

Abstract
Hyperkinetic Jak2 tyrosine kinase signaling has been implicated in several hematological disorders including the myeloproliferative neoplasms (MPNs). Effective Jak2 inhibitors can thus have significant therapeutic potential. Here, using structure based virtual screening, we identified a benzothiophene derived Jak2 inhibitor named A46. We hypothesized that this compound would inhibit Jak2-V617F mediated pathologic cell growth. To test this, A46 was analyzed for its ability to i) inhibit recombinant Jak2 protein catalysis ii) suppress Jak2-mediated pathogenic cell growth in vitro iii) inhibit the aberrant ex vivo growth of Jak2-V617F expressing primary human bone marrow cells and iv) inhibit Jak2-mediated pathogenesis in vivo. To this end, we found that A46 selectively inhibited Jak2-V617F protein when compared to wild type Jak2 protein. The drug also selectively inhibited the proliferation of Jak2-V617F expressing cells in both a time- and dose-dependent manner and this correlated with decreased Jak2 and STAT5 phosphorylation within treated cells. The Jak2-V617F cell growth inhibition correlated with an induction of cell cycle arrest and promotion of apoptosis. A46 also inhibited the pathologic growth of primary Jak2-V617F expressing bone marrow cells, ex vivo. Lastly, using a mouse model of Jak2-V617F mediated MPN, A46 significantly reduced the splenomegaly and megakaryocytic hyperplasia in the spleens of treated mice and the levels of IL-6 in the plasma. Collectively, our data demonstrate that the benzothiophene based compound, A46, suppresses Jak2-mediated pathogenesis, thereby making it a potential candidate drug against Jak2-mediated disorders.
Keywords: Jak2 kinase, V617F, Small Molecule Inhibitor, Benzothiophene, Myeloproliferative Neoplasms
INTRODUCTION
Jak2, a member of the Janus family of cytoplasmic tyrosine kinases, is ubiquitously expressed and is a key mediator of signal transduction and gene transcription. A variety of cytokines, growth factors and GPCR ligands can activate Jak2, trigger the canonical Jak/STAT signaling cascade and cause physiological effects such as regulation of cell survival, development, growth, and proliferation.
The critical role of Jak2 in embryonic development was demonstrated by gene deletion studies in which Jak2 knock-out mice were found to be embryonic lethal due to absence of definitive erythropoiesis that led to profound anemia [1, 2]. Conversely, constitutive activation of Jak2 promotes aberrant cell proliferation and can lead to the development of hematological malignancies and myeloproliferative neoplasms (MPNs) [3–5]. Philadelphia chromosome negative MPNs include three pathogenetically related disorders; polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). These disorders arise from a transformed hematopoietic stem cell and are characterized by increased production of blood cells of the myeloid origin.
A somatic Jak2 mutation (Jak2-V617F) was identified in ~90% of PV patients and about 50% of patients with ET and PMF [6–10]. In hematopoietic stem cells, a valine to phenylalanine substitution at codon 617 (V617F) in the pseudokinase domain of Jak2 results in a constitutively active Jak-STAT signaling pathway which in turn promotes the cytokine independent growth of these cells. The presence of this mutation has also been detected in other hematological malignancies such as acute myeloid leukemia, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia [11]. The co-expression of Jak2-V617F and an ectopic erythropoietin receptor (EpoR) in the IL-3-dependent hematopoietic cell line, Ba/F3, is sufficient to confer cytokine independent growth [12]. Subsequently, it has also been shown that expression of this mutation in both murine bone marrow transplant models [13, 14] as well as transgenic models [15–18] is sufficient for the development of MPN-like phenotypes in recipient mice. These reports strongly suggest that the Jak2-V617F mutation plays a causative role in the pathogenesis of myeloproliferative neoplasms.
The presence of the Jak2-V617F mutation in a majority of MPN patients suggests that identification of Jak2 specific inhibitors is an important step towards developing an effective targeted therapy for MPNs. Our laboratory has been actively involved in the identification of Jak2 inhibitors and has previously reported three small molecules with anti-Jak2 activity [19–23]. Here, we used structure-based drug design to identify a novel benzothiophene based structure, 1-benzothiophen-2-yl-(4-dimethylaminophenyl)methanol (termed as A46), and show that this compound suppresses Jak2-mediated pathological cell growth in vitro, ex vivo, and in vivo.
MATERIALS AND METHODS
Drugs
The small molecule A46 was obtained from the National Cancer Institute/Developmental Therapeutics Program (NCI/DTP), solubilized in dimethyl sulfoxide (DMSO) at a concentration of 10 mM and stored at −20°C. PP2, a c-Src inhibitor, was purchased from Calbiochem, solubilized in DMSO at a concentration of 10 mM and also stored at −20°C.
In Vitro Kinase Assay
Approximately 300 ng of recombinant protein was incubated in 40 μl of in vitro kinase reaction buffer (60 mM HEPES pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 3 μM Na3VO4, 2.5 mM dithiothreitol and 1 mM ATP) for 30 min at room temperature, either in the presence or the absence of inhibitors, as indicated. Kinase reactions were terminated by the addition of SDS-sample buffer and were subsequently analyzed by western blotting with the indicated antibodies as described below. Jak2-WT/JH1+JH2 (wild-type Jak2), Jak2-V617F/JH1+JH2 (mutant Jak2), and c-Src recombinant proteins were all purchased from Invitrogen.
Cell Culture
HEL and Raji cells were purchased from the American Type Culture Collection (ATCC) and CMK cells from the German Collection of Microorganisms and Cell Cultures, DSMZ. SET-2 cells were a kind gift from Dr. Gary Reuther at the Moffitt Cancer Center and Research Institute, Tampa, FL. Cells were cultured in RPMI 1640 (Mediatech) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin and L-glutamine at 37°C and 5% CO2.
Cell Proliferation Assay
HEL, Raji, CMK and SET-2 cells were plated in 96-well plates at a concentration of approximately 5 × 104 cells per well and treated with either 0.25% DMSO or A46 for the indicated periods of time or concentrations. Cell viability was assessed by trypan blue exclusion staining and hemocytometer.
ELISA
HEL cells, treated with either 0.25% DMSO or increasing doses of A46 for 48 hrs, were lysed and analyzed by ELISA for detection of phospho-Jak2 and phospho-STAT5 protein levels. For this, Jak2 [pY1007/pY1008] and STAT5b [pY699] ELISA kits (Invitrogen) were used according to the manufacturer’s protocol.
Cell Cycle Assay
The CycleTEST™ PLUS DNA Reagent Kit (BD Biosciences) was used to analyze the DNA obtained from HEL cells suspensions. The cells were first incubated with 0.25% DMSO or increasing doses of A46 for 48 hrs and then analyzed using a FACSCalibur flow cytometer (BD Biosciences) as per the manufacturer’s protocol.
Apoptosis Assay
Induction of apoptosis in HEL cells was determined with the FITC AnnexinV Apoptosis Detection Kit (BD Pharmigen) as per the manufacturer’s protocol. The cells were incubated with either 0.25% DMSO or increasing doses of A46 for 48 hrs, stained with AnnexinV and Propidium Iodide (PI) and then analyzed using a FACSCalibur flow cytometer (BD Biosciences) to determine the percentage of cells undergoing apoptosis.
Western Blotting
HEL cells were treated with the indicated concentrations of A46 for 24 hrs. The cells (~107) were then lysed in 0.8 ml of ice-cold RIPA buffer and protein concentration was determined using a Bradford assay (Bio-Rad). Protein samples (~50 μg) were separated by gel electrophoresis. The separated proteins were then transferred to nitrocellulose membranes and blotted with the indicated antibodies. All the antibodies used were purchased from Cell Signaling Technology except for those noted below; the anti-cyclin D1 was from Santa Cruz Biotechnology, the anti-Jak2 pY1007/pY1008 antibody was from Invitrogen, the anti-c-Src pY418 was obtained from BioSource. A cocktail of polyclonal antibodies from Santa Cruz Biotechnology and BD Transduction Laboratories and a cocktail of antibodies from Millipore and BioSource were used to detect total c-Src and total Jak2 protein, respectively.
Colony Formation Assay
Following an Institutional Review Board approved protocol, a residual bone marrow aspirate was obtained from a de-identified Jak2-V617F positive female diagnosed with Essential Thrombocythemia. The marrow-derived mononuclear cells were first washed in Iscove’s Modified Dulbecco’s Medium (IMDM) and then cultured in human methylcellulose complete medium lacking erythropoietin and thrombopoietin (R&D Systems). Cells were plated in 35-mm petri dishes at a concentration of approximately 4 × 104 cells/ml in the absence or presence of increasing doses of A46. Thrombopoietin (50 ng/ml) was added to the indicated samples. The dishes were incubated at 37°C and 5% CO2 in a humidified atmosphere for 14 days following which the number of colony forming units-megakaryocytes (CFU-Megs) were counted.
Clonogenic Assay
Bone marrow cells from wild type or Jak2-V617F transgenic mice [16] were harvested, cultured ex vivo in Iscove’s Modified Dulbecco’s Medium (IMDM), and treated ex vivo with the indicated concentrations of A46 for 24 hrs. Post treatment, the drug was extensively washed away and the cells (approximately 3 × 104 cells/35 mm dish) were plated in MethoCult medium (Stem Cell Technologies) containing recombinant cytokines, but lacking erythropoietin. Recombinant mouse erythropoietin (R&D Systems) was added at a final concentration of 50 ng/ml where indicated. Five days later, the number of erythroid burst forming units were counted and plotted as a function of treatment group.
In Vivo Analysis
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Florida. Animals were maintained in accordance with NIH standards established in the Guidelines for the Care and Use of Experimental Animals. Briefly, 6–8 month old transgenic mice homozygous for the Jak2-V617F mutation generated on a C57BL/6 background were used to assess the in vivo efficacy of A46. Age-matched non-transgenic C57BL/6 mice were used as wild type controls. The Jak2-V617F allele was detected by genomic PCR using primers 5′-TACAACCTCAGTGGGACAAAGAAGAAC and 5′-CCATGCCAACTGTTTAGCAACTTCA. Transgenic mice received daily IP injections of either DMSO vehicle control (n=3) or 1 mg/kg/day of A46 (n=3) for 17 days, at which time the mice were euthanized. For histological analysis, spleens were fixed overnight in buffered formalin, dehydrated through a graded ethanol series, paraffin-embedded and sectioned (4 μm). Tissue sections were stained with hematoxylin and eosin (H&E) and observed under a light microscope for overall morphology and quantitative assessment of megakaryocytic hyperplasia. At euthanasia, terminal blood samples were obtained and placed into tubes containing potassium salt of ethylenediamine tetraacetic acid (EDTA). The blood was centrifuged at 10,000 × g for 10 minutes and the level of IL-6 in each sample was measured using a commercially available mouse IL-6 ELISA kit (Invitrogen).
Statistical Analysis
Results are expressed as mean +/− SEM. For statistical evaluation of time-dependent responses to A46, a two-way analysis of variance (ANOVA) was used. For the analysis of inhibition of phosphorylation, induction of cell cycle arrest, induction of apoptosis and suppression of pathologic cell growth and clonogenic potential ex vivo, a Student’s t-test was employed. Data were assumed to be statistically significant when p < 0.05.
RESULTS
A46 specifically inhibits Jak2 kinase activity
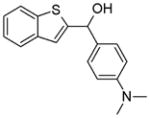
Using an in silico structure based drug design approach, we screened a library of small molecules from the NCI/DTP repository as described previously [20] and identified a small molecule, termed A46, which bound the ATP-binding pocket of Jak2 with a favorable energy score. Table 1 lists the chemical name, NSC number, chemical structure, and molecular weight of this compound.
Table 1. Chemical characteristics of A46.
A46 is shown here along with its chemical name, NSC #, chemical structure, and molecular weight.
| Compound Name | Chemical Name | NSC # | Structure | Molecular Weight |
|---|---|---|---|---|
| A46 | 1-benzothiophen-2-yl-(4-dimethylaminophenyl)methanol | 40282 |
|
283.3874 |
To determine whether this compound had any Jak2-V617F inhibitory potential, we first conducted in vitro kinase assays using recombinant Jak2-V617F protein. After termination of the reactions, kinase activity was determined via anti-phospho-Jak2 (pY1007/pY1008) western blot analysis. In the absence of ATP, the Jak2-V617F protein had an inherent level of autophosphorylation, and this was increased with the addition of ATP (Fig. 1A). Exposure to A46 decreased the kinase activity of Jak2-V617F in a dose-dependent manner.
Figure 1. A46 selectively inhibits Jak2-V617F protein in a cell-free system.

Recombinant Jak2-JH1+JH2-V617F (mutant) (A) and Jak2-JH1+JH2 (wild-type) (B) proteins were incubated in a kinase reaction buffer, either with DMSO or with the indicated doses of A46. The kinase reactions were then separated on an SDS-PAGE and immunoblotted with a phospho-Jak2 (pY1007/pY1008) antibody (A & B, top panels). The membranes were stripped and reprobed with an anti-Jak2 antibody to demonstrate equal loading of protein across all lanes (A & B, bottom panels). Shown is one of four representative results for each. C. Densitometric analysis was done on four representative Western blots to quantify A46-mediated inhibition of Jak2-V617F and Jak2 wild-type phosphorylation at the Tyr1007 residue. The ratio of phosphorylated Jak2 to total Jak2 was expressed as % control and plotted as a function of treatment condition. Shown are the means ± S.D. of four independent experiments. D. Recombinant c-Src protein was incubated in a kinase reaction buffer, either with DMSO or with the indicated doses of PP2 and A46. The kinase reactions were then separated on an SDS-PAGE and immunoblotted with a phospho-Src (pY418) antibody (D, top panel) or with an anti-Src antibody to demonstrate equal loading of protein across all lanes (D, bottom panel). Shown is one of three representative results. E. Densitometric analysis was done on the Western blots to quantify A46-mediated inhibition of c-Src phosphorylation at the Tyr418 residue. The ratio of phosphorylated Src to total Src was expressed as % control and plotted as a function of treatment condition. Shown are the means ± S.D. of three independent experiments.
To determine whether this inhibitory potential was unique to Jak2-V617F, we repeated the kinase reactions, but this time used Jak2 wild type protein. We found that A46 also inhibited the kinase activity of Jak2 wild type, but with what appeared to be less potency (Fig. 1B). To quantify these data, the blots representing Figs. 1A and 1B were subjected to densitometry and the data were then plotted as a function of A46 concentration. We found that A46 induced a dose-dependent decrease in the phosphorylation levels of both Jak2-V617F and Jak2 wild type proteins with an IC50 of ~0.1 μM and ~1 μM, respectively (Fig. 1C). These data thereby suggest that A46 is about 10-fold more selective for inhibiting the Jak2-V617F mutant protein when compared to the wild-type protein.
We next wanted to determine whether this compound was selective for suppressing Jak2 autophosphorylation when compared to other ubiquitous, cytoplasmic tyrosine kinases. For this, recombinant c-Src protein was subjected to similar in vitro kinase reactions in the presence of either A46 or PP2, an inhibitor of Src family kinases. Fig. 1D is a representative blot showing the phospho-c-Src levels and Fig. 1E is a quantification of all blots normalized to total protein. We found that while PP2 reduced c-Src kinase activity in a dose-dependent manner, A46 was without effect. These data therefore demonstrate that A46 selectively inhibits Jak2 tyrosine kinase when compared to c-Src.
Overall, the data in Fig. 1 indicate that A46 preferentially inhibits Jak2-V617F when compared to Jak2 wild type. Furthermore, A46 has no effect on c-Src tyrosine kinase at doses that maximally inhibit Jak2-V617F.
A46 inhibits Jak2-V617F-dependent cell proliferation
We next wanted to test the ability of this compound to suppress Jak2-V617-mediated cell proliferation. For this, we used the human erythroleukemia (HEL 92.1.7) cell line, which is homozygous for the Jak2-V617F mutation [24] and has a transformed proliferative phenotype that is driven by constitutively active Jak2-V617F signaling [25]. HEL cells were treated either with DMSO or with 10 μM of A46 for increasing periods of time. Viable cell numbers for each treatment were determined using trypan blue exclusion and a hemocytometer. When compared to vehicle treated cells, we found that A46 significantly reduced viable cell numbers in a time-dependent manner (Fig. 2A).
Figure 2. A46 inhibits Jak2-V617F-dependent cell proliferation.

A. HEL cells were treated with either DMSO or 10 μM A46 for 0, 24, 48 or 72 hours. Viable cell numbers for each treatment were determined by trypan blue exclusion staining using a hemocytometer. Each sample was measured in triplicate. Shown is one of two sets of representative results. *p < 0.05 with respect to DMSO. B. HEL cells were treated with 10 μM A46 for 0, 8, 12, 24, 48 and 72 hours. At the end of each treatment, the cells were washed, placed in fresh medium and cultured in the absence of the inhibitor for an additional 72 hours. The number of viable cells in each sample was then determined. Each sample was measured in triplicate. Shown are the means ± S.E. from two independent experiments. HEL, SET-2, Raji and CMK cells were treated with increasing doses of A46 for 48 (C) or 72 (D) hours. The percent viable cells from each condition were determined either by trypan blue exclusion staining. Each sample was measured in triplicate.
We next wanted to determine if the suppressive effects of A46 treatment on HEL cell growth were reversible. For this, HEL cells were first exposed to 10 μM of A46 for 0, 8, 12, 24, 48 and 72 hrs. At the end of each time point, the drug was washed away and the cells were resuspended in fresh growth medium. They were then allowed to grow for an additional 72 hrs in the absence of the inhibitor. Viable cell numbers were determined at the end of the 72 hour recovery period. We found that ~19 hrs of initial exposure to A46 prevented 50% of the cells from recovering from the treatment (Fig. 2B). For cells that had been treated with A46 for 48 hrs or more, fewer than 20% were able to recover after removal of the drug (Fig. 1B), suggesting that after a finite time of initial treatment, the effect of the drug on HEL cell growth is largely irreversible.
To determine the specificity of A46 for Jak2-V617F in cultured cells, the drug was applied to four different cell lines and cell viability was assessed. The cell lines were (i) HEL which are homozygous for the Jak2-V617F mutation, (ii) Raji which are a Burkitt’s lymphoma cell line that harbors a translocation between the c-Myc gene on chromosome 8 and the heavy chain locus on chromosome 14, (iii) SET-2 which are essential thrombocythemia cells and are heterozygous for the Jak2-V617F mutation, and (iv) CMK which are acute megakaryocytic leukemia cells that harbor a Jak3-A572V mutation. Each cell line was treated with increasing doses of A46 for 48 and 72 hours. A46 treatment for 48 hours caused a dose-dependent decrease in HEL (Fig. 2C) and SET2 (Fig. 2C) cell viability, had intermediate effect on the viability of CMK cells (Fig. 2C), but had no effect on the viability of Raji cells (Fig. 2C). Similarly, we found that 72 hrs of A46 treatment markedly inhibited the growth of the Jak2-V617F expressing HEL (Fig. 2D) and SET-2 (Fig. 2D) cells in a dose dependent manner with a GI50 of ~ 400 nM for both cell types. The viability of Raji cells (Fig. 2D) was, however, not affected by A46 treatment (GI50 > 25,000 nM). On the other hand, the Jak3-A572V expressing CMK cells had intermediate susceptibility to the drug (Fig. 2D) with a GI50 of ~2,500 nM, suggesting that A46 is selective for Jak2-V617F expressing cell lines over Jak3 and c-Myc mutated cell lines.
Collectively, the data in Fig. 2 demonstrate that A46 selectively inhibits Jak2-V617F dependent cell growth, and this inhibitory effect on HEL cell growth is largely irreversible after a determinate period of initial drug exposure.
A46 specifically inhibits the Jak/STAT signaling pathway
Phosphorylation of Jak2 on tyrosine residues 1007/1008 is concomitant with increased Jak2 kinase activity. Activated Jak2 can in turn phosphorylate and activate downstream signaling targets such as STAT5. Hence, we next wanted to determine if the A46 mediated reduction in HEL cell numbers directly correlated with the suppression of the Jak/STAT signaling pathway. This study is important in order to exclude the possibility that A46 exerts its cell growth inhibitory effect via mechanisms that are independent of the Jak/STAT signaling pathway. For this, we treated HEL cells with increasing concentrations of A46 for 48 hours and then measured the levels of phospho-Jak2 and phospho-STAT5 proteins in these cells using an ELISA-based assay. It is important to note that the phospho-protein levels were measured 48 hrs after drug exposure rather than 72 hrs, as far fewer viable cells exist at the longer time point. We observed that A46 significantly reduced levels of both phospho-Jak2 (Fig. 3A) and phospho-STAT5 (Fig. 3B) proteins in a dose-dependent manner, and the reductions in levels of phosphorylated proteins of the Jak/STAT signaling pathway correlated well with the A46-mediated reductions in HEL cell numbers.
Figure 3. A46 inhibits the Jak2/STAT5 phosphorylation.

HEL cells were treated with 0, 0.09, 0.3, 0.9, 3, 9 and 30 μM of A46 for 48 hours. The cell lysates were then analyzed by ELISA for the measurement of phospho-Jak2 [pY1007/pY1008] (A) and phospho-STAT5 [pY699] (B) protein levels. Each condition was run in triplicate. Shown are the means ± S.D. of two independent experiments.
Thus, the data in Fig. 3 indicate that A46 inhibits HEL cell growth by selectively down regulating the phosphorylation of key signaling molecules of the aberrant Jak/STAT signaling pathway; namely, Jak2-V617F and STAT5.
A46 induces G1/S cell cycle arrest in HEL cells
To determine the mechanism of A46-mediated suppression of HEL cell growth, we first analyzed the cell cycle distribution of cells as a function of drug treatment. Here, we treated HEL cells with increasing doses of A46 for 48 hours and then performed cell cycle analysis via flow cytometry. Again, analysis was performed 48 hours after drug exposure because far fewer viable cells exist at longer time points. We found that exposure to A46 increased the percentage of cells in G1 phase and correspondingly decreased the percentage of cells in S phase, and this effect was dose-dependent (Fig. 4A).
Figure 4. A46 arrests HEL cells in G1 phase of the cell cycle.

HEL cells were treated with 0, 0.1, 0.3, 1, 3, 10 and 30 μM of A46. A. After 48 hrs of treatment, cell cycle distribution of treated cells was determined as a function of drug treatment using flow cytometry. Shown are the means ± S.E. of two independent experiments. B. Following 24 hrs of treatment with the indicated concentrations of A46, cells were lysed and analyzed by immunoblotting with an anti-cyclin D1 (top) or an anti-β-actin antibody (bottom). Shown is one of two representative results.
Cell cycle progression is driven and regulated by the cyclic expression of cyclin/CDK complexes [26]. Cyclin D1 levels play a critical role in promoting the progression of the cell cycle from G1 to S phase [27] and is also known to be a downstream target of STAT5 transcriptional regulation [28]. Therefore, in order to confirm the induction of G1 phase arrest in these cells, we performed anti-cyclin D1 western blot analysis on HEL cells that had been treated with increasing doses of A46 for 24 hours. We found that A46 treatment decreased the levels of cyclin D1 protein in a dose-dependent manner (Fig. 4B, upper panel), thereby confirming the A46-induced arrest of HEL cells in the G1 phase of the cell cycle. Analysis of the same samples with an anti β-actin antibody was used as a loading control (Fig. 4B, bottom panel).
Overall, the data in Fig. 4 demonstrate that A46 treatment induces G1/S cell cycle arrest in HEL cells which correlates with the down regulation of cyclin D1.
A46 induces apoptosis in HEL cells
The Jak/STAT signaling pathway is known to have direct effects on cell survival and apoptosis [28]. Having shown that A46 inhibited HEL cell growth (Fig 2) and induced cell cycle arrest (Fig. 4), we next wanted to determine if this drug causes apoptotic cell death in HEL cells. For this, we used Annexin V/Propidium Iodide (PI) double staining and flow cytometry. The treatment conditions were the same as in Fig. 4A. The data in Fig. 5A show representative apoptosis assay staining profiles from one experiment while Fig 5B is a quantitative graph of two independent experiments showing the percentage of cells in early apoptosis (Annexin V positive/PI negative) plotted as a function of treatment condition. These data show that A46 induces apoptosis in HEL cells in a dose-dependent manner.
Figure 5. A46 induces apoptosis in HEL cells.

HEL cells were treated with 0, 0.1, 0.3, 1, 3, 10 and 30 μM of A46 for 48 hours, stained with annexin V-FITC and propidium iodide and then analyzed by flow cytometry to determine the level of apoptosis in the treated cells. A. Shown are representative flow cytometry profiles from one of two independent experiments. B. Quantification of the percentage of cells in early apoptosis as a function of drug treatment. Shown are the means ± S.E. of two independent experiments.
Cleavage of procaspases converts these proteins into their functionally active forms. Active caspases can then act on and cleave their substrates, such as the DNA repair enzyme PARP, thereby affecting the integrity of the cell and triggering apoptosis [29]. Cleavage of procaspases and PARP are therefore considered hallmarks of apoptotic induction. In order to confirm the induction of apoptosis by A46, we analyzed the effect of A46 treatment on the cleavage of procaspase 3 and its downstream substrate, PARP. HEL cells were treated with increasing doses of A46 for 24 hours and the protein levels of procaspase-3, PARP, and β-actin were then determined. We found that A46 induced a dose-dependent cleavage of PARP and a decrease in procaspase 3 expression, but had no effect on β-actin levels (Fig. 6A). As such, these data corroborate the Annexin V/PI apoptosis assay in Fig. 5.
Figure 6. A46-induced apoptosis is mediated by the down regulation/cleavage of Bcl-2 family proteins Bim, Bax and Bid.

HEL cells were treated with 0, 0.1, 0.3, 1, 3, 10 and 30 μM of A46 for 24 hours. The whole cell lysates were then analyzed by western blotting with a series of antibodies as indicated: A. anti-poly(ADP-ribose) polymerase (PARP), anti-caspase 3 and anti-β-actin, B. anti-Bim, -Bax, -Bid, -Bcl-2 and -β-actin. Shown are the results from one of two independent experiments for each.
Having confirmed the ability of A46 to induce apoptosis in HEL cells, we next wanted to determine the mechanism by which this drug causes apoptotic cell death. Apoptosis is regulated by members of the Bcl-2 family, many of which are direct downstream gene targets of the Jak/STAT signaling pathway [28]. Therefore, we next monitored the expression of Bcl-2 family members in HEL cells exposed to different doses of A46 for 24 hours via western blot analysis. We observed an A46-induced dose-dependent decrease in expression BimEL, a pro-apoptotic member of the Bcl-2 family (Fig. 6B). Although Bim is a pro-apoptotic protein, its disappearance/down regulation has been shown to be associated with the generation of cleaved pro-apoptotic forms that positively regulate and amplify apoptotic signaling [30, 31]. Another pro-apoptotic Bcl-2 family protein, Bax, was down regulated in response to A46 treatment, but this decrease was not statistically significant (Fig. 6B). Cleavage of Bax in response to apoptotic cues is not uncommon and is known to enhance its cell death function [32, 33]. Additionally, we found that A46 decreased the levels of the inactive precursor form of Bid (Fig. 6B), which is yet another pro-apoptotic protein that is activated upon cleavage [34]. Levels of anti-apoptotic Bcl-2 protein did not change with A46 treatment (Fig. 6B). Lastly, the samples were blotted with an anti β-actin antibody to demonstrate equal protein loading across all lanes (Fig. 6B).
In summary, the data in Figs. 5 and 6 indicate that A46 induces apoptotic cell death in HEL cells via the down regulation/cleavage of Bcl-2 family proteins Bim and Bid. Thus, from the data in Figs. 4, 5, and 6, we conclude that A46 inhibits HEL cell proliferation by arresting the cells at G1/S transition and inducing apoptosis.
A46 suppresses cytokine-independent pathologic cell growth of Jak2-V617F positive bone marrow cells, ex vivo
We next wanted to determine the ability of A46 to inhibit the pathologic cell growth of patient-derived Jak2-V617F positive bone marrow cells, ex vivo. For this, mononuclear cells isolated from the bone-marrow of a female ET patient, who was Jak2-V617F positive, were cultured in the presence of increasing doses of A46. Hematopoietic progenitor cells isolated from a normal individual are unable to grow in the absence of exogenously added cytokines. However, the V617F positive progenitor cells grow under such conditions because the Jak2-V617F mutation confers cytokine-independent growth [35, 36]. The ET patient derived bone marrow cells were also cultured in the presence or absence of the exogenously added thrombopoietin. The results show that treatment of the mutant bone marrow cells with A46 significantly suppressed the Jak2-V617F, pathologic cell growth, in a dose-dependent manner (Fig. 7A). We also observed that exposure of this patient’s marrow derived cells to exogenous thrombopoietin significantly increased the number of colonies formed (Fig. 7A), and this was also inhibited by A46.
Figure 7. A46 suppresses cytokine-independent pathologic cell growth of Jak2-V617F positive bone marrow cells, ex vivo.

A. Patient-derived bone marrow mononuclear cells were cultured in semisolid medium in the presence of increasing doses of A46, with or without thrombopoietin (TPO). At the end of 14 days, the numbers of megakaryocyte colony-forming units (CFU-Megs) were counted and plotted as a function of treatment condition. Each condition was measured in duplicate. *p < 0.05 with respect to 0 μM A46 (−TPO), #p < 0.05 with respect to 0 μM A46 (+TPO), and §p < 0.05 (+ TPO) vs. (−TPO). B and C. Bone marrow aspirates obtained from either Jak2-WT or Jak2-V617F transgenic mice were treated with 0, 2.5, or 25 μM A46 for 24 hours, ex vivo. The drug was then washed away and the cells were cultured in drug-free semi-solid medium for an additional 5 days. The numbers of erythroid burst-forming units (BFU-E) in each sample were then plotted as a function of genotype. Each point was measured in duplicate. *, p<0.05 vs. − EPO 0 μM control;, p<0.05 vs. + EPO 0 μM control; #, p<0.05 vs. − EPO, within same drug treatment group.
To better understand the Jak2 inhibitory properties of A46, we also carried out clonogenic assays whereby bone marrow cells were isolated from wild type and Jak2-V617F transgenic mice and then cultured ex vivo in the presence of increasing concentrations of A46, either with or without recombinant erythropoietin. We found that cells taken from wild type mice had a visible clonogenic growth potential and this was increased with erythropoietin (Fig. 7B). Furthermore, A46 decreased the clonogenic growth potential of these cells by 50% – 67% at 25 μM. In contrast to this, cells harvested from the marrow of Jak2-V617F mice exhibited a higher degree of clonogenic growth potential, an insensitivity to exogenously added erythropoietin, but a higher sensitivity to A46 (Fig. 7C).
As such, data in Fig. 7 demonstrate that A46 inhibits Jak2-V617F-dependent pathologic cell growth of patient derived bone marrow cells and preferentially suppresses the clonogenic growth potential of Jak2-V617F-positive, bone marrow cells from MPN mice.
A46 attenuates the myeloproliferative phenotype in Jak2-V617F transgenic mice
To demonstrate in vivo efficacy of A46, Jak2-V617F transgenic MPN mice [16] were treated with either vehicle solution or 1 mg/kg/day of A46 for 17 days, at which time the mice were euthanized and several efficacy endpoints were determined. First, A46 treatment resulted in a significant reduction in the spleen to body weight ratio when compared to DMSO vehicle treated animals (Fig. 8A). Second, a common characteristic of the MPN phenotype is megakaryocytic hyperplasia [37]. Fig. 8B shows representative spleen sections for each condition while Fig. 8C shows the average number of megakaryocytes per 0.043 mm2 of spleen tissue. We found that MPN transgenic mice treated with vehicle control solution had marked megakaryocytic hyperplasia when compared to wild type controls, and this was significantly reduced with A46 treatment. Lastly, increased levels of interleukin-6 (IL-6) and/or its cognate receptor have previously been implicated in the pathogenesis of the MPNs [1, 38, 39]. Therefore, we measured IL-6 levels in the plasma of the mice at euthanasia. We found that when compared to wild type mice, the MPN mice receiving vehicle control solution had significantly elevated levels of IL-6, and this was significantly reduced with A46 treatment (Fig. 8D).
Figure 8. A46 treatment alleviates Jak2-V617F mediated pathogenesis in vivo.
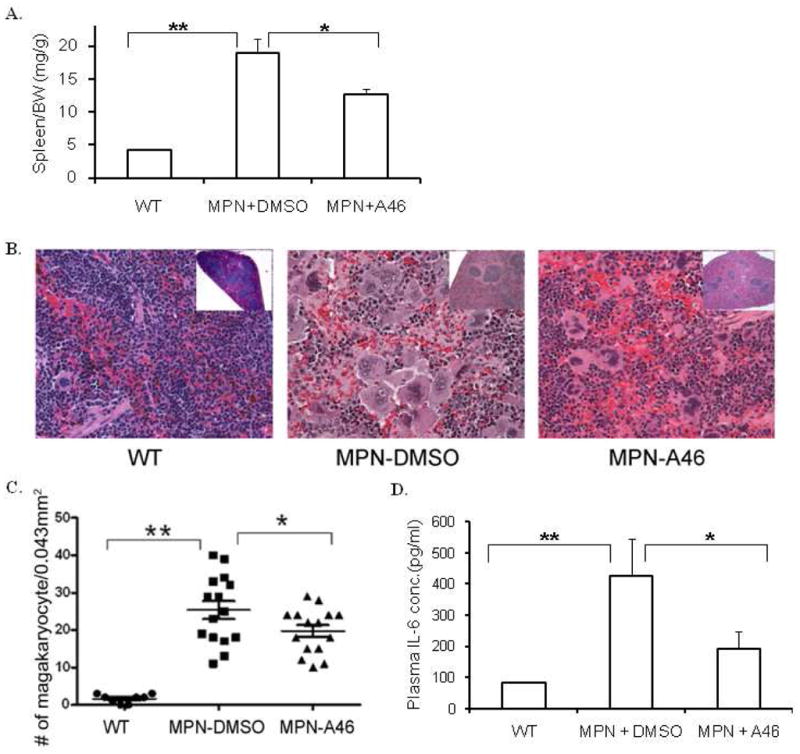
A. Spleen weight to body weight ratios of wild-type, vehicle treated and A46 treated MPN transgenic animals. B. Representative images of H&E stained spleen histological sections from each of these different treatment groups at 100X (Inset images: 4X). C. Quantification of the number of megakaryocytes in the spleen plotted as a function of the treatment group. D. IL-6 levels in the plasma plotted as a function of the treatment group. *p<0.05, **p<0.01.
Overall, data in Fig. 8 demonstrate the in vivo therapeutic efficacy of A46 in alleviating the MPN phenotype in Jak2-V617F expressing transgenic mice.
DISCUSSION
Aberrant Jak2 kinase activity has been linked to human disorders including several hematological malignancies and the MPNs [3–5]. A Jak2 gain-of-function mutation (Jak2-V617F) has been detected in over 90% of patients with PV and greater than 50% of patients with ET and PMF [6–10]. Jak2-V617F-negative MPN patients often carry other activating mutations either in Jak2 [40–42] or in upstream signaling molecules, such as the thrombopoietin receptor, MPL [43, 44]. The critical role of Jak2 in the pathogenesis of these disorders coupled with the fact that there are no curative treatment options currently available for MPN patients, has generated a considerable amount of interest in developing Jak2 inhibitors as potential therapeutics for MPNs.
In this study, we characterized a novel benzothiophene inhibitor of Jak2 that was identified by in silico structure based drug screening. We show that this compound preferentially inhibits Jak2-V617F protein when compared to Jak2 wild type protein (~10 fold) and it has no effect on c-Src protein (Fig. 1). It also inhibits Jak2-V617F mediated cell proliferation both time and dose dependently (Fig. 2A, 2C & 2D) and this inhibitory effect of the drug on HEL cell growth is largely irreversible after ~48 hrs of initial exposure (Fig. 2B). An important parameter used in evaluating the effectiveness and safety of a drug is its specificity since more selective drugs can be safely administered at higher doses without causing severe side-effects. In the past, Jak2 inhibitors have been reported to have non-specific off target effects. For example, AG490, one of the first Jak2 inhibitors identified, was a potent Jak2 inhibitor, but had poor specificity [45–47]. Our cell based data here show that the Jak2-V617F positive cell lines, HEL and SET2, are significantly more sensitive to inhibition by A46 (GI50 ~400 nM, Fig. 2D) when compared to Jak3-dependent CMK cells (GI50 ~2,500 nM, Fig. 2D) or c-myc-dependent Raji cells (GI50 > 25,000 nM, Fig. 2D), thereby suggesting that A46 selectively inhibits the proliferation of Jak2-V617F-dependent cell lines while having little to no effect on other cells lines whose proliferation is driven by Jak2-independent mechanisms.
We have also shown that the A46-induced cell growth inhibition correlates with direct suppression of the Jak/STAT signaling (Fig. 3A & 3B) which eliminates the possibility that A46 might be exerting its cell growth inhibitory effect via mechanisms that are independent of this pathway. Our results indicate that the mechanism by which A46 suppresses Jak2-V617F-mediated HEL cell proliferation is via the induction of both G1/S cell cycle arrest (Fig. 4) and apoptosis (Fig. 5). Deregulation of Jak/STAT signaling promotes cell proliferation and blocks apoptosis resulting in disease pathogenesis. Hence, a better understanding of the mechanism of Jak2-inhibition induced cell death may lead to the development of more effective therapeutic strategies for treating MPN patients, including combination therapies of Jak2 inhibitor and apoptotic modulator mimetics. Here, we also show that A46 induces apoptotic cell death in HEL cells via the down regulation/cleavage of Bcl-2 family proteins Bim and Bid. There are conflicting reports on the effect of apoptosis induction on the expression of the proapoptotic protein Bim; some studies suggest that Bim is up regulated during apoptosis [48, 49] whereas others suggest that Bim is cleaved during apoptosis generating active and proapoptotic cleaved fragments [30, 31].
Our in vivo data demonstrate the therapeutic efficacy of A46 in MPN transgenic mice expressing the Jak2-V617F mutant protein (Fig. 8). We show that in these animals, A46 treatment is able to significantly reduce MPN-associated pathologic symptoms such as a high spleen to body weight ratio (Fig. 8A), megakaryocytic hyperplasia (Fig. 8B & 8C) and elevated IL-6 levels (Fig. 8D). Moreover, the data in Fig. 7 demonstrate that exposure of Jak2-V617F bone marrow cells to A46 subsequently alter the clonogenic growth potential of those cells. Overall, these data indicate efficacy of A46 in the spleen, peripheral blood, and bone marrow.
Jak2 small molecule inhibitors currently represent a diverse number of chemical structures including pyrazines, pyrimidines, azaindoles, aminoindazoles, deazapurines, stilbenes, benzoxazoles, and quinoxalines [50]. Our work here is significant in that it is the first report indicating that benzothiophene based compounds, such as A46, possess Jak2 inhibitory potential. Benzothiophenes are heterocyclic structures known to have pro- and anti-estrogenic [51], anti-lipoxygenase [52] and anti-fungal properties [53]. Not surprisingly, several approved pharmaceuticals including raloxifene, zileuton, and sertaconazole have benzothiophene based chemical structures. Molecules with a benzothiophene core have also been reported to inhibit tubulin, cysteine and serine proteases, herpes simplex virus type I replication and opioid receptor analgesics [54]. These heterocyclic structures are relatively stable and their reactive site allows for subsequent derivatization, suggesting that A46 is quite amenable to future lead optimization. As such, benzothiophenes may be a new class of scaffolds for Jak2 small molecule inhibitors.
In summary, our data collectively show that the novel benzothiophene small molecule inhibitor of Jak2, A46, inhibits Jak2-V617F-mediated pathological cell growth in vitro, ex vivo, and in vivo. As such, this compound may perhaps serve as a lead therapeutic agent for the treatment of Jak2-V617F mediated pathogenesis.
Acknowledgments
This work was supported by National Institutes of Health Award R01-HL67277 and a Pilot Project Award from the University of Florida Clinical and Translational Science Institute.
Footnotes
Conflict of interest disclosure: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 3.Peeters P, Raynaud SD, Cools J, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- 4.Griesinger F, Hennig H, Hillmer F, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329–333. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 5.Lacronique V, Boureux A, Valle VD, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 6.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 8.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 9.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Zhao R, Xing S, Li Z, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jelinek J, Oki Y, Gharibyan V, et al. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005;106:3370–3373. doi: 10.1182/blood-2005-05-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102:18962–18967. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 14.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marty C, Lacout C, Martin A, et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood. 2010;116:783–787. doi: 10.1182/blood-2009-12-257063. [DOI] [PubMed] [Google Scholar]
- 16.Xing S, Wanting TH, Zhao W, et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood. 2008;111:5109–5117. doi: 10.1182/blood-2007-05-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115:3589–3597. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Spensberger D, Ahn JS, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116:1528–1538. doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandberg EM, Ma X, He K, Frank SJ, Ostrov DA, Sayeski PP. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of jak2 tyrosine kinase autophosphorylation [correction of autophophorylation] J Med Chem. 2005;48:2526–2533. doi: 10.1021/jm049470k. [DOI] [PubMed] [Google Scholar]
- 20.Sayyah J, Magis A, Ostrov DA, Allan RW, Braylan RC, Sayeski PP. Z3, a novel Jak2 tyrosine kinase small-molecule inhibitor that suppresses Jak2-mediated pathologic cell growth. Mol Cancer Ther. 2008;7:2308–2318. doi: 10.1158/1535-7163.MCT-08-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiss R, Polgar T, Kirabo A, et al. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett. 2009;19:3598–3601. doi: 10.1016/j.bmcl.2009.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majumder A, Govindasamy L, Magis A, et al. Structure-Function Correlation of G6, a Novel Small Molecule Inhibitor of Jak2: Indispensability Of The Stilbenoid Core. Journal of Biological Chemistry. 2010;285:31399–31407. doi: 10.1074/jbc.M110.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirabo A, Embury J, Kiss R, et al. The stilbenoid tyrosine kinase inhibitor, G6, suppresses Jak2-V617F-mediated human pathological cell growth in vitro and in vivo. J Biol Chem. 2011;286:4280–4291. doi: 10.1074/jbc.M110.200774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quentmeier H, MacLeod RA, Zaborski M, Drexler HG. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia. 2006;20:471–476. doi: 10.1038/sj.leu.2404081. [DOI] [PubMed] [Google Scholar]
- 25.Walz C, Crowley BJ, Hudon HE, et al. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–18183. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 26.Ivanchuk SM, Rutka JT. The cell cycle: accelerators, brakes, and checkpoints. Neurosurgery. 2004;54:692–699. doi: 10.1227/01.neu.0000109534.28063.5d. discussion 699–700. [DOI] [PubMed] [Google Scholar]
- 27.Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15:158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- 28.Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–6737. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, Harvey NL. Role of multiple cellular proteases in the execution of programmed cell death. FEBS Lett. 1995;375:169–173. doi: 10.1016/0014-5793(95)01186-i. [DOI] [PubMed] [Google Scholar]
- 30.Chen D, Zhou Q. Caspase cleavage of BimEL triggers a positive feedback amplification of apoptotic signaling. Proc Natl Acad Sci U S A. 2004;101:1235–1240. doi: 10.1073/pnas.0308050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Bougie P, Oliver L, Le Gouill S, Bataille R, Amiot M. Melphalan-induced apoptosis in multiple myeloma cells is associated with a cleavage of Mcl-1 and Bim and a decrease in the Mcl-1/Bim complex. Oncogene. 2005;24:8076–8079. doi: 10.1038/sj.onc.1208949. [DOI] [PubMed] [Google Scholar]
- 32.Wood DE, Newcomb EW. Cleavage of Bax enhances its cell death function. Exp Cell Res. 2000;256:375–382. doi: 10.1006/excr.2000.4859. [DOI] [PubMed] [Google Scholar]
- 33.Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 34.Gross A, Yin XM, Wang K, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 35.Axelrad AA, Eskinazi D, Correa PN, Amato D. Hypersensitivity of circulating progenitor cells to megakaryocyte growth and development factor (PEG-rHu MGDF) in essential thrombocythemia. Blood. 2000;96:3310–3321. [PubMed] [Google Scholar]
- 36.Xiong H, Chen ZF, Liang QC, et al. Inhibition of DNA methyltransferase induces G2 cell cycle arrest and apoptosis in human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5 signalling. J Cell Mol Med. 2009;13:3668–3679. doi: 10.1111/j.1582-4934.2009.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaleskas VM, Krause DS, Lazarides K, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu RY, Fan C, Garcia R, Jove R, Zuckerman KS. Constitutive activation of the JAK2/STAT5 signal transduction pathway correlates with growth factor independence of megakaryocytic leukemic cell lines. Blood. 1999;93:2369–2379. [PubMed] [Google Scholar]
- 39.Wang Y, Morella KK, Ripperger J, et al. Receptors for interleukin-3 (IL-3) and growth hormone mediate an IL-6-type transcriptional induction in the presence of JAK2 or STAT3. Blood. 1995;86:1671–1679. [PubMed] [Google Scholar]
- 40.Zhang SJ, Li JY, Li WD, Song JH, Xu W, Qiu HX. The investigation of JAK2 mutation in Chinese myeloproliferative diseases-identification of a novel C616Y point mutation in a PV patient. Int J Lab Hematol. 2007;29:71–72. doi: 10.1111/j.1365-2257.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 41.Schnittger S, Bacher U, Kern W, Schroder M, Haferlach T, Schoch C. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia. 2006;20:2195–2197. doi: 10.1038/sj.leu.2404325. [DOI] [PubMed] [Google Scholar]
- 42.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 45.Meydan N, Grunberger T, Dadi H, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 46.Kleinberger-Doron N, Shelah N, Capone R, Gazit A, Levitzki A. Inhibition of Cdk2 activation by selected tyrphostins causes cell cycle arrest at late G1 and S phase. Exp Cell Res. 1998;241:340–351. doi: 10.1006/excr.1998.4061. [DOI] [PubMed] [Google Scholar]
- 47.Gu Y, Zou Y, Aikawa R, et al. Growth hormone signalling and apoptosis in neonatal rat cardiomyocytes. Mol Cell Biochem. 2001;223:35–46. doi: 10.1023/a:1017941625858. [DOI] [PubMed] [Google Scholar]
- 48.Will B, Siddiqi T, Jorda MA, et al. Apoptosis induced by JAK2 inhibition is mediated by Bim and enhanced by the BH3 mimetic ABT-737 in JAK2 mutant human erythroid cells. Blood. 2010;115:2901–2909. doi: 10.1182/blood-2009-03-209544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubert J, Qian Z, Andraos R, Guthy DA, Radimerski T. Bim and Mcl-1 exert key roles in regulating JAK2V617F cell survival. BMC Cancer. 2011;11:24. doi: 10.1186/1471-2407-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baskin R, Majumder A, Sayeski PP. The recent medicinal chemistry development of Jak2 tyrosine kinase small molecule inhibitors. Curr Med Chem. 2010;17:4551–4558. doi: 10.2174/092986710794182953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuchs-Young R, Glasebrook AL, Short LL, et al. Raloxifene is a tissue-selective agonist/antagonist that functions through the estrogen receptor. Ann N Y Acad Sci. 1995;761:355–360. doi: 10.1111/j.1749-6632.1995.tb31392.x. [DOI] [PubMed] [Google Scholar]
- 52.Carter GW, Young PR, Albert DH, et al. 5-lipoxygenase inhibitory activity of zileuton. J Pharmacol Exp Ther. 1991;256:929–937. [PubMed] [Google Scholar]
- 53.Palacin C, Tarrago C, Ortiz JA. Sertaconazole: pharmacology of a gynecological antifungal agent. Int J Gynaecol Obstet. 2000;71 (Suppl 1):S37–46. doi: 10.1016/s0020-7292(00)00351-9. [DOI] [PubMed] [Google Scholar]
- 54.Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]