

Abstract
During embryonic development of the visual system, retinal ganglion cells (RGCs) project their axons towards the brain, passing through the optic chiasm. Axons are guided on this journey by molecular cues in the environment. The heparan sulphate sulphotransferase (Hst) enzymes Hs2st and Hs6st1 are each known to be required for specific aspects of axon guidance in the developing visual system, as revealed by studies of Hs2st−/− and Hs6st1−/− mutant embryos. However, it remained possible that these two enzymes have additional, overlapping, functions in RGC axon guidance; but that no effect is manifest in single mutant embryos, because the other enzyme is sufficient to fulfil the shared function. To investigate this possibility, we generated a set of Hs2st;Hs6st1 double mutant embryos that had reduced gene dosage of each of these Hsts, reasoning that any additional phenotypes in these animals would indicate the presence of functional overlap. We first characterised the structure of the mutant Hs6st1 locus, identifying the insertion site of the gene trap vector, to allow us to genotype compound mutants reliably. We found that Hs2st−/−;Hs6st1−/− mutants that lack both enzymes died prior to E15.5. As the optic chiasm has not formed by this stage, we were unable to determine the effect of complete loss of Hs2st and Hs6st1 on chiasm formation. However, compound mutant embryos lacking one Hst and heterozygous for the other were viable. We found that RGC axon guidance defects in such compound mutants were no more severe than those found in the single mutant embryos. We also found that expression of the Hs6st1 isoform Hs6st3 overlaps with that of Hs6st1 in the developing visual system, suggesting that some Hs6st activity remains present in this region of Hs6st1−/− mutant embryos.
Keywords: axon guidance, heparan sulphate proteoglycan, heparan sulphate sulphotransferase, Hs2st, Hs6st1, optic chiasm
Introduction
The retinal ganglion cell (RGC) axon projection of the visual system has been a particularly good model for elucidating developmental mechanisms used by axons to navigate through the developing nervous system (Mann et al. 2004; Inatani, 2005; Erskine & Herrera, 2007; Haupt & Huber, 2008). RGCs project their axons from the back of the retina to form the optic nerves. The optic nerves from each eye meet at the ventral surface of the hypothalamus, forming the characteristic X-shaped structure of the optic chiasm. Here, RGC axons are sorted into the optic tracts that project dorso-caudally, carrying the axons towards their targets in the dorsal lateral geniculate nucleus and the superior colliculus.
Heparan sulphate proteoglycans are cell surface and extracellular matrix macromolecules composed of a core protein and a variable number of covalently attached glycosaminoglycan (GAG) side-chains. Widely distributed and structurally diverse, heparan sulphate proteoglycans (HSPGs) have been shown to interact selectively with a myriad of extracellular proteins and thereby affect many biological processes. The GAG side-chains of HSPGs vary in length and are subjected to extensive post-translational modifications, including sulphation (Esko & Lindahl, 2001; Esko & Selleck, 2002; Lin & Perrimon, 2002; Gorsi & Stringer, 2007). Sulphation is carried out by heparan sulphate sulphotransferases, including 2-O sulphotransferase (Hs2st) and 6-O sulphotransferase (Hs6st), which sulphate the 2-O position of uronic acid and the 6-O position of glucosamine, respectively. Sulphotransferase reactions typically do not go to completion, contributing to the enormous potential diversity in HS structures that confers enormous potential for regulating multiple biological activities (Turnbull et al. 2001).
Animals carrying inactivating mutations in specific HSPG-modifying enzymes have shown that specific HSPG modifications are required for regulation of important developmental processes (Bullock et al. 1998; Ringvall et al. 2000; Shworak et al. 2002; Li et al. 2003; McLaughlin et al. 2003; Pan et al. 2006; Habuchi et al. 2007; Hu et al. 2009), including axon guidance events (Inatani et al. 2003; Johnson et al. 2004; Lee & Chien, 2004; Steigemann et al. 2004; Grobe et al. 2005; Pratt et al. 2006; Irie et al. 2008; Conway et al. 2011). Previously, we examined development of the optic chiasm in mutant mouse embryos lacking either of the heparan sulphate sulphotransferases Hs2st and Hs6st1. We found that these mutants have distinct phenotypes at the optic chiasm, mediated at least in part by effects on Slit/Robo signalling. The distinct phenotypes found in Hs2st−/− and Hs6st1−/− mutants are consistent with distinct functions for 2-O and 6-O sulphated HSPGs in guiding RGC axons (Pratt et al. 2006; Conway et al. 2011). However, it remained possible that the enzymes encoded by these genes have other activities whose effects are not seen in the single mutant embryos because they are compensated for by the function of the other enzyme. To test this hypothesis, we generated a set of compound mutants in which the gene dosage of each Hst is lowered, in the absence of the other, and determined whether this revealed additional phenotypes not observed in either of the single mutants. We found that Hs2st−/−;Hs6st1−/− mutants die before E15.5, despite the fact that each single mutant can survive until at least the time of birth. Although this early embryonic lethality precluded analysis of the optic chiasm in doubly homozygous mutants, we were able to examine compound mutants that lacked three out of the four hst alleles. We found that these had no more severe defects than either single mutant. There are two additional isoforms of Hs6st1 in the mouse, encoded by the Hs6st2 and Hs6st3 genes. We examined the expression of all three Hs6st genes in the developing visual system and found that the expression domain of Hs6st3 overlaps significantly with that of Hs6st1, and may therefore be able to compensate for the loss of Hs6st1 in the developing visual system.
Materials and methods
Animals
Hs6st1LacZ-IRES-hPLAP (Hs6st1+/−) mice were maintained on a pigmented CBA background by crossing wild-type CBA females with Hs6st1+/− males (Leighton et al. 2001; Mitchell et al. 2001). Hs2stLacZ (Hs2st+/−) mice were maintained on a pigmented CBA background by crossing wild-type CBA females with Hs2st+/− males (Bullock et al. 1998; Leighton et al. 2001). Compound Hs2st+/−/Hs6st1+/− mutants were generated by crossing Hs2st+/− and Hs6st1+/− animals. The day that the vaginal plug was detected was designated E0.5, and the day of birth as P0.
Genotyping
Hs2st animals were genotyped using a multiplex PCR strategy as described previously (Pratt et al. 2006). Hs6st1 mutant animals were initially identified using the activity of the LacZ reporter in the gene trap vector as described previously (Pratt et al. 2006), and subsequently using the strategy outlined in Fig. 1. Primer sequences: P1: 5′ATGGTGACTGTGACCCACAA3′; P2: 5′GGGATATAGGGGACCTTGGA3′ (179 bp) and hP1: 5′ACAGCTGCCAGGATCCTAAA3′; hP2: 5′CCCACCTTGGCTGCAGTCAT3′ (328 bp). Reaction parameters were: 30 cycles of 30 s at 94 °C; 30 s at 58 °C; 30 s at 72 °C.
Fig. 1.
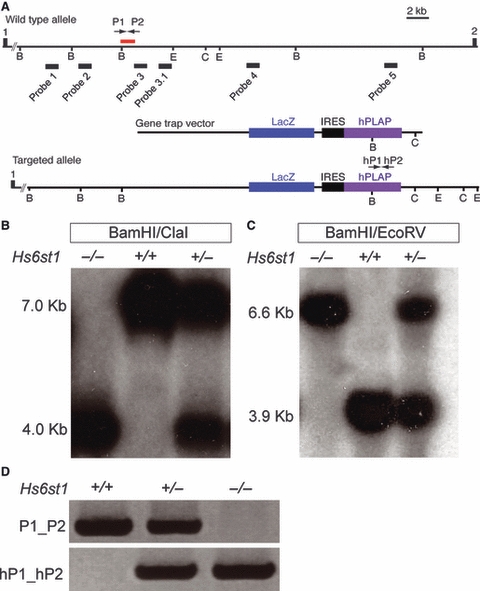
Characterisation of the Hs6st1 gene trap allele. (A) Schematic representation of the Hs6st1-gene trapped locus. Hs6st1 consists of two exons separated by a 35-kb intron. The gene trap vector has inserted into intron1 and abolishes wild-type Hs6st1 transcripts. (B) Southern blot analysis on BamHI/ClaI digested genomic DNA from wild-type, Hs6st1+/− and Hs6st1−/− tissue using radiolabelled DNA probes 1–5. Probe 3 hybridised to a 7-kb restriction fragment in wild-type and a 4-kb restriction fragment in Hs6st1 mutants, indicating that the gene trap vector had inserted into the 7-kb BamHI–ClaI restriction fragment in the Hs6st1 allele. (C) Probe 3 hybridised to a 3.9-kb BamHI/EcoRV restriction fragment in wild-type and a 6.6-kb fragment in Hs6st1 mutants. (D) PCR primers P1/P2 amplify a 205-bp region of Hs6st1 intron1 at the gene trap insertion site. This PCR product was absent in Hs6st1−/− mutants, but present in wild-type and Hs6st1+/− heterozygotes. Primers hP1 and hP2 amplify a 316-bp region within the hPLAP coding sequence, which is present in Hs6st1+/− and Hs6st1−/− mutants, but absent in wild-types.
DNA probes for Southern blotting
Using Primer3 software (http://frodo.wi.mit.edu/primer3/), we designed six sets of PCR primer pairs that amplified specific segments of Hs6st1 intron1. These primer pairs were used to generate the following DNA probes:
Probe1: 5′CCTGTCATTTGCTGGCTGTTG3′, 5′CTAAACGCTGGAAAGGGTTGC3′;
Probe2: 5′CCCCAGGCTCCATCTCTATGTTAG3′, 5′CAAGTCCCCCATTTCCAGTAGTG3′;
Probe 3: 5′TGGAGAAGACAGTGTTGTGGGC3′, 5′CAGCAGAATGAGGCAGTCAAGC3′;
Probe 3.1: 5′-TATCCATCTCTGCTCCCTGACTGC-3′, 5′GCTGAATAAACCCTTTCTTCCCC-3′;
Probe 4: 5′GAGCCTGAATGAATGAACCCTCCTTC3′, 5′CCTGTCCTGACTTCCTTTGGTG3′;
Probe 5: 5′GGGGTGTCGTGGCTTTTTTC3′, 5′TGCTTCCGTGTGAGTCAGGATG3′.
Polymerase chain reaction products were gel-purified and extracted using a Qiagen Gel Extraction kit according to manufacturer's instructions before cloning into plasmid pCR2.1 using an Invitrogen TA cloning kit. Probe fragments were excised using EcoRI (New England Biolabs) and labelled with 32P-dCTP using Rediprime II Random Probe labelling system (Amersham), following the manufacturer's instructions.
Southern blotting
Genomic DNA was extracted from the liver of E15.5 embryos using phenol-chloroform and digested with BamHI and ClaI overnight at 37 °C and run on a 0.8% agarose gel for 16 h at 40 V in 1 × TBE buffer without ethidium bromide. Subsequent steps were carried out as described previously (Rose et al. 2002).
In situ hybridisation
Hs2st primers were designed using primer3 software to create an RNA antisense riboprobe complementary to the 3′-UTR of the Hs2st transcript: 5′TGTGAACGGCTTATGCACTC3′, 5′CTGTGACAGCGACTTTCCAA3′. Hs6st1, Hs6st2 and Hs6st3 riboprobe plasmids were kindly provided by K. Izvolsky (Sedita et al. 2004). E15.5 brains were fixed in 4% paraformaldehyde (PFA)/phosphate-buffered saline (PBS) pH 9.5 overnight at 4 °C and sectioned horizontally at 100 μm using a vibratome. Sections were mounted on Superfrost Plus (VWR International) slides and dried at 37 °C overnight. In situ hybridisation for Hs2st and Hs6st1 was performed using digoxigenin-labelled antisense probes, as described by Erskine et al. (2000).
DiI tract tracing
E15.5 heads were fixed in 4% PFA/PBS overnight at 4 °C with shaking. The following day, 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI) crystals were packed into the exposed retina of one eye and allowed to diffuse for 6 weeks in 4% PFA/PBS at room temperature. Heads were then placed in 4% low-melting agarose/ddH2O and sectioned horizontally at 200 μm using a vibratome. Sections were cleared in 1 : 1 glycerol : PBS overnight at 4 °C, and then further cleared in 9 : 1 glycerol : PBS containing the nuclear counter-stain TOPRO3 (1 μL mL−1) overnight at 4 °C. Sections were mounted in Vectashield on glass slides. Images were captured using either an epifluorescense microscope (with TRITC filter) with digital camera (Leica Microsystems, Germany) or a TCS NT confocal microscope (Leica Microsystems). DiI back-labelled RGCs were quantified by counting RGC bodies observed at × 20 objective on serial sections as described previously (Pratt et al. 2006).
Results
Mapping the gene trap insertion site in the Hs6st1 locus facilitates polymerase chain reaction (PCR) genotyping of mutants
In order to genotype Hs2st;Hs6st1 compound mutant embryos reliably, we first had to design a PCR genotyping strategy to identify Hs6st1 mutant embryos. We therefore sought to identify the precise insertion site of the gene trap vector in the Hs6st1 locus. The insertion site had previously been localised to the 35-kb intron1 of Hs6st1 (Fig. 1A; Leighton et al. 2001; Mitchell et al. 2001; Pratt et al. 2006). We used Southern blotting to narrow down the insertion site more precisely. We generated five specific DNA probes distributed across Hs6st1 intron1 (designated as probes 1–5, Fig. 1A) and used them to screen for BamHI/ClaI restriction fragments whose length was altered in mutant embryos, indicating that they contained the insertion site. Probe 3 hybridised to a 7.0-kb BamHI/ClaI fragment in Hs6st1+/−, and Hs6st1−/− mutants and a 4.0-kb fragment in wild type Hs6st1+/+ DNAs (Fig. 1B), while the other four probes hybridised to identically sized fragments in all genotypes (not shown). This showed that the insertion site lies within a 7-kb BamHI/ClaI fragment of intron1. We further narrowed down the insertion site to a 3.9-kb BamHI/EcoRV fragment, again using probe 3 (Fig. 1C). A further probe, derived from the 3′ region of the 3.9-kb BamHI/EcoRV fragment (probe 3.1, Fig. 1A), gave rise to an identical hybridisation pattern on BamHI/EcoRV digested DNA to that obtained with probe 3 (not shown), allowing us to deduce that the gene trap vector had inserted on the 5′ side of probe 3, thus narrowing down the insertion site to an 1100-bp region of Hs6st1 intron1, indicated by a red bar in Fig. 1A.
Screening a series of primers designed in and around this 1100-bp region identified a PCR primer pair (P1/P2) that produced a 205-bp product in wild-type and Hs6st1+/− embryos (Fig. 1A,D). This product could not be detected in Hs6st1−/− mutant embryos, indicating that the primers likely flanked the insertion site. However, despite numerous attempts we were unable to identify a primer pair that would amplify a fragment encompassing intron1 sequences 5′ to the insertion site and the gene trap vector sequence (i.e. including the precise insertion site). This was most likely due to DNA rearrangement at the insertion that altered the sequence of intron1. Instead, we used a primer pair (designated hP1/hP2) designed to identify the gene trap-specific hPLAP reporter, which produced a 316-bp PCR product in Hs6st1+/− and Hs6st1−/− mutant embryos (Fig. 1D). This combination of PCR reactions allows unambiguous genotyping of the Hs6st1 mutants (Fig. 1D).
Only a small percentage of Hs6st1−/− mutants survive postnatally
Inter-crossing Hs2st+/−;Hs6st1+/− animals should give rise to Hs2st−/−;Hs6st1−/− double mutant progeny at a frequency of one in 16. This frequency would improve substantially if it were possible instead to use parent animals that were homozygous for one of the mutant alleles. This is not possible for Hs2st−/− mutants, which die around the time of birth (Bullock et al. 1998). However, the postnatal viability of the Hs6st1 homozygous mutants was unknown. We therefore inter-crossed Hs6st1+/− mice and genotyped their progeny either at E14.5 or at postnatal day (P)21 (Table 1). Hs6st1−/− mutants were found at the expected ratio at E14.5, but only three of 100 mice genotyped at P21 were Hs6st1−/− mutants. This under-representation was highly statistically significant (chi-squared test, P < 0.001). We did not examine the cause(s) underlying the poor postnatal survival of Hs6st1−/− mutant animals. However, mice homozygous for a null allele of Hs6st1 generated by gene-targeting have been reported to have a similarly low postnatal survival rate, attributed to defects in placental development (Habuchi et al. 2007).
Table 1.
Genotypes of E14.5 and P21 progeny obtained from Hs6st1+/− × Hs6st1+/− inter-crosses
| Age | No. of animals | Genotype | Observed (%) | Expected (%) |
|---|---|---|---|---|
| E14.5 | 50 | Wild-type | 13 (25) | 13 (25) |
| Hs6st1+/− | 24 (48) | 25 (50) | ||
| Hs6st1−/− | 13 (25) | 13 (25) | ||
| P21 | 100 | Wild-type | 26 (26) | 25 (25) |
| Hs6st1+/− | 71 (71) | 50 (50) | ||
| Hs6st1−/− | 3 (3)* | 25 (25) |
Chi-squared analysis P < 0.001.
Hs2st−/−;Hs6st1−/− double mutants die before E15.5
Hs2st+/−;Hs6st1+/− mice were inter-crossed and their progeny were PCR genotyped at E15.5 (Table 2). Mendelian laws predict that 1/16 of embryos resulting from such a cross should be Hs2st−/−;Hs6st1−/− double mutants; however, none of the 159 E15.5 embryos generated had this genotype. Chi-squared analysis showed that the difference between the observed and expected numbers of Hs2st−/−;Hs6st1−/− embryos was statistically significant (P < 0.05; Table 2). None of the other possible genotypes from this cross were under-represented. Hs2st−/−;Hs6st1+/− embryos were, however, slightly over-represented, although any biological relevance of this is unclear.
Table 2.
Genotypes of E15.5 progeny obtained by intercrossing Hs2st+/−;Hs6st1+/− mice
| Age | No. of animals | Genotype | Observed (%) | Expected (%) |
|---|---|---|---|---|
| E15.5 | 159 | Wild-type | 10 (6.3) | 10 (6.3) |
| Hs2st+/+;Hs6st1+/− | 19 (11.9) | 20 (12.6) | ||
| Hs2st+/+;Hs6st1−/− | 12 (7.5) | 10 (6.3) | ||
| Hs2st+/−;Hs6st1+/+ | 17 (10.7) | 20 (12.6) | ||
| Hs2st+/−;Hs6st1+/− | 41 (25.8) | 40 (24.5) | ||
| Hs2st+/−;Hs6st1−/− | 20 (12.6) | 20 (12.6) | ||
| Hs2st−/−;Hs6st1+/+ | 10 (6.3) | 10 (6.3) | ||
| Hs2st−/−;Hs6st1+/− | 30 (18.9)* | 20 (12.6) | ||
| Hs2st−/−;Hs6st1−/− | 0 (0)* | 10 (6.3) |
Chi-squared analysis P < 0.05.
The failure of Hs2st−/−;Hs6st1−/− mutants to survive to E15.5 prevented analysis of axon guidance at the optic chiasm in embryos lacking both of these enzymes. However, it remained possible that the reduced gene dosage of either Hst found in heterozygous embryos may reveal additional phenotypes when combined with homozygous mutation of the other Hst. We next examined the optic chiasms in a series of Hs2st;Hs6st1 compound mutants to look for possible heterozygous effects.
No evidence for overlapping functions between Hs2st and Hs6st1 in the formation of ectopic retino-retinal projections
Loss of Hs6st1 sulphation leads to a dramatic increase in the number of RGC axons mis-projecting from one eye to the other (Pratt et al. 2006). This phenotype can be quantitated reliably, and we have used it previously to investigate interactions between Hst and Slit genes (Conway et al. 2011). We measured the extent of ectopic inter-retinal projections in Hs2st;Hs6st1 compound heterozygous mutants by injecting DiI into one eye of E15.5 embryos and subsequently counting back-labelled RGC bodies in serial sections of the contralateral retina (Fig. 2). We found no significant increase in the number of misprojecting RGC axons in Hs2st−/−;Hs6st1+/+ mutants, Hs2st−/−;Hs6st1+/− double mutants or Hs2st+/−;Hs6st1+/− double mutants when compared with wild-types (Fig. 2E). As expected, RGC counts revealed a significant increase in the number of RGC axons misprojecting to the opposite eye in Hs2st+/+;Hs6st1−/− mutants and Hs2st+/−;Hs6st1−/− double mutants, compared with wild types. However, no significant difference was found between Hs2st+/+;Hs6st1−/− and Hs2st+/−;Hs6st1−/− double mutants (Fig. 2E).
Fig. 2.
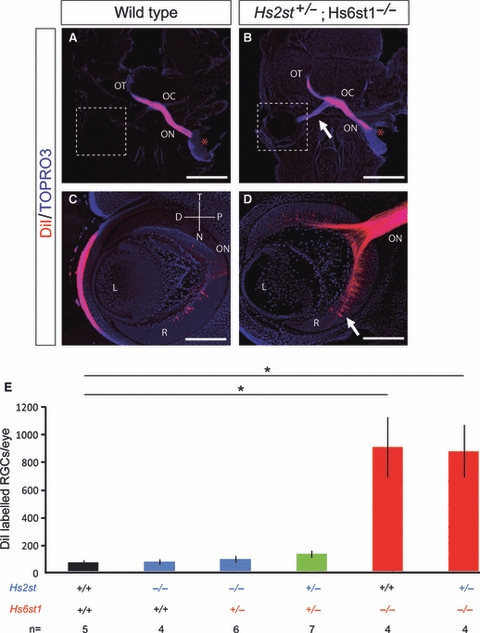
Quantitation of inter-retinal projection in Hs2st;Hs6st1 compound mutants. Retinal ganglion cell (RGC) axons were labelled by placing DiI (red*) unilaterally into the retina of E15.5 embryos and their trajectories observed in 200-μm horizontal sections. Nuclei were counterstained with TOPRO3 (blue). (A) In wild-type embryos, few RGC axons mis-navigated from one eye to the other, compared with the much more conspicuously labelled contralateral optic nerve in Hs2st+/−;Hs6st1−/− mutants (B, white arrow). (C, D) Higher power views showing back-labelled RGC bodies in the contralateral retina of labelled wild-type (C) and Hs2st+/−;Hs6st1−/− mutant (D) embryos. Many more back-labelled RGCs are visible in the mutant retina (white arrow). (E) Quantitative analysis of the number of back-labelled RGC bodies in the contralateral eye (mean ± SEM) showing no statistically significant change in the magnitude of the retino-retinal projection in Hs2st−/− mutants 65 ± 17 (n = 4), Hs2st+/−;Hs6st1+/− double mutants 119 ± 25 (n = 7) or Hs2st−/−;Hs6st1+/− double mutants 84 ± 25 (n = 6) when compared with wild-type 59 ± 18 (n = 5). While there was a statistically significant increase in the number of back-labelled RGC bodies in Hs6st1−/− mutants 894 ± 219 (n = 4) and Hs2st+/−;Hs6st1−/− double mutants 865 ± 191 (n = 4) when compared with the wild-type [Kruskal–Wallis one-way anova on ranks, pair-wise multiple comparisons (Dunn's method), P < 0.05], there was no significant difference when they were compared with each other. Asterisks indicate statistically significant differences. D, distal; L, lens; N, nasal; OC, optic chiasm; ON, optic nerve; OT, optic tract; P, proximal; R, retina; T, temporal. Scale bars: 400 μm (A,B); 100 μm (C,D).
No evidence for overlapping functions between Hs2st and Hs6st1 at the optic chiasm
We showed previously that many RGC axons in Hs2st−/− mutants wander inappropriately along the ventral midline of the brain (Pratt et al. 2006). Subsequently, we showed that some RGC axons also project ectopically into the pre-optic area in Hs2st−/− mutants (Conway et al. 2011). Together, the effect of these two phenotypes is to increase the rostro-caudal extent of the optic chiasm. Further, in Hs6st1−/− mutants, ectopic axon navigation along the caudal midline (arrow in Fig. 3C) leads to an apparent rostro-caudal widening of the optic chiasm. To look for evidence of overlapping function between Hs2st and Hs6st1, we therefore compared the width of the optic chiasm at the anatomical midline in compound Hs2st/Hs6st1 mutants with that in Hs2st−/− and Hs6st1−/− single mutants (Fig. 3). RGC axons were labelled by injecting DiI into one eye of E15.5 embryos, and the rostro-caudal extent of the optic chiasm (indicated by a dotted red line in Fig. 3B–E) was measured. As expected, we found a significant increase in the rostro-caudal width of the chiasm in Hs2st−/−;Hs6st1+/+ mutants, compared with wild-types (Fig. 3B,F). Both Hs2st−/−;Hs6st1+/+ mutants and Hs2st−/−;Hs6st1+/− double mutants showed an ectopic projection into the pre-optic area (arrow in Fig. 3B,D), but no significant differences were observed at the optic chiasm between these two genotypes (Fig. 3B,D). The rostro-caudal width of the optic chiasm was significantly increased in both Hs2st+/+; Hs6st1−/− and Hs2st+/−;Hs6st1−/− mutants compared with wild-types, due to ectopic RGC axon navigation along the caudal midline of the ventral diencephalon (white arrows in Fig. 3C,E). However, no significant differences were found between the optic chiasms of these two genotypes (Fig. 3C,E,F).
Fig. 3.
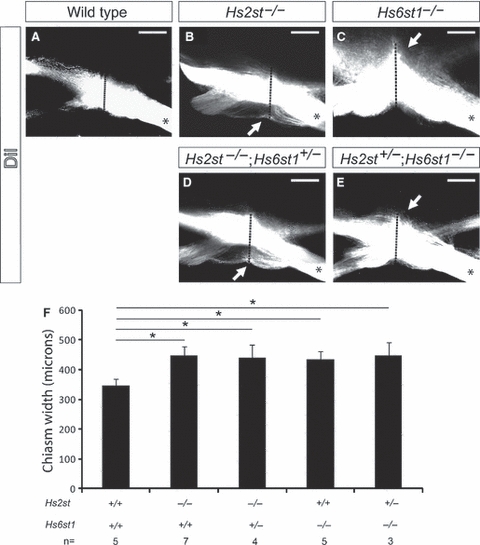
Quantitation of optic chiasm width in Hs2st;Hs6st1 compound mutants. Labelling of optic nerve following unilateral DiI injection (denoted by *) into E15.5 eyes. Labelled axons appear white in these grey-scale images and reveal the anatomy of the optic chiasm in (A) wild-type, (B) Hs2st−/− and (C) Hs6st1−/− embryos. In Hs2st−/− mutants, the optic chiasm appeared wider along the rostro-caudal midline due to the formation of an ectopic tract rostral to the chiasm (white arrow in B). In mutants lacking Hs6st1 sulphation the optic chiasm appeared wider along the rostro-caudal midline due to ectopic axon navigation along the caudal midline of the ventral diencephalon (white arrow in C). The width of the optic chiasm, taken to consist of any DiI RGC axon that could be detected using confocal microscopy, was measured along the midline (red dotted line). (F) Graph showing the width (mean ± SEM) of the optic chiasm in wild-type and compound mutant embryos. The optic chiasm was significantly wider in both the Hs6st1−/− mutant (435 ± 25 μm, n = 5) and Hs2st−/− mutant (448 ± 30 μm, n = 7) than in wild-types [345 ± 51 μm, n = 5; one way anova, multiple comparisons vs. a control group (Holm–Sidak method), P < 0.05]. The optic chiasm in Hs2st−/−;Hs6st1+/− mutants (n = 4) was 438 ± 45 μm wide, and in Hs2st+/−;Hs6st1−/− mutants (n = 3) it was 447 ± 44 μm wide. These values are not significantly different to those obtained in Hs2st−/− and Hs6st1−/− single mutants, respectively. OC, optic chiasm; ON, optic nerve; OT, optic tract. Scale bars: 200 μm.
Expression of Hs6st isoforms in the developing retina and optic chiasm
There are three Hs6st isoforms in mouse, encoded by separate genes; Hs6st1, Hs6st2 and Hs6st3 (Habuchi et al. 2000), each of which is expressed in a different pattern during embryogenesis (Sedita et al. 2004). We previously described the expression of Hs6st1 in the developing retina and optic chiasm, based on expression of the β-galactosidase reporter contained in the gene trap vector (Pratt et al. 2006), but the expression patterns of Hs6st2 and Hs6st3 in the retina and at the optic chiasm have not been reported. We determined the expression patterns of Hs6st1, Hs6st2 and Hs6st3 in the retina and at the optic chiasm at E14.5 by in situ hybridisation. Hs6st1 RNA was detected in the RGC layer, the region of the optic nerve closest to the eye, and in a distinct pattern at the optic chiasm (Fig. 4A), similar to that seen for the β-galactosidase reporter (Pratt et al. 2006). In contrast, we found no Hs6st2 expression in the RGC layer and very little at the optic chiasm (Fig. 4B). Hs6st3 was expressed at high levels in the RGC layer and at a slightly lower level around the optic chiasm (Fig. 4C). It is therefore likely that, even in the absence of Hs6st1, some 6-O sulphation of HSPGs is still present in the developing visual system.
Fig. 4.
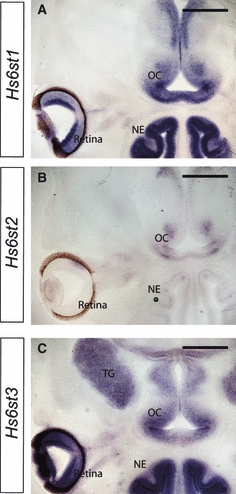
Expression patterns of Hs6st1, Hs6st2 and Hs6st3 in the retina and optic chiasm at E14.5. RNA in situ hybridisation showing expression of three Hs6st isoforms in 100-μm horizontal sections of wild-type embryos. (A) Hs6st1 is expressed in the RGC layer as well as in a distinct pattern at the ventral diencephalon where the optic chiasm will form with particularly high expression where the optic nerve first encounters the ventral diencephalon. (B) No Hs6st2 expression was detected in the retina, and little expression was observed at the ventral diencephalon. (C) High expression of Hs6st3 was seen in the RGC layer, with slightly lower levels expressed at the ventral diencephalon. OC, optic chiasm; NE, nasal epithelium; TG, trigeminal ganglion. Scale bars: 400 μm.
Discussion
Hs2st and Hs6st1 each have unique functions in axon guidance at the optic chiasm, indicating that 2-O and 6-O sulphation have separate roles in influencing axon navigation responses at the optic chiasm (Pratt et al. 2006; Conway et al. 2011). However, the question of whether Hs2st and Hs6st1 may also have overlapping functions in mediating axon guidance at the optic chiasm has not previously been addressed. We aimed to create Hs2st−/−;Hs6st1−/− animals to investigate this possibility. As part of our strategy to maximise recovery of double mutants, we first characterised the survival rate of Hs6st1−/− animals. We found that Hs6st1−/− embryos were present at expected frequencies at E14.5, but only 3% of those offspring that survived to weaning (P21) were Hs6st1−/−. Another group has reported the generation and characterisation of a conventional Hs6st1−/− knockout mouse line (Habuchi et al. 2007). These authors found that their Hs6st1−/− embryos were present in normal numbers at E14.5, but at P21 only 4% of C57Bl/6 offspring were Hs6st1−/−. Those Hs6st1−/− animals that did survive to adulthood were smaller than wild-type littermates, but were outwardly healthy and fertile (Habuchi et al. 2007). The late embryonic or early postnatal lethality observed in these Hs6st1−/− mutants was attributed to a ∼50% reduction in the number of foetal micro-vessels in the labyrinthine zone of the placenta. Gross observations of the placenta in our Hs6st1−/− embryos did show a dark red discoloration that was not seen in the placentas of wild-type embryos (data not shown), consistent with the possibility that the high incidence of postnatal lethality in our Hs6st1−/− mutants is also the result of placental defects.
We found that Hs2st−/−;Hs6st1−/− embryos did not survive until E15.5, preventing any analysis of axon guidance at the optic chiasm in these double mutants. Lethality is a common theme in animals carrying mutations in genes that encode HSPG-modifiying enzymes, indicating the importance of HSPG modifications in embryonic development (Bullock et al. 1998; Lin et al. 2000; Li et al. 2003; Grobe et al. 2005; Stickens et al. 2005; Habuchi et al. 2007). The finding that Hs2st−/−;Hs6st1−/− animals die early in development, while Hs2st−/− and Hs6st1−/− animals can survive to birth and beyond is consistent with the possibility that these genes have overlapping functions in development – for example, Hs2st and Hs6st1 may both be required in the same critically important developmental pathway, and the presence of at least one copy of either of the two genes is sufficient for the pathway to function. However, it is also possible that Hs2st function is required for one developmental pathway while Hs6st1 is required for another; a loss of both pathways resulting in early embryonic death.
While the failure to generate Hs2st−/−;Hs6st1−/− double mutants prevented us from unambiguously determining whether or not Hs2st and Hs6st1 have overlapping roles in guiding axons at the optic chiasm, it was still possible that compound mutants lacking one Hst and heterozygous for the other may reveal additional phenotypes. There are strong precedents for heterozygous phenotypes in mutants affecting HS structure. For example, heterozygous mutation of Ext1 (which encodes an HS biosynthetic enzyme) combined with loss of Slit2 function, confers an optic chiasm phenotype intermediate in severity between those of Ext1+/+;Slit2−/− and Ext1−/−;Slit2−/− mutants (Inatani et al. 2003). Heterozygous effects have also been reported in both Hs2st+/− and Hs6st1+/− mutants. HS extracted from Hs2st+/− animals has decreased bioactivity in cell assays involving fibroblast growth factor (FGF) signalling activation, despite the presence of some 2-O sulphation, suggesting the importance of Hs2st gene dosage in establishing normal biological activity (Guimond et al. 2009). Further, Hs6st1+/−;Slit2+/− compound heterozygotes revealed synergistic interaction between Hs6st1 and Slit2 during formation of the corpus callosum (Conway et al. 2011). However, we have not identified an Hs2st+/− or Hs6st1+/− phenotype at the optic chiasm. Further, we found no differences at the optic chiasm between Hs2st−/−;Hs6st1+/+ and Hs2st−/−;Hs6st1+/− embryos or between Hs2st+/+;Hs6st1−/− and Hs2st+/−;Hs6st1−/− embryos. While the evidence to date suggests that Hs2st and Hs6st1 have unique, non-overlapping functions in optic chiasm formation, we cannot rule out the possibility that a single copy of either Hs2st or Hs6st1 is sufficient to maintain a signalling pathway that requires both Hs2st and Hs6st1 function without characterising Hs2st−/−;Hs6st1−/− double mutants. As a further complication, we found that the Hs6st3 gene, encoding an isoform of Hs6st, is also expressed in RGCs and around the presumptive optic chiasm and therefore has the potential to provide 6-O sulphation in the developing visual system of mutants lacking Hs6st1, although this must be insufficient to allow normal development of the optic chiasm.
A recently published study found evidence of overlapping functions between Hs2st and Hs6st1 during mouse lacrimal gland development using a conditional inactivation strategy (Qu et al. 2011). They found that loss of Hs2st alone had no effect on lacrimal gland development at E14.5, while about 20% of Hs6st1 conditional mutants lacked lacrimal glands at the same stage. Conditional inactivation of both Hs2st and Hs6st1 resulted in approximately 60% of embryos lacking lacrimal glands. The authors demonstrated that Hs2st and Hs6st1 activity are required together for optimal activity of FGF10 signalling during lacrimal gland development (Qu et al. 2011).
The present study demonstrates that the combined activities of Hs2st and Hs6st1 are critical for normal development; their absence leads to embryonic death before E15.5. In future work, it will be interesting to discover the embryonic stage at which Hs2st−/−;Hs6st1−/− double mutants die, and to establish the cause. Determining the extent of any functional overlap between Hs2st and Hs6st1 at the optic chiasm will require the use of conditional mutant animals.
Acknowledgments
We are grateful to K. Izvolsky for providing plasmids for in situ hybridisation probes. We thank the Biological Research Resources staff at the University of Edinburgh for expert animal husbandry. Work in the authors’ laboratories is funded by the Medical Research Council (MRC), and the Biotechnology and Biological Sciences Research Council (BBSRC).
References
- Bullock SL, Fletcher JM, Beddington RS, et al. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway CD, Howe KM, Nettleton NK, et al. Heparan sulfate sugar modifications mediate the functions of slits and other factors needed for mouse forebrain commissure development. J Neurosci. 2011;31:1955–1970. doi: 10.1523/JNEUROSCI.2579-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erskine L, Herrera E. The retinal ganglion cell axon's journey: insights into molecular mechanisms of axon guidance. Dev Biol. 2007;308:1–14. doi: 10.1016/j.ydbio.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Erskine L, Williams SE, Brose K, et al. Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. J Neurosci. 2000;20:4975–4982. doi: 10.1523/JNEUROSCI.20-13-04975.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Gorsi B, Stringer SE. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol. 2007;17:173–177. doi: 10.1016/j.tcb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, et al. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimond SE, Puvirajesinghe TM, Skidmore MA, et al. Rapid purification and high sensitivity analysis of heparan sulfate from cells and tissues: toward glycomics profiling. J Biol Chem. 2009;284:25 714–25 722. doi: 10.1074/jbc.M109.032755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi H, Tanaka M, Habuchi O, et al. The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J Biol Chem. 2000;275:2859–2868. doi: 10.1074/jbc.275.4.2859. [DOI] [PubMed] [Google Scholar]
- Habuchi H, Nagai N, Sugaya N, et al. Mice deficient in heparan sulfate 6-O-sulfotransferase-1 exhibit defective heparan sulfate biosynthesis, abnormal placentation, and late embryonic lethality. J Biol Chem. 2007;282:15 578–15 588. doi: 10.1074/jbc.M607434200. [DOI] [PubMed] [Google Scholar]
- Haupt C, Huber AB. How axons see their way – axonal guidance in the visual system. Front Biosci. 2008;13:3136–3149. doi: 10.2741/2915. [DOI] [PubMed] [Google Scholar]
- Hu Z, Wang C, Xiao Y, et al. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- Inatani M. Molecular mechanisms of optic axon guidance. Naturwissenschaften. 2005;92:549–561. doi: 10.1007/s00114-005-0042-5. [DOI] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, et al. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- Irie F, Okuno M, Matsumoto K, et al. Heparan sulfate regulates ephrin-A3/EphA receptor signaling. Proc Natl Acad Sci USA. 2008;105:12 307–12 312. doi: 10.1073/pnas.0801302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KG, Ghose A, Epstein E, et al. Axonal heparan sulfate proteoglycans regulate the distribution and efficiency of the repellent slit during midline axon guidance. Curr Biol. 2004;14:499–504. doi: 10.1016/j.cub.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Lee JS, Chien CB. When sugars guide axons: insights from heparan sulphate proteoglycan mutants. Nat Rev Genet. 2004;5:923–935. doi: 10.1038/nrg1490. [DOI] [PubMed] [Google Scholar]
- Leighton PA, Mitchell KJ, Goodrich LV, et al. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature. 2001;410:174–179. doi: 10.1038/35065539. [DOI] [PubMed] [Google Scholar]
- Li JP, Gong F, Hagner-McWhirter A, et al. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking L-iduronic acid and in neonatal lethality. J Biol Chem. 2003;278:28 363–28 366. doi: 10.1074/jbc.C300219200. [DOI] [PubMed] [Google Scholar]
- Lin X, Perrimon N. Developmental roles of heparan sulfate proteoglycans in Drosophila. Glycoconj J. 2002;19:363–368. doi: 10.1023/A:1025329323438. [DOI] [PubMed] [Google Scholar]
- Lin X, Wei G, Shi Z, et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol. 2000;224:299–311. doi: 10.1006/dbio.2000.9798. [DOI] [PubMed] [Google Scholar]
- Mann F, Harris WA, Holt CE. New views on retinal axon development: a navigation guide. Int J Dev Biol. 2004;48:957–964. doi: 10.1387/ijdb.041899fm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin D, Karlsson F, Tian N, et al. Specific modification of heparan sulphate is required for normal cerebral cortical development. Mech Dev. 2003;120:1479–1486. doi: 10.1016/j.mod.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, et al. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Pan Y, Woodbury A, Esko JD, et al. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- Pratt T, Conway CD, Tian NM, et al. Heparan sulphation patterns generated by specific heparan sulfotransferase enzymes direct distinct aspects of retinal axon guidance at the optic chiasm. J Neurosci. 2006;26:6911–6923. doi: 10.1523/JNEUROSCI.0505-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Carbe C, Tao C, et al. Lacrimal gland development and Fgf10-Fgfr2b signaling is controlled by 2-O and 6-O sulfated heparan sulfate. J Biol Chem. 2011;286:14 435–14 444. doi: 10.1074/jbc.M111.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringvall M, Ledin J, Holmborn K, et al. Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J Biol Chem. 2000;275:25 926–25 930. doi: 10.1074/jbc.C000359200. [DOI] [PubMed] [Google Scholar]
- Rose K, Mason JO, Lathe R. Hybridisation parameters revisited: solutions containing SDS. BioTechniques. 2002;33:54–58. doi: 10.2144/02331st01. [DOI] [PubMed] [Google Scholar]
- Sedita J, Izvolsky K, Cardoso WV. Differential expression of heparan sulfate 6-O-sulfotransferase isoforms in the mouse embryo suggests distinctive roles during organogenesis. Dev Dyn. 2004;231:782–794. doi: 10.1002/dvdy.20173. [DOI] [PubMed] [Google Scholar]
- Shworak NW, HajMohammadi S, de Agostini AI, et al. Mice deficient in heparan sulfate 3-O-sulfotransferase-1: normal hemostasis with unexpected perinatal phenotypes. Glycoconj J. 2002;19:355–361. doi: 10.1023/A:1025377206600. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Molitor A, Fellert S, et al. Heparan sulfate proteoglycan syndecan promotes axonal and myotube guidance by slit/robo signaling. Curr Biol. 2004;14:225–230. doi: 10.1016/j.cub.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Stickens D, Zak BM, Rougier N, et al. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 2005;132:5055–5068. doi: 10.1242/dev.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]