygdala-related behaviors in adulthood. Sexual behaviors, which are in part regulated by the medial amygdala, are demasculinized in male rats exposed to DEX in utero (Holson et al., 1995). On the contrary, postnatal treatment with GCs and GC secretogogues including DEX, corticosterone, and adrenocorticotropic hormone (ACTH), fail to demasculize sexual behavior and in some cases have been reported to cause hyper-masculinization (Kamphuis et al., 2004; Politch and Herrenkohl, 1984). Regulation of the HPA axis is also differentially affected depending on the developmental timing of GC exposure. Prenatal DEX exposure has been shown to cause a hyperactivation of the HPA axis in adulthood in which corticosterone levels following a stressor are elevated during the recovery phase (Shoener et al., 2006), suggesting a loss of negative feedback. In contrast, postnatal DEX blunts stress-induced corticosterone and ACTH responses in adult rats (Kamphuis et al., 2002; Felszeghy et al., 2000).
Unlike sexual behaviors and HPA axis regulation, emotional behaviors appear to be similarly effected in rodents following both pre- and postnatal GC treatments. GC exposure either in utero or postnatal results in increased anxiety-related behaviors (Hossain et al., 2008; Nagano et al. 2008; Pitzer and Schmidt, 2009), an elevated startle response (Hossain et al., 2008; Kjaer et al., 2011; Ferguson et al., 2001), and increased depressive-like behaviors (Roque et al., 2011; Felszeghy et al., 1993). Together, these behavioral findings suggest that brain development is also differentially affected by the timing of GC exposure. Furthermore, enhanced cell loss in the amygdala following DEX treatment may be one mechanism whereby permanent changes occur that underlie these differences.
Correlating with the diminished sexual behavior reported in rats treated in utero with GCs, our results show increased apoptosis of cells within the medial amygdala, a region known to regulate rodent sexual behavior (Wood, 1996). The medial amygdala also receives afferents from hypothalamic nuclei and has projections to other nuclei of the amygdala, bed nucleus of the stria terminalis, and hypothalamus, thus placing it in a critical location for regulating a number of amygdala functions (Alheid, 2003). The basomedial amygdala is functionally considered to be a part of the basolateral complex which is involved in controlling fear responses, depression, and anxiety-like behaviors (Martinez et al., 2011; Goosens and Maren, 2001; Hale et al., 2006). Therefore, an elevation in cell death within this region may contribute to programming of emotional behavioral deficits reported in prenatal DEX treated rodents (Hossain et al., 2008; Nagano et al. 2008; Roque et al., 2011).
Postnatal GC treatment specifically induced apoptosis within the central amygdala, which may be one mechanism through which HPA functions are altered in similarly treated adult rodents (Kamphuis et al., 2004; Politch and Herrenkohl, 1984). The central nucleus also represents the major output center of the amygdala as it receives input from other amygdala sub-regions and projects to regions throughout the brain including areas of the cortex, hypothalamus, and midbrain regions (Fudge and Tucker, 2009; Akmaev et al., 2004). Through this complex pattern of connections the central amygdala is positioned to regulate a variety of functions including emotional behaviors as well as autonomic functions (Al Maskati & Zbrozyna 1989), which are altered by DEX treatment early in life (Pitzer and Schmidt, 2009; Hou and Slotkin, 1989; de Vries et al., 2007). Several other mechanisms including alterations in neuronal migration, differentiation, and decreased cell proliferation (Fukumoto et al., 2009; Noorlander et al., 2008) and a variety of other brain regions (eg. hippocampus, hypothalamus) also likely contribute to the development of GC-related behavioral disorders.
Increases in the number of caspase-3 positive cells within the amygdala of pre- and postnatal DEX treated rats also correlated with an increase in the pro-apoptotic gene Bax. On the contrary no significant differences were found in the anti-apoptotic gene Bcl2. These results indicate that DEX acts by altering expression of apoptosis related genes of the intrinsic pathway, specifically by increasing expression of the pro-apoptotic gene Bax. Bax has previously been demonstrated to contribute to GC mediated apoptosis particularly within the adult hippocampus (Almeida et al., 2000) however, to our knowledge this is the first evidence indicating an involvement of Bax in GC induced apoptosis within the amygdala. Furthermore, we also provide novel evidence that GC administration during the perinatal period can significantly increase Bax expression, although administration of GCs in early development have been shown to elevate expression of pro-apoptotic genes Bnip3 and Puma (Sandau and Handa, 2007; Noguchi et al., 2008).
Co-localization studies to identify the phenotype of cells in the amygdala that undergo apoptosis revealed that approximately 90% of cleaved caspase-3-ir cells also expressed the neuronal marker NeuN, indicating that it is virtually all neurons, not glia that are undergoing apoptosis. We also observed that cells expressing cleaved caspase-3-ir often displayed a weaker NeuN signal relative to cells not undergoing apoptosis. This observation introduces the possibility that some cleaved-caspase-3 positive cells may have a diminished protein expression and as a result, they cannot be phenotypically identified using existing technology resulting in a lower estimate of the percentage of these cells that were truly neuronal. Nevertheless, the DEX induced increase in cleaved caspase-3-ir within the amygdala may result in a lasting decrease in neuron number and consequently could have long-term consequences on amygdala related behaviors.
In these studies we detected a subpopulation of cleaved caspase-3 ir cells in the amygdala that also expressed immunoreactivity for subsets of GABAergic neurons that express CBPs (calbindin and calretinin). Co-localization was greater at P7 (postnatal tx) whereas there was very little co-localization at P0 (prenatal tx). The low level of co-localization in P0 animals likely reflects the lower expression of CBPs at this developmental age and the potential inability of our immunocytochemical technique to pick up weakly expressing CBP neurons. In our studies, calbindin expressing cells were minimal at this time point with some expression in the basolateral division and scattered immunoreactivity within the basomedial, medial, and central divisions. On the contrary, P7 calbindin-ir cells were much more densely distributed. Calretinin positive cells were very sparse at P0 with only a few cells visible within the basomedial and medial subdivisions, while at P7 calretinin-ir was abundant. The lower distribution of CBPs at P0 indicates that many cells in the amygdala have not yet begun to express these phenotypes. Therefore, the numerous apoptotic cells within the P0 amygdala may indeed be neurons that were programmed to express CBPs but had not yet begun to express the phenotype.
Postnatal DEX-treated rats also showed a greater percentage of co-localization of cleaved caspase-3 with CBPs (calbindin and calretinin combined) than did vehicle treated controls indicating that GCs may preferentially induce apoptosis within this subtype of amygdala neurons. Consequently, the number of CBP neurons in the amygdala may also be permanently altered by DEX treatment. GABAergic neurons in the amygdala play a critical role in the regulation of anxiety/stress related behaviors (Maciag et al., 2010; Helmeke et al., 2008; Yilmazer-Hanke et al., 2002) therefore, a selective loss of a specific subtype of these cells (CBPs) suggests a mechanism through which postnatal GCs can alter these behaviors. However, since the percentage of P7 dual labeled cleaved caspase-3/CBP cells within the amygdala was approximately 25% this indicates that for the majority of apoptotic cells the phenotypes and specific vulnerability to GC induced apoptosis remains unknown.
Together, these findings indicate that developmental DEX treatment can increase apoptosis in select regions and perhaps cell types of the amygdala, and these effects are dependent on the timing of treatment and sex. These alterations in neuronal apoptosis may contribute to the lasting effects of early GC exposure in adult behaviors associated with emotional regulation, hormone secretion, and sexual function (Hossain et al., 2008; Nagano et al., 2008; Holson et al., 1995).
Highlights.
>dexamethasone induces apoptosis in the perinatal amygdala >Effects are timing, sex, and subregion dependent >Dependent on increases in Bax expression.
Acknowledgments
The authors acknowledge Drs. Stuart Tobet and Jill Goldstein for providing helpful input in the design of these studies and the preparation of the manuscript. Also thanks to Alicia Quihuis and Anthony Lacaganina for their expert technical assistance. Support for these studies was provided by United States Public Health Service Grants NS039951 and MH082679 (RJH).
Footnotes
Disclosure Statement: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akmaev IG, Kalimullina LB, Sharipova LA. The central nucleus of the amygdaloid body of the brain: cytoarchitectonics, neuronal organization, connections. Neurosci Behav Physiol. 2004;34(6):603–10. doi: 10.1023/b:neab.0000028292.14402.ad. [DOI] [PubMed] [Google Scholar]
- Alheid GF. Extended amygdala and basal forebrain. Ann N Y Acad Sci. 2003;985:185–205. doi: 10.1111/j.1749-6632.2003.tb07082.x. [DOI] [PubMed] [Google Scholar]
- al Maskati HA, Zbrozyna AW. Cardiovascular and motor components of the defence reaction elicited in rats by electrical and chemical stimulation in amygdala. J Auton Nerv Syst. 1989;28(2):127–31. doi: 10.1016/0165-1838(89)90085-4. [DOI] [PubMed] [Google Scholar]
- Almeida OF, Condé GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, Meyer M, Holsboer F, Michaelidis TM. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J. 2000;14(5):779–90. doi: 10.1096/fasebj.14.5.779. [DOI] [PubMed] [Google Scholar]
- Arya V, Demarco VG, Issar M, Hochhaus G. Contrary to adult, neonatal rats show pronounced brain uptake of corticosteroids. Drug Metab Dispos. 2006;34(6):939–42. doi: 10.1124/dmd.105.007419. [DOI] [PubMed] [Google Scholar]
- Barrington KJ. Postnatal steroids and neurodevelopmental outcomes: a problem in the making. Pediatrics. 2001;107(6):1425–6. doi: 10.1542/peds.107.6.1425. [DOI] [PubMed] [Google Scholar]
- Berdel B, Moryś J. Expression of calbindin-D28k and parvalbumin during development of rat's basolateral amygdaloid complex. Int J Dev Neurosci. 2000;18(6):501–13. doi: 10.1016/s0736-5748(00)00024-1. [DOI] [PubMed] [Google Scholar]
- Brabham T, Phelka A, Zimmer C, Nash A, López JF, Vázquez DM. Am J Physiol Regul Integr Effects of prenatal dexamethasone on spatial learning and response to stress is influenced by maternal factors. Comp Physiol. 2000;279(5):R1899–909. doi: 10.1152/ajpregu.2000.279.5.R1899. [DOI] [PubMed] [Google Scholar]
- Breedlove SM, Arnold AP. Hormone accumulation in a sexually dimorphic motor nucleus of the rat spinal cord. Science. 1980;210(4469):564–6. doi: 10.1126/science.7423210. [DOI] [PubMed] [Google Scholar]
- Broad KD, Curley JP, Keverne EB. Increased apoptosis during neonatal brain development underlies the adult behavioral deficits seen in mice lacking a functional paternally expressed gene 3 (Peg3) Dev Neurobiol. 2009;69(5):314–25. doi: 10.1002/dneu.20702. [DOI] [PubMed] [Google Scholar]
- Cintra A, Solfrini V, Bunnemann B, Okret S, Bortolotti F, Gustafsson JA, Fuxe K. Prenatal development of glucocorticoid receptor gene expression and immunoreactivity in the rat brain and pituitary gland: a combined in situ hybridization and immunocytochemical analysis. Neuroendocrinology. 1993;57(6):1133–47. doi: 10.1159/000126480. [DOI] [PubMed] [Google Scholar]
- Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA. Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol. 1994;341:95–116. doi: 10.1002/cne.903410109. [DOI] [PubMed] [Google Scholar]
- Crane J, Armson A, Brunner M, De La Ronde S, Farine D, Keenan-Lindsay L, Leduc L, Schneider C, Van Aerde J Executive Committee of the Society of Obstetricians and Gynaecologists of Canada. Antenatal corticosteroid therapy for fetal maturation. J Obstet Gynaecol Can. 2003;25(1):45–52. doi: 10.1016/s1701-2163(16)31081-7. [DOI] [PubMed] [Google Scholar]
- de Vries A, Holmes MC, Heijnis A, Seier JV, Heerden J, Louw J, Wolfe-Coote S, Meaney MJ, Levitt NS, Seckl JR. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest. 2007;117(4):1058–67. doi: 10.1172/JCI30982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duksal F, Kilic I, Tufan AC, Akdogan I. Effects of different corticosteroids on the brain weight and hippocampal neuronal loss in rats. Brain Res. 2009;1250:75–80. doi: 10.1016/j.brainres.2008.10.051. [DOI] [PubMed] [Google Scholar]
- Felszeghy K, Bagdy G, Nyakas C. Blunted pituitary-adrenocortical stress response in adult rats following neonatal dexamethasone treatment. J Neuroendocrinol. 2000;12(10):1014–21. doi: 10.1046/j.1365-2826.2000.00551.x. [DOI] [PubMed] [Google Scholar]
- Felszeghy K, Sasvári M, Nyakas C. Behavioral depression: opposite effects of neonatal dexamethasone and ACTH-(4-9) analogue (ORG 2766) treatments in the rat. Horm Behav. 1993;27(3):380–96. doi: 10.1006/hbeh.1993.1028. [DOI] [PubMed] [Google Scholar]
- Ferguson SA, Paule MG, Holson RR. Neonatal dexamethasone on day 7 in rats causes behavioral alterations reflective of hippocampal, but not cerebellar, deficits. Neurotoxicol Teratol. 2001;23(1):57–69. doi: 10.1016/s0892-0362(00)00115-x. [DOI] [PubMed] [Google Scholar]
- Frye CA. Estradiol tends to improve inhibitory avoidance performance in adrenalectomized male rats and reduces pyknotic cells in the dentate gyrus of adrenalectomized male and female rats. Brain Res. 2001;889(1-2):358–63. doi: 10.1016/s0006-8993(00)03236-4. [DOI] [PubMed] [Google Scholar]
- Frye CA, McCormick CM. Androgens are neuroprotective in the dentate gyrus of adrenalectomized female rats. Stress. 2000a;3(3):185–94. doi: 10.3109/10253890009001122. [DOI] [PubMed] [Google Scholar]
- Frye CA, McCormick CM. The neurosteroid, 3alpha-androstanediol, prevents inhibitory avoidance deficits and pyknotic cells in the granule layer of the dentate gyrus induced by adrenalectomy in rats. Brain Res. 2000b;855(1):166–70. doi: 10.1016/s0006-8993(99)02208-8. [DOI] [PubMed] [Google Scholar]
- Fudge JL, Tucker T. Amygdala projections to central amygdaloid nucleus subdivisions and transition zones in the primate. Neuroscience. 2009;159(2):819–41. doi: 10.1016/j.neuroscience.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K, Morita T, Mayanagi T, Tanokashira D, Yoshida T, Sakai A, Sobue K. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol Psychiatry. 2009;14(12):1119–31. doi: 10.1038/mp.2009.60. [DOI] [PubMed] [Google Scholar]
- Giachino C, Canalia N, Capone F, Fasolo A, Alleva E, Riva MA, Cirulli F, Peretto P. Maternal deprivation and early handling affect density of calcium binding protein-containing neurons in selected brain regions and emotional behavior in periadolescent rats. Neuroscience. 2007;145(2):568–78. doi: 10.1016/j.neuroscience.2006.12.042. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem. 2001;8(3):148–55. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme R, Renaud C, Dommergues M, Mitanchez D. Repeat doses of prenatal corticosteroids for women at risk of preterm birth: a difficult consensus. J Gynecol Obstet Biol Reprod (Paris) 2009;38(6):459–68. doi: 10.1016/j.jgyn.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Hale MW, Bouwknecht JA, Spiga F, Shekhar A, Lowry CA. Exposure to high- and low-light conditions in an open-field test of anxiety increases c-Fos expression in specific subdivisions of the rat basolateral amygdaloid complex. Brain Res Bull. 2006;71(1-3):174–82. doi: 10.1016/j.brainresbull.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF. Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Exp Neurol. 1996;140(1):43–52. doi: 10.1006/exnr.1996.0113. [DOI] [PubMed] [Google Scholar]
- Helmeke C, Ovtscharoff W, Jr, Poeggel G, Braun K. Imbalance of immunohistochemically characterized interneuron populations in the adolescent and adult rodent medial prefrontal cortex after repeated exposure to neonatal separation stress. Neuroscience. 2008;152(1):18–28. doi: 10.1016/j.neuroscience.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann N Y Acad Sci. 2004;1018:35–45. doi: 10.1196/annals.1296.004. [DOI] [PubMed] [Google Scholar]
- Holson RR, Gough B, Sullivan P, Badger T, Sheehan DM. Prenatal dexamethasone or stress but not ACTH or corticosterone alter sexual behavior in male rats. Neurotoxicol Teratol. 1995;17(4):393–401. doi: 10.1016/0892-0362(94)00074-n. [DOI] [PubMed] [Google Scholar]
- Hossain A, Hajman K, Charitidi K, Erhardt S, Zimmermann U, Knipper M, Canlon B. Prenatal dexamethasone impairs behavior and the activation of the BDNF exon IV promoter in the paraventricular nucleus in adult offspring. Endocrinology. 2008;149(12):6356–65. doi: 10.1210/en.2008-0388. [DOI] [PubMed] [Google Scholar]
- Hou QC, Slotkin TA. Effects of prenatal dexamethasone or terbutaline exposure on development of neural and intrinsic control of heart rate. Pediatr Res. 1989;26(6):554–7. doi: 10.1203/00006450-198912000-00005. [DOI] [PubMed] [Google Scholar]
- Jones RA. Randomized, controlled trial of dexamethasone in neonatal chronic lung disease: 13- to 17-year follow-up study: I. Neurologic, psychological, and educational outcomes. Pediatrics. 2005;116(2):370–8. doi: 10.1542/peds.2004-1818. [DOI] [PubMed] [Google Scholar]
- Jyotika J, McCutcheon J, Laroche J, Blaustein JD, Forger NG. Deletion of the Bax gene disrupts sexual behavior and modestly impairs motor function in mice. Dev Neurobiol. 2007;67(11):1511–9. doi: 10.1002/dneu.20525. [DOI] [PubMed] [Google Scholar]
- Kamphuis PJ, Bakker JM, Broekhoven MH, Kunne C, Croiset G, Lentjes EG, Tilders FJ, van Bel F, Wiegant VM. Enhanced glucocorticoid feedback inhibition of hypothalamo-pituitary-adrenal responses to stress in adult rats neonatally treated with dexamethasone. Neuroendocrinology. 2002;76(3):158–69. doi: 10.1159/000064526. [DOI] [PubMed] [Google Scholar]
- Kamphuis PJ, Croiset G, Bakker JM, Van Bel F, Van Ree JM, Wiegant VM. Neonatal dexamethasone treatment affects social behaviour of rats in later life. Neuropharmacology. 2004;47(3):461–74. doi: 10.1016/j.neuropharm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Karemaker R, Kavelaars A, ter Wolbeek M, Tersteeg-Kamperman M, Baerts W, Veen S, Samsom JF, Visser GH, van Bel F, Heijnen CJ. Neonatal dexamethasone treatment for chronic lung disease of prematurity alters the hypothalamus-pituitary-adrenal axis and immune system activity at school age. Pediatrics. 2008;121(4):e870–8. doi: 10.1542/peds.2007-2454. [DOI] [PubMed] [Google Scholar]
- Kemppainen S, Pitkänen A. Distribution of parvalbumin, calretinin, and calbindin-D(28k) immunoreactivity in the rat amygdaloid complex and colocalization with gamma-aminobutyric acid. J Comp Neurol. 2000;426(3):441–67. doi: 10.1002/1096-9861(20001023)426:3<441::aid-cne8>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Khaing ZZ, Weickert CS, Weinberger DR, Lipska BK. Differential DNA damage in response to the neonatal and adult excitotoxic hippocampal lesion in rats. Eur J Neurosci. 2000;12(12):4424–33. doi: 10.1046/j.0953-816x.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- Kishi T, Tsumori T, Yokota S, Yasui Y. Topographical projection from the hippocampal formation to the amygdala: a combined anterograde and retrograde tracing study in the rat. J Comp Neurol. 2006;496(3):349–68. doi: 10.1002/cne.20919. [DOI] [PubMed] [Google Scholar]
- Kitraki E, Alexis MN, Papalopoulou M, Stylianopoulou F. Glucocorticoid receptor gene expression in the embryonic rat brain. Neuroendocrinology. 1996;63(4):305–17. doi: 10.1159/000126971. [DOI] [PubMed] [Google Scholar]
- Kjaer SL, Hougaard KS, Tasker RA, MacDonald DS, Rosenberg R, Elfving B, Wegener G. Influence of diurnal phase on startle response in adult rats exposed to dexamethasone in utero. Physiol Behav. 2011;102(5):444–52. doi: 10.1016/j.physbeh.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Kreider ML, Tate CA, Cousins MM, Oliver CA, Seidler FJ, Slotkin TA. Lasting effects of developmental dexamethasone treatment on neural cell number and size, synaptic activity, and cell signaling: critical periods of vulnerability, dose-effect relationships, regional targets, and sex selectivity. Neuropsychopharmacology. 2006;31(1):12–35. doi: 10.1038/sj.npp.1300783. [DOI] [PubMed] [Google Scholar]
- Lajic S, Nordenström A, Hirvikoski T. Long-term outcome of prenatal dexamethasone treatment of 21-hydroxylase deficiency. Endocr Dev. 2011;20:96–105. doi: 10.1159/000321228. [DOI] [PubMed] [Google Scholar]
- Legaz I, Olmos L, Real MA, Guirado S, Dávila JC, Medina L. Development of neurons and fibers containing calcium binding proteins in the pallial amygdala of mouse, with special emphasis on those of the basolateral amygdalar complex. J Comp Neurol. 2005;488(4):492–513. doi: 10.1002/cne.20608. [DOI] [PubMed] [Google Scholar]
- Li WZ, Li WP, Yao YY, Zhang W, Yin YY, Wu GC, Gong HL. Glucocorticoids increase impairments in learning and memory due to elevated amyloid precursor protein expression and neuronal apoptosis in 12-month old mice. Eur J Pharmacol. 2010;628(1-3):108–15. doi: 10.1016/j.ejphar.2009.11.045. [DOI] [PubMed] [Google Scholar]
- Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50(4):515–25. [PubMed] [Google Scholar]
- Maciag D, Hughes J, O'Dwyer G, Pride Y, Stockmeier CA, Sanacora G, Rajkowska G. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol Psychiatry. 2010;67(5):465–70. doi: 10.1016/j.biopsych.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolova A, Manolov S. Prenatal development of the rat amygdaloid complex: an electron microscopic study. Adv Exp Med Biol. 1991;296:21–8. doi: 10.1007/978-1-4684-8047-4_3. [DOI] [PubMed] [Google Scholar]
- Martinez RC, Carvalho-Netto EF, Ribeiro-Barbosa ER, Baldo MV, Canteras NS. Amygdalar roles during exposure to a live predator and to a predator-associated context. Neuroscience. 2011;172:314–28. doi: 10.1016/j.neuroscience.2010.10.033. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Luine VN, Plapinger L, de Kloet ER. Putative estrogen and glucocorticoid receptors in the limbic brain. J Steroid Biochem. 1975;6(6):971–7. doi: 10.1016/0022-4731(75)90337-4. [DOI] [PubMed] [Google Scholar]
- McGimsey WC, Cidlowski JA, Stumpf WE, Sar M. Immunocytochemical localization of the glucocorticoid receptor in rat brain, pituitary, liver, and thymus with two new polyclonal antipeptide antibodies. Endocrinology. 1991;129(6):3064–72. doi: 10.1210/endo-129-6-3064. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res. 1996;26(3):235–69. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- Nagano M, Ozawa H, Suzuki H. Prenatal dexamethasone exposure affects anxiety-like behaviour and neuroendocrine systems in an age-dependent manner. Neurosci Res. 2008;60(4):364–71. doi: 10.1016/j.neures.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15(10):1582–92. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorlander CW, Visser GH, Ramakers GM, Nikkels PG, de Graan PN. Prenatal corticosteroid exposure affects hippocampal plasticity and reduces lifespan. Dev Neurobiol. 2008;68(2):237–46. doi: 10.1002/dneu.20583. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y. Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience. 1993;57(4):1061–76. doi: 10.1016/0306-4522(93)90049-l. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 7. San Diego: Elsevier Academic Press; 2007. [Google Scholar]
- Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20(4):633–47. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- Pitzer M, Schmidt MH. Neonatal exposure to fenoterol and betamethasone: effects on the behavioral development in the rat. Int J Neurosci. 2009;119(10):1548–71. doi: 10.1080/00207450802323947. [DOI] [PubMed] [Google Scholar]
- Politch JA, Herrenkohl LR. Postnatal ACTH and corticosterone: effects on reproduction in mice. Physiol Behav. 1984;32(3):447–52. doi: 10.1016/0031-9384(84)90261-0. [DOI] [PubMed] [Google Scholar]
- Ritzén EM. Prenatal dexamethasone treatment of fetuses at risk for congenital adrenal hyperplasia: benefits and concerns. Semin Neonatol. 2001;6(4):357–62. doi: 10.1053/siny.2001.0071. [DOI] [PubMed] [Google Scholar]
- Rondi-Reig L, Mariani J. To die or not to die, does it change the function? Behavior of transgenic mice reveals a role for developmental cell death. Brain Res Bull. 2002;57(1):85–91. doi: 10.1016/s0361-9230(01)00639-6. [DOI] [PubMed] [Google Scholar]
- Roque S, Oliveira TG, Nobrega C, Barreira-Silva P, Nunes-Alves C, Sousa N, Palha JA, Correia-Neves M. Interplay between Depressive-Like Behavior and the Immune System in an Animal Model of Prenatal Dexamethasone Administration. Front Behav Neurosci. 2011;5:4. doi: 10.3389/fnbeh.2011.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995a;52(4):701–6. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- Sandau US, Handa RJ. Glucocorticoids exacerbate hypoxia-induced expression of the pro-apoptotic gene Bnip3 in the developing cortex. Neuroscience. 2007;144(2):482–94. doi: 10.1016/j.neuroscience.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckl JR. Glucocorticoid programming of the fetus; adult phenotypes and molecular mechanisms. Mol Cell Endocrinol. 2001;185(1-2):61–71. doi: 10.1016/s0303-7207(01)00633-5. [DOI] [PubMed] [Google Scholar]
- Seidel K, Helmeke C, Poeggel G, Braun K. Repeated neonatal separation stress alters the composition of neurochemically characterized interneuron subpopulations in the rodent dentate gyrus and basolateral amygdala. Dev Neurobiol. 2008;68(9):1137–52. doi: 10.1002/dneu.20651. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001;50(9):651–8. doi: 10.1016/s0006-3223(01)01263-x. [DOI] [PubMed] [Google Scholar]
- Shoener JA, Baig R, Page KC. Prenatal exposure to dexamethasone alters hippocampal drive on hypothalamic-pituitary-adrenal axis activity in adult male rats. Am J Physiol Regul Integr Comp Physiol. 2006;290(5):R1366–73. doi: 10.1152/ajpregu.00757.2004. [DOI] [PubMed] [Google Scholar]
- Tobe I, Ishida Y, Tanaka M, Endoh H, Fujioka T, Nakamura S. Effects of repeated maternal stress on FOS expression in the hypothalamic paraventricular nucleus of fetal rats. Neuroscience. 2005;134(2):387–95. doi: 10.1016/j.neuroscience.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Tottenham N, Hare TA, Quinn BT, McCarry TW, Nurse M, Gilhooly T, Millner A, Galvan A, Davidson MC, Eigsti IM, Thomas KM, Freed PJ, Booma ES, Gunnar MR, Altemus M, Aronson J, Casey BJ. Prolonged institutional rearing is associated with atypically large amygdala volume and difficulties in emotion regulation. Dev Sci. 2010;13(1):46–61. doi: 10.1111/j.1467-7687.2009.00852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi MT, Florenzano F, Latini L, Molinari M. Remote cell death in the cerebellar system. Cerebellum. 2009;8(3):184–91. doi: 10.1007/s12311-009-0107-7. [DOI] [PubMed] [Google Scholar]
- Vos AA, Bruinse HW. Congenital adrenal hyperplasia: do the benefits of prenatal treatment defeat the risks? Obstet Gynecol Surv. 2010;65(3):196–205. doi: 10.1097/OGX.0b013e3181d61046. [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Hojo R, Kuroda Y, Fujimaki H. Estrogen modulates Bcl-2 family protein expression in the sexually dimorphic nucleus of the preoptic area of postnatal rats. Neurosci Lett. 2008;432(1):58–63. doi: 10.1016/j.neulet.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Veiga S, Garcia-Segura LM, Azcoitia I. Neuroprotection by the steroids pregnenolone and dehydroepiandrosterone is mediated by the enzyme aromatase. J Neurobiol. 2003;56(4):398–406. doi: 10.1002/neu.10249. [DOI] [PubMed] [Google Scholar]
- Whalen PJ, Phelps EA. The human amygdala. New York, NY: Guilford press; 2009. [Google Scholar]
- Wiltgen BJ, Godsil BP, Peng Z, Saab F, June HL, Linn ML, Cook JM, Houser CR, O'Dell TJ, Homanics GE, Fanselow MS. The alpha1 subunit of the GABA(A) receptor modulates fear learning and plasticity in the lateral amygdala. Front Behav Neurosci. 2009;3:37. doi: 10.3389/neuro.08.037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RI. Estradiol, but not dihydrotestosterone, in the medial amygdala facilitates male hamster sex behavior. Physiol Behav. 1996;59(4-5):833–41. doi: 10.1016/0031-9384(95)02204-x. [DOI] [PubMed] [Google Scholar]
- Yi SJ, Masters JN, Baram TZ. Glucocorticoid receptor mRNA ontogeny in the fetal and postnatal rat forebrain. Mol Cell Neurosci. 1994;5(5):385–93. doi: 10.1006/mcne.1994.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmazer-Hanke DM, Faber-Zuschratter H, Linke R, Schwegler H. Contribution of amygdala neurons containing peptides and calcium-binding proteins to fear-potentiated startle and exploration-related anxiety in inbred Roman high- and low-avoidance rats. Eur J Neurosci. 2002;15(7):1206–18. doi: 10.1046/j.1460-9568.2002.01945.x. [DOI] [PubMed] [Google Scholar]
- Yilmazer-Hanke DM, Hantsch M, Hanke J, Schulz C, Faber-Zuschratter H, Schwegler H. Neonatal thyroxine treatment: changes in the number of corticotropin-releasing-factor (CRF) and neuropeptide Y (NPY) containing neurons and density of tyrosine hydroxylase positive fibers (TH) in the amygdala correlate with anxiety-related behavior of wistar rats. Neuroscience. 2004;124(2):283–97. doi: 10.1016/j.neuroscience.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Yu S, Patchev AV, Wu Y, Lu J, Holsboer F, Zhang JZ, Sousa N, Almeida OF. Depletion of the neural precursor cell pool by glucocorticoids. Ann Neurol. 2010;67(1):21–30. doi: 10.1002/ana.21812. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Gonzalez-Burgos G, Povysheva NV, Kroner S, Lewis DA, Krimer LS. Localization of calcium-binding proteins in physiologically and morphologically characterized interneurons of monkey dorsolateral prefrontal cortex. Cereb Cortex. 2005;15:1178–1186. doi: 10.1093/cercor/bhh218. [DOI] [PubMed] [Google Scholar]
- Zhang LX, Levine S, Dent G, Zhan Y, Xing G, Okimoto D, Kathleen Gordon M, Post RM, Smith MA. Maternal deprivation increases cell death in the infant rat brain. Brain Res Dev Brain Res. 2002;133(1):1–11. doi: 10.1016/s0926-6410(01)00118-5. [DOI] [PubMed] [Google Scholar]
- Zhou R, Wang S, Zhu X. Prenatal ethanol exposure attenuates GABAergic inhibition in basolateral amygdala leading to neuronal hyperexcitability and anxiety-like behavior of adult rat offspring. Neuroscience. 2010;170(3):749–57. doi: 10.1016/j.neuroscience.2010.07.055. [DOI] [PubMed] [Google Scholar]