

Abstract
The largest genetic risk for late-onset Alzheimer’s disease (AD) resides at the apolipoprotein E gene (APOE) locus, which has three common alleles (ε2, ε3, ε4) that encode three isoforms (apoE2, apoE3, apoE4). The very strong association of the APOE ε4 allele with AD risk and its role in the accumulation of amyloid β and animal models solidify the biological relevance of apoE isoforms but do not provide mechanistic insight. The innate immune response is consistently observed in AD and is a likely contributor to neuronal injury and response to injury. Here we review emerging data showing that apoE isoform regulation of multiple facets of the innate immune response in the brain may alter AD not only through amyloid β-dependent mechanisms, but also through other, amyloid β-independent mechanisms.
Introduction
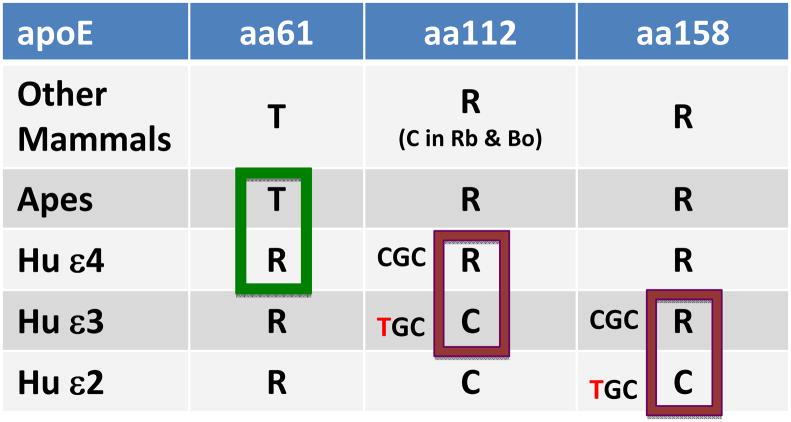
Alzheimer’s disease (AD) has rare autosomal dominant forms. However, the form of AD causing a major public health problem is late-onset AD, which is not caused by dominantly inherited mutations but rather by genes conferring genetic risk in combination with other processes that are currently ill-defined. Although the etiology and pathogenesis of late-onset AD remains obscure, a key component of all forms of AD appears to be accumulation of Aβ peptides: endoproteolytic fragments of the product of the amyloid precursor protein gene, APP. The risk of the common late-onset form of AD, likely arising from modulated age of onset, has been repeatedly associated with the apolipoprotein E gene (APOE) following pioneering work that now has been replicated in genome-wide association studies (GWAS) for AD [1–3]. The strength of the association of the APOE locus with AD is orders of magnitude greater than that of other loci. Humans have three common alleles of APOE, unlike most other mammals who possess only one allele. AD risk is greatest with inheritance of ε4 allele, less with ε3 allele, and least with ε2 allele, and there is a gene dosage effect. The corresponding human apoE isoforms are 299-amino acid proteins that differ in amino acids at positions 112 and 158 (Table) [4] and possess very well-characterized isoform-specific actions in a variety of biological contexts. ApoE is an integral constituent of many lipid transport lipoproteins, playing key roles in both particle assembly and structure, as well as receptor-mediated lipoprotein uptake via the family of cell surface LDL receptors. ApoE is synthesized principally in the liver, with a discrete pool synthesized, secreted and maintained in the CNS.
Table 1. Common isoforms of human apolipoprotein E.
Common allelic variants of the apolipoprotein E gene (APOE), called ε2, ε3, and ε4, encode for three different 299 amino acid proteins called apoE2, apoE3, and apoE4 that differ only at amino acid positions 112 and 158. For comparison, corresponding amino acids for the sequence of most other mammals and for apes are included. Most other mammals, including apes, have threonine (T) at position 61 and arginine (R) at positions 112 and 158; the exceptions are rabbit (Rb) and bovine (Bo) apoE that have cysteine (C) at position 112. Humans differ from other mammals in having R at position 61. Human apoE4 has R at positions 112 and 158, making it the “simian” form of human apoE. ApoE3 differs from apoE4 by a single amino acid substitution: C for R at position 112 that derives from the transition of CGC to TGC in the codon for this amino acid. Human apoE2 differs from apoE3 by the same transition in the codon for amino acid 158. Thus, human apoE apparently is evolving from the “simian” apoE4 to apoE2 with progressive substitutions of C for R at positions 112 and 158.
|
Genetic associations establish biological relevance but not mechanism of action. Although the contribution to AD risk of neighboring chromosome 19 genes for translocase of outer mitochondrial membrane (TOMM) 40 and apoC-I has not been formally excluded [5, 6], the vast majority of work has focused on APOE. The model that has emerged from studies to elucidate mechanism of action is that apoE isoform-dependent effects, with apoE4 > apoE3 > apoE2, lead to increased aggregation and deposition, decreased clearance, or both, of Aβ peptides in the cerebrum [7].
In spite of the wealth of mechanistic data for apoE isoform-specific effects in experimental models of AD and in human neuroimaging and cerebrospinal fluid (CSF) biomarker studies, our knowledge is incomplete [8–16].
How apoE isoforms lead to altered Aβ aggregation and deposition or clearance is not entirely clear. Several groups have suggested that differential binding of apoE isoforms to Aβ influences these processes through receptor-mediated mechanisms, thereby modulating biological activity of Aβ peptides [17–19]. However, the fundamentals of apoE isoform-dependent binding to Aβ peptides in vivo are just beginning to be explored and appear to have quite different characteristics than in vitro [20].
Here we review evidence in support of an additional, or perhaps alternate, mechanism of action: that apoE isoforms differentially regulate the innate immune response to Aβ peptides and other relevant activators in AD, which in turn influences Aβ peptide aggregation, deposition, and clearance.
Innate immunity and AD
Innate immunity is the portion of the immune system that defends the host through non-pathogen-specific pathways. In contrast, T cells, B cells, and antibody-secreting plasma cells of the adaptive arm mobilize in response to specific antigenic determinants. This characteristic restriction of specific pathogen recognition by adapative immune system effectors arises through complex genetic reorganization during development (Figure 1). The net effect of the innate immune response may be beneficial, deleterious, or variable depending on the repertoire of components activated and the intensity, duration, and repetition of the response. It is not surprising then that some of the work in this area has revealed components of the innate immune response that suppress processes of AD or neuron damage, while other components have the opposite effects in experimental models (reviewed in [21]). Two major components of the innate immune response are the complement cascade and a class of pattern recognition receptors called Toll-like receptors (TLRs).
Figure 1. Innate and Adaptive Immunity.

Innate immune system effectors include components of the complement cascade (C1q, C3(a), C5(a)) as well as professional phagocytes (macrophages, tissue-specific dendritic cells). The adaptive arm of the immune system includes T cells, B cells, and the antibody-secreting plasma cells. Functions of innate immune system effectors are not burdened by genetic restriction as seen in adaptive responses.
The complement system comprises over 30 proteins and protein fragments that are activated and greatly amplified through a tightly regulated proteolytic cascade [22]. The complement system is designed to assist with opsonization and phagocytosis, chemotaxis, and modification and lysis of the pathogen by the membrane attack complex (MAC). The complement system can be activated by three upstream pathways—classical, alternative, and mannose-binding lectin binding—which in turn are activated by different endogenous and exogenous molecules. Activation via any of the three pathways leads to C3 convertase-catalyzed hydrolysis of C3 to C3a and C3b, promoting further amplification of the complement cascade. More distal components of the complement cascade include C5a, a potent chemotaxis factor, and C5b, which initiates assembly of MAC. Aβ has been shown to directly bind C1q, activating the complement cascade via the classical pathway. In addition, critical players in the alternative complement pathway, namely C3 and C5, as well as their activated forms and cognate receptors, are implicated in altered plaque deposition in AD mouse models, likely through changes to AB clearance by microglia [23]. Some GWAS also have associated the complement receptor 1 gene (CR1) with increased AD risk [1, 2]. While not as strong as APOE, the association between CR1 and AD reached genome-wide significance [2] and has been replicated and proposed to be related to a particular CR1 isoform [24]. CR1 regulates the complement cascade by modulating phagocytosis and activating distal complement components.
In the CNS, microglia appear to be the major source of complement proteins, whose transcription is increased in regions affected by AD [25]. Numerous groups have reported the association of Aβ plaques and neurofibrillary tangles in AD brain specimens with complement proteins in the classical pathway, particularly the MAC [26–29]. In addition to these observational data, functional data on the role of complement in AD pathogenesis comes from biochemical and in vivo studies. Interestingly, the classical complement pathway, traditionally activated by antigen-antibody complexes, can also be activated by other molecules, including Aβ [30]. In vivo studies with mouse models reveal that the perturbation of specific complement proteins can either promote or suppress the pathogenesis of Aβ deposition. For example, when C3 activation was suppressed, either by genetic ablation (C3−/−) [31] or by expression of soluble complement receptor-related protein y (sCrry; a complement inhibitor) [32], neuronal damage and Aβ accumulation increased. Conversely, when interferon-γ enhanced the activation of the complement system in another transgenic model of AD, Aβ deposition decreased [33]. In contrast to these studies indicating that the activation of some complement proteins may be useful in suppressing some AD processes, a transgenic mouse model of AD lacking C1q showed neuroprotection and fewer activated microglia without modification of Aβ [34]. In a different transgenic model of AD a C5a receptor (CD88) antagonist reduced Aβ accumulation and microglial activation [23], likely by binding to microglial CD88 [35] Future studies will be needed to elucidate whether the net effect of complement is to promote or suppress AD pathogenesis in vivo.
TLRs comprise a family of plasma and nuclear membrane glycoprotein receptors that recognize exogenous structures present in microorganisms and endogenous structures produced from tissue injury or disease [36]. Once activated, these pattern recognition receptors coordinate gene transcription through several pathways, including those involving NF-κB, p38 MAP kinase (K), to initiate innate immune responses. Key effectors of the innate immune response include the inducible form of cyclooxygenase (COX2), tumor necrosis factor α (TNFα), interleukin 6 (IL-6), and inducible nitric oxide synthase (iNOS), among many other protein molecules. Not all of these molecules are activated directly by TLRs but rather follow in waves of derivative activation. For example, secretion of TNFα and IL-6 following TLR3 or TLR4 activation is dependent upon microglial activation of PGE2 receptor subtype 1 (EP1) [37]. Moreover, while ample data support a role for TNFα as a key effector of microglial-mediated neuron damage, recent work also indicates that increased expression of TNFα in mouse hippocampus suppresses Aβ accumulation in a model of AD [38]; however, it is not clear whether this constitutes neuroprotection in this model.
Microglia express TLR2, TLR3, TLR4, and TLR9 and respond with varying degrees of effectiveness to selective activation of each receptor [37]. Myeloid differentiation factor 88 (MyD88) is a required signal transducing adaptor protein for TLR2, TLR4, and TLR9, but not TLR3. The activation of microglial TLR2 damages the brain in experimental animal models of multiple sclerosis [39, 40], head trauma [41, 42], cerebral ischemia [43], and AD [44, 45]. The activation of microglial TLR4 injures brain tissue in models of AD [45–47] and head trauma [41] In transgenic models of AD the activation of microglial TLR9, TLR2, or MyD88 suppresses Aβ oligomerization and reduces Aβ accumulation while also improving cognitive performance [34, 48–50]. Direct activation of microglial TLR3 or TLR4 produces paracrine damage to neurons in cell culture and in vivo [51–53].
Aggregated Aβ peptides activate microglia in large part through CD14/TLR4-dependent mechanisms [54]. Conversely, microglial CD14 activation modulates Aβ deposition [55]. One group has associated a polymorphism in TLR4 with risk for AD [56]. Moreover, since mRNA is an endogenous ligand for TLR3 [57], nucleic-acid mediated activation of TLR3 may potentially occur in neurodegenerative diseases such as AD, Parkinson’s disease, and amyotrophic lateral sclerosis [58], in which mRNA accumulates in pathologic lesions.
Taken together, observations from human autopsy material and GWAS point to a potential role for the innate immune response in AD pathogenesis. Experimental studies from animal models highlight multifaceted mechanisms by which specific components of innate immunity not only act as indirect effectors of neuron damage, but also influence the amount and form of Aβ accumulation. The overall effect of innate immune activation or suppression in AD likely will vary between individuals and brain regions, representing a delicate balance between activation sufficient to suppress Aβ accumulation and activation that unleashes neurotoxicity (Figure 2).
Figure 2. Interaction between Aβ, Innate Immune Response, and APOE.

Schematic of complex interaction between Aβ and the innate immune response and subsequent effect on Aβ aggregation, deposition and clearance. Other activators contribute as well, resulting both direct and indirect effects on neurons. There is evidence that apoE isoforms differentially regulate these processes, and thus the extent of neuron damage and Aβ burden.
ApoE isoforms in the regulation of innate immunity
While associations between APOE and amyotrophic lateral sclerosis, multiple sclerosis, Parkinson’s disease, stroke, vascular dementia, and other disorders have been proposed, the strongest evidence to date is between APOE and disease risk for AD [7]. ApoE isoforms have been proposed to possess Aβ-dependent and Aβ-independent mechanisms by which they influence the initiation and progression of AD [59]. Immune activation is a prominent feature of AD in autopsy material, in which microglia invade and surround senile plaques. Intense APOE immunoreactivity colocalizes with plaque-associated amyloid and microglia [60], suggesting a role for apoE in regulating the innate immune response in AD. The following sections review data supporting a role for apoE isoforms as key regulators of the innate immune response in three areas: (1) microglial activation, (2) immune-mediated toxicity to neurons, and (3) potential modulators of Aβ peptide deposition and clearance.
1. Microglial Activation
Most investigations of apoE isoform-specific regulation of the innate immune response in brain have focused on microglial activation, since these are the major resident immune effector cells in the CNS. Primary adult rat microglia treated with HDL-like particles were used by Chen and colleagues to investigate apoE-specific differences in inflammatory mediators secreted by microglia [61]. ApoE3- and apoE4-containing HDL-like particles were partially purified by size exclusion chromatography from conditioned medium of HEK cells stably transfected with cDNA encoding either apoE isoform. HEK-apoE4 and human HDL spiked with recombinant apoE4 dramatically induces microglial PGE2 and IL-1β secretion compared to HEK-apoE3.
Microglia from targeted replacement (TR) APOE mice show similar isoform effects. TR APOE mice derived by homologous recombination were developed by Sullivan and colleagues and contain chimeric genes consisting of mouse 5’ regulatory sequence continuous with mouse (noncoding) exon 1 followed by human exons (and introns) 2–4 to produce TR APOE2/2, TR APOE3/3, or TR APOE4/4 mice [62, 63]. Using these mice, Brown, Colton, and colleagues have shown that that activated TR APOE4/4 microglia secrete more NO than TR APOE3/3 microglia [64, 65] and have expanded their studies to include peritoneal macrophages and PIC, a selective activator of TLR3 [66]. TR APOE4/4 primary microglia display enhanced activation, including increased secretion of TNFα and IL-6, with subsequent induction of NOS2 expression and activity; similar effects were seen in peritoneal macrophages from TR APOE3/3 and TR APOE4/4 mice. They also generated mice that express only one copy of APOE3 (TR APOE3/0) whose macrophages display an intermediate pro-inflammatory status between TR APOE3/3 and TR APOE4/4. In combination, these data confirm the pro-inflammatory nature of TR APOE4/4 microglia/macrophages compared to TR APOE3/3, suggesting inherent differences in the activity of apoE3 and apoE4 following activation of these cells by TLR3 or TLR4 ligands. They further suggest that gene dosage may be critical in the regulation of these components of the innate immune response.
This previous study [66] also included in vivo experiments using peripheral LSP injection to activate systemic innate immunity and then examined responses in mouse cerebral cortex. Using TR APOE3/3 and TR APOE4/4 mice, they observed significantly greater TNFα and IL-12 transcription of genes for TNF in APOE4/4 mice than APOE3/3 by quantitative PCR. While this important experiment demonstrates the relevance of APOE alleles in systemic immune activation, interpretation of the results is limited by the complexities of transduction of peripheral inflammation to central inflammation [69]. The reduced immune response observed in cerebral cortex from TR APOE3/3 mice might be due to reduced peripheral immune activation, reduced efficiency of signal transduction from periphery to CNS, or the inherent differences in microglial activation demonstrated in cell culture experiments.
In another in vivo study, Ophir and colleagues used C57Bl/6 transgenic (tg) mice that lacked apoE but expressed a single copy of APOE3 or APOE4 [67] to investigate whether regulation of brain inflammation is affected by APOE [68]. Immune activation was achieved by intracerebroventricular (ICV) injection of LPS, and gliosis was assessed by immunohistochemistry followed by morphometry [68]. Maximal hippocampal gliosis is observed 3 days after LPS injection in 3-month-old mice, with similar results in 6-month-old mice but much less activation in 12-month-old mice. LPS activation leads to increased hippocampal apoE production that is not isoform-specific. Interestingly, the authors observed that astrogliosis in younger mice is apoE-dependent and greater in apoE3 tg mice than apoE4 tg mice. In contrast, microglial activation is apoE-independent.
2. Innate Immune-Mediated Toxicity To Neurons
While some components of the innate immune response are neurotrophic or protective, intense or protracted activation of the innate immune response leads to elaboration of neurotoxic species. The exact composition of the neurotoxic effectors is not clear; thus, understanding apoE effects on the innate immune response includes determining whether differences in microglial activation manifest as differences in neurotoxicity. Our group tested the hypothesis that apoE isoform modulation of glial innate immune response may differentially alter bystander or paracrine damage to neurons [70]. Using mixed primary co-cultures of cerebral microglia from the TR mice mentioned above [62, 63] and cerebral cortical neurons from either wild type (wt) or apoE−/− mice, we observed that paracrine damage to neurons from microglia is greatest with TR APOE4/4, intermediate with TR APOE3/3, and least with TR APOE2/2 microglia [70]. Importantly, there is no difference among TR APOE cultures in expression of TLR4 or binding of LPS by apoE isoforms. We confirmed the results mentioned above [64, 65] that activated TR APOE4/4 microglia secrete more NO than TR APOE3/3 microglia and extended them to show that activated TR APOE2/2 are no different from wt [70]. However, in our study we also determined that detectable increase in medium NO occurs after paracrine neurotoxicity, thereby questioning the relevance of NO as neurotoxic effector in this model. Similarly, when microglial secretion of 10 cytokines and chemokines is screened, only TNFα and IL-6 are significantly elevated coincident with neurotoxicity. Both show TR APOE4/4 > TR APOE3/3 > TR APOE2/2. A P38-MAPK inhibitor ablates TR APOE-dependent microglial TNFα and IL-6 secretion and protects neurons. Using primary cultures of TNFα astrocytes for comparison, we also observed TR APOE-dependent differences in cytokine secretion; however, these are NF-κB- and not p38-MAPK-dependent [71].
While primary cultures of enriched cell types, including cocultures, have the advantage of being a relatively pure population of cells, they also have several disadvantages with respect to understanding CNS physiology, since interactions and direct connections among cells is lost. This can be addressed not only in vivo but also in organotypic cultures that retain physiologically relevant proportions of cells and their local contacts. The studies by us described in the previous paragraph were repeated using organotypic cultures of hippocampus from TR APOE mice activated with the same preparation of LPS [70]. The same rank order for hippocampal pyramidal neuron damage and TNFα and IL-6 secretion is observed, with TR APOE4/4 > TR APOE3/3 > TR APOE2/2. Importantly, pharmacologic inhibition of p38MAPK suppresses these TR APOE-dependent differences and protects neurons from the bystander damage caused by microglial activation in organotypic cultures.
These studies indicate that some important elements in this neurotoxic response are apoE isoform-dependent: p38MAPK, PGE2, NO, TNFα, and IL-6. Activated microglia-mediated neurotoxicity in multiple models is greatest with TR APOE4, followed by TR APOE3, and least with TR APOE2, and apparently is related to expression of apoE isoforms, since in at least some models the difference among TR APOE cells can be blocked with low density lipoprotein receptor-related protein associated protein 1 (RAP).
Interestingly, while LPS is commonly used to activate specifically the innate immune response, the source and purity of the LPS preparation used can be critical to its specificity of activation [72]. In culture, several groups have shown that high quality LPS preparations have no demonstrable effect on neurons because these cells lack functional CD14 and TLR4. In an in vivo study using direct activation of CD14/TLR4 co-receptors in cerebrum by ICV injection of LPS into TR APOE mice, we demonstrated specificity of TLR4 activation by observing no effect of ICV LPS in mice genetically lacking CD14 or TLR4 but full effect in mice lacking TLR2 [73]. Damage to hippocampal pyramidal neurons was assessed following activation of microglial CD14/TLR4 by measuring changes in dendrite length using Golgi staining. We and others have found that TR APOE4/4 mice have slightly but significantly shorter dendrites at baseline (6 weeks of age), a result confirmed by others [74]. Following exposure to ICV LPS, there is comparable loss of dendrite length at 24 hr among the three TR APOE mice.
Recovery of dendrite length over the next 48 hr is greater in TR APOE2/2 than TR APOE3/3 mice, while TR APOE4/4 mice show failure of dendrite regeneration. Accompanying cell culture experiments suggested that the enhanced neurotrophic effect observed in TR APOE2/2 is RAP-dependent [73].
Overall, the in vivo studies largely validate the cell culture studies and indicate that apoE isoform-specific modulation of microglial innate immunity and the resulting damage to neurons is modulated by expression of different apoE isoforms.
3. Aβ Peptide Modulation
Sequential proteolysis of the amyloid precursor protein to generate Aβ peptides is promiscuous at the C terminus and generates a family of peptides that vary in the number of amino acids; the most abundant contain 40 or 42 amino acids. One group investigated the modulatory role of apoE isoforms in Aβ40-mediated complement activation [75]. Aβ40 of unclear aggregation state adhered to the plastic of a microwell followed by addition of human serum displays robust activation of C3. ApoE3, apoE4, or BSA substituted for Aβ40 results in no C3 activation. When Aβ40 is followed by apoE isoforms and a polyclonal apoE antibody to detect binding, apoE2 and apoE4 appear to bind more rapidly and perhaps more avidly than apoE3. While apoE4 bound to Aβ40 increases Aβ-mediated complement activation by about 25%, there was no significant effect of apoE2 or apoE3 in this in vitro assay.
Microglia possess many actions, one of which is activation to a neurotoxic phenotype, as reviewed above. Microglia also are the major phagocytic cell in brain. We explored in vitro possible mechanisms to explain apparent decreased clearance of Aβ peptides in animal models that express apoE4 [7]. Using a phagocytosis assay in which we demonstrated that the PGE2 receptor subtype 2 (EP2) suppressed mouse microglial phagocytosis of Aβ peptides from AD brain sections [76], we attempted to determine if microglia from TR APOE mice might also show differential phagocytosis. We found there was no difference in microglial phagocytosis when comparing TR APOE4, TR APOE3, and TR APOE2 mice (unpublished data). Microglia are migratory cells and part of their overall effectiveness resides in their ability to move towards lesions or areas of damage. Indeed, microglia migrate toward Aβ deposits in brain in mice and apparently do so in humans with AD. Recently we demonstrated that microglial migration in vitro is highly TR APOE-dependent. Using standard activators of microglial migration, C5a and ATP, we showed that rank order for microglial migration in vitro was TR APOE3/3 > TR APOE4/4 ≈ TR APOE2/2 for both activators [77]. Interestingly, the mechanisms for TR APOE-dependent differences in microglia migration differed for complement-mediated activation vs. ATP with the former related to RAP-dependent processes and the latter to alteration in second messenger signaling. While these results do not follow the order of genetic risk for AD they do mirror the results for complement activation. Moreover, these results clearly demonstrate apoE isoform-specific effects on microglial migration, which in turn might influence the processes that underlie Aβ clearance.
Conclusion
Accumulation of Aβ peptides in cerebrum appears to be a central event in the initiation or progression of AD. Aβ peptides are pleiotropic neurotoxins that can directly damage neurons; they also can activate microglia to adopt a neurotoxic phenotype, and thus indirectly damage neurons.
ApoE also is a pleiotropic molecule with multiple actions, at least some of which vary among its three common isoforms. Several laboratories have now reproducibly shown that microglial activation to a neurotoxic phenotype is apoE isoform-dependent and greatest with apoE4 expressing cells, less with apoE3, and least with apoE2. Indeed, this apoE isoform-dependent microglial activation translates into apoE isoform-dependent (microglial-mediated) damage to neurons in primary culture, organotypic culture, and in vivo. Thus, under conditions when the innate immune response has become neurotoxic, greatest neuron damage is observed in those models expressing apoE4, less in those expressing apoE3, and least in those expressing apoE2.
As reviewed earlier, expression of different APOE alleles influences Aβ peptide accumulation in brain. One mechanism underlying this effect may be direct apoE isoform-specific binding to and subsequent apoE receptor-mediated events. We have reviewed data to support another mechanism: apoE isoform-specific modulation of components of the innate immune response that modulate the amount and quality of Aβ deposition. We return to the fact that genetic associations define biological relevance but not mechanism of action. Perhaps the outstandingly strong association of APOE alleles with risk of AD derives from multiple apoE isoform-specific mechanisms that include both direct binding of Aβ and modulation of the innate immune response.
Highlights.
Largest genetic risk for late-onset Alzheimer’s disease resides at the apoE gene
Innate immune response is consistently observed in AD and a likely contributor
apoE isoforms regulate multiple facets of innate immune response in brain
apoE may alter AD through both Aβ-dependent and independent mechanisms
Acknowledgments
This work was supported by AG05136, AG00258, and ES16754 as well as the Nancy and Buster Alvord Endowment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
C. Dirk Keene, Email: cdkeene@uw.edu.
Eiron Cudaback, Email: eiron@uw.edu.
Xianwu Li, Email: xli@u.washington.edu.
Kathleen S. Montine, Email: kmontine@uw.edu.
Thomas J. Montine, Email: tmontine@uw.edu.
References
- 1.Harold D, Abraham R, Hollingworth P, Sims R, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JC, Heath S, Even G, Campion D, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 3.Han MR, Schellenberg GD, Wang LS. Genome-wide association reveals genetic effects on human Abeta42 and tau protein levels in cerebrospinal fluids: a case control study. BMC Neurol. 2010;10:90. doi: 10.1186/1471-2377-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257:2518–21. [PubMed] [Google Scholar]
- 5.Yu CE, Seltman H, Peskind ER, Galloway N, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89:655–65. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petit-Turcotte C, Stohl SM, Beffert U, Cohn JS, et al. Apolipoprotein C-I expression in the brain in Alzheimer’s disease. Neurobiol Dis. 2001;8:953–63. doi: 10.1006/nbdi.2001.0441. [DOI] [PubMed] [Google Scholar]
- 7**.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011;10:241–52. doi: 10.1016/S1474-4422(10)70325-2. An assessment of evidence for associations of APOE with risk and outcomes for various neurological diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtzman DM, Bales KR, Tenkova T, Fagan AM, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fagan AM, Watson M, Parsadanian M, Bales KR, et al. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–18. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 11*.Bales KR, Liu F, Wu S, Lin S, et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–9. doi: 10.1523/JNEUROSCI.0887-09.2009. In an AD mouse model crossed with TR APOE mice, E4 mice have increased Aβ and decreased apoE levels relative to E2 or E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang GC, Insel PS, Tosun D, Schuff N, et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010;75:1976–81. doi: 10.1212/WNL.0b013e3181ffe4d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Reiman EM, Chen K, Liu X, Bandy D, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:6820–5. doi: 10.1073/pnas.0900345106. Imaging studies of cognitively normal older adults show fibrillar Aβ burden is highest in those with two copies of the APOE epsilon 4 allelle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunderland T, Mirza N, Putnam KT, Linker G, et al. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry. 2004;56:670–6. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 15**.Morris JC, Roe CM, Xiong C, Fagan AM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. Confirms Aβ-APOE association but finds no effect of APOE on CSF tau levels, which is used as a biomarker of neurofibrillary tangles in brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu F, Vitek MP, Colton CA, Previti ML, et al. Human apolipoprotein E redistributes fibrillar amyloid deposition in Tg-SwDI mice. J Neurosci. 2008;28:5312–20. doi: 10.1523/JNEUROSCI.1042-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manelli AM, Stine WB, Van Eldik LJ, LaDu MJ. ApoE and Abeta1–42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci. 2004;23:235–46. doi: 10.1385/JMN:23:3:235. [DOI] [PubMed] [Google Scholar]
- 18.Bales KR, Dodart JC, DeMattos RB, Holtzman DM, et al. Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv. 2002;2:363–75. 39. doi: 10.1124/mi.2.6.363. [DOI] [PubMed] [Google Scholar]
- 19.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, et al. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–6. [PubMed] [Google Scholar]
- 20.Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, et al. Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-beta in human Alzheimer brain. PLoS One. 2011;6:e14586. doi: 10.1371/journal.pone.0014586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–39. doi: 10.1038/nri2565. The dual nature—beneficial and detrimental—of the innate immune response must be considered in therapeutic strategies. [DOI] [PubMed] [Google Scholar]
- 22.Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90. doi: 10.1016/j.it.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Fonseca MI, Ager RR, Chu SH, Yazan O, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009;183:1375–83. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.24. epub ahed of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol. 1999;154:927–36. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zanjani H, Finch CE, Kemper C, Atkinson J, et al. Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord. 2005;19:55–66. doi: 10.1097/01.wad.0000165506.60370.94. [DOI] [PubMed] [Google Scholar]
- 27.Afagh A, Cummings BJ, Cribbs DH, Cotman CW, et al. Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol. 1996;138:22–32. doi: 10.1006/exnr.1996.0043. [DOI] [PubMed] [Google Scholar]
- 28.Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation. 2008;5:9. doi: 10.1186/1742-2094-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webster S, Lue LF, Brachova L, Tenner AJ, et al. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging. 1997;18:415–21. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H, Burdick D, Glabe CG, Cotman CW, et al. beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol. 1994;152:5050–9. [PubMed] [Google Scholar]
- 31.Maier M, Peng Y, Jiang L, Seabrook TJ, et al. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–41. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyss-Coray T, Yan F, Lin AH, Lambris JD, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A. 2002;99:10837–42. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184:5333–43. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richard KL, Filali M, Prefontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci. 2008;28:5784–93. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ager RR, Fonseca MI, Chu SH, Sanderson SD, et al. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J Neurochem. 2010;113:389–401. doi: 10.1111/j.1471-4159.2010.06595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Cudaback E, Keene CD, Breyer RM, et al. Suppressed microglial E prostanoid receptor 1 signaling selectively reduces tumor necrosis factor alpha and interleukin 6 secretion from toll-like receptor 3 activation. Glia. 2011;59:569–76. doi: 10.1002/glia.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakrabarty P, Herring A, Ceballos-Diaz C, Das P, et al. Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. doi: 10.1186/1750-1326-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sloane JA, Batt C, Ma Y, Harris ZM, et al. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107:11555–60. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farez MF, Quintana FJ, Gandhi R, Izquierdo G, et al. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol. 2009;10:958–64. doi: 10.1038/ni.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koedel U, Merbt UM, Schmidt C, Angele B, et al. Acute brain injury triggers MyD88-dependent, TLR2/4-independent inflammatory responses. Am J Pathol. 2007;171:200–13. doi: 10.2353/ajpath.2007.060821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Babcock AA, Wirenfeldt M, Holm T, Nielsen HH, et al. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J Neurosci. 2006;26:12826–37. doi: 10.1523/JNEUROSCI.4937-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abe T, Shimamura M, Jackman K, Kurinami H, et al. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41:898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jana M, Palencia CA, Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol. 2008;181:7254–62. doi: 10.4049/jimmunol.181.10.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009. Study demonstrating that microglial activation by fibrillar Abeta requires TLR2, TLR4, and CD14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin JJ, Kim HD, Maxwell JA, Li L, et al. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michaud JP, Richard KL, Rivest S. MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2011;6:5. doi: 10.1186/1750-1326-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29:1846–54. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doi Y, Mizuno T, Maki Y, Jin S, et al. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid {beta} neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am J Pathol. 2009;175:2121–32. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin J, Shie FS, Liu J, Wang Y, et al. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J Neuroinflammation. 2007;4:2. doi: 10.1186/1742-2094-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin L, Wu X, Block ML, Liu Y, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–62. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Field R, Campion S, Warren C, Murray C, et al. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fassbender K, Walter S, Kuhl S, Landmann R, et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004;18:203–5. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- 55.Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer’s disease pathology by influencing the brain’s inflammatory milieu. J Neurosci. 2010;30:15369–73. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minoretti P, Gazzaruso C, Vito CD, Emanuele E, et al. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci Lett. 2006;391:147–9. doi: 10.1016/j.neulet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 57.Kariko K, Ni H, Capodici J, Lamphier M, et al. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–50. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 58.Ginsberg SD, Galvin JE, Chiu TS, Lee VM, et al. RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathol. 1998;96:487–94. doi: 10.1007/s004010050923. [DOI] [PubMed] [Google Scholar]
- 59.Huang Y. Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16:287–94. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Uchihara T, Duyckaerts C, He Y, Kobayashi K, et al. ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci Lett. 1995;195:5–8. doi: 10.1016/0304-3940(95)11763-m. [DOI] [PubMed] [Google Scholar]
- 61.Chen S, Averett NT, Manelli A, Ladu MJ, et al. Isoform-specific effects of apolipoprotein E on secretion of inflammatory mediators in adult rat microglia. J Alzheimers Dis. 2005;7:25–35. doi: 10.3233/jad-2005-7104. [DOI] [PubMed] [Google Scholar]
- 62.Sullivan PM, Mezdour H, Aratani Y, Knouff C, et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–80. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 63.Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest. 1998;102:130–5. doi: 10.1172/JCI2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown CM, Wright E, Colton CA, Sullivan PM, et al. Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic Biol Med. 2002;32:1071–5. doi: 10.1016/s0891-5849(02)00803-1. [DOI] [PubMed] [Google Scholar]
- 65.Colton CA, Brown CM, Cook D, Needham LK, et al. APOE and the regulation of microglial nitric oxide production: a link between genetic risk and oxidative stress. Neurobiol Aging. 2002;23:777–85. doi: 10.1016/s0197-4580(02)00016-7. [DOI] [PubMed] [Google Scholar]
- 66**.Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–60. doi: 10.1016/j.neurobiolaging.2007.11.014. APOE-related differences in microglial activation may arise from both isoform differences and gene dose effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu PT, Schmechel D, Rothrock-Christian T, Burkhart DS, et al. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis. 1996;3:229–45. doi: 10.1006/nbdi.1996.0023. [DOI] [PubMed] [Google Scholar]
- 68.Ophir G, Meilin S, Efrati M, Chapman J, et al. Human apoE3 but not apoE4 rescues impaired astrocyte activation in apoE null mice. Neurobiol Dis. 2003;12:56–64. doi: 10.1016/s0969-9961(02)00005-0. [DOI] [PubMed] [Google Scholar]
- 69.Nadeau S, Rivest S. Endotoxemia prevents the cerebral inflammatory wave induced by intraparenchymal lipopolysaccharide injection: role of glucocorticoids and CD14. J Immunol. 2002;169:3370–81. doi: 10.4049/jimmunol.169.6.3370. [DOI] [PubMed] [Google Scholar]
- 70.Maezawa I, Nivison M, Montine KS, Maeda N, et al. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 2006;20:797–9. doi: 10.1096/fj.05-5423fje. [DOI] [PubMed] [Google Scholar]
- 71.Maezawa I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006;3:10. doi: 10.1186/1742-2094-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rutledge HR, Jiang W, Yang J, Warg LA, et al. Gene expression profiles of RAW264.7 macrophages stimulated with preparations of LPS differing in isolation and purity. Innate Immun. 2011 doi: 10.1177/1753425910393540. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 73.Maezawa I, Zaja-Milatovic S, Milatovic D, Stephen C, et al. Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J Neuroinflammation. 2006;3:21. doi: 10.1186/1742-2094-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317–22. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGeer PL, Walker DG, Pitas RE, Mahley RW, et al. Apolipoprotein E4 (ApoE4) but not ApoE3 or ApoE2 potentiates beta-amyloid protein activation of complement in vitro. Brain Res. 1997;749:135–8. doi: 10.1016/s0006-8993(96)01324-8. [DOI] [PubMed] [Google Scholar]
- 76.Shie FS, Breyer RM, Montine TJ. Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol. 2005;166:1163–72. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cudaback E, Li X, Montine KS, Montine TJ, et al. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011 doi: 10.1096/fj.10-176891. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]