

Abstract
Diseases of the coronary circulation remain the leading cause of death in Western society despite impressive advances in diagnosis, pharmacotherapy and post-event management. Part of this statistic likely stems from a parallel increase in the prevalence of obesity and metabolic dysfunction, both significant risk factors for coronary disease. Obesity and diabetes pose unique challenges for the heart and their impact on the coronary vasculature remain incompletely understood. The vascular endothelium is a major interface between arterial function and the physical and chemical components of blood flow. Proper function of the endothelium is necessary to preserve hemostasis, maintain vascular tone and limit the extent of vascular diseases such as atherosclerosis. Given its central role in vascular health, endothelial dysfunction has been the source of considerable research interest in diabetes and obesity. In the current review, we will examine the pathologic impact of obesity and diabetes on coronary function and the extent to which these two factors impact endothelial function.
Diseases of the coronary circulation remain the leading cause of death in Western society despite impressive advances in diagnosis, pharmacotherapy and post-event management. Part of this statistic likely stems from a parallel increase in the prevalence of obesity and metabolic dysfunction, significant risk factors for coronary disease. Obesity and diabetes pose unique challenges for the heart. For example, obesity increases cardiac output to service a large corpus, increasing the denominator of the perfusion/function relationship. While hyperlipidemia is well-recognized as a coronary risk factor, diabetes is characterized more as a triglyceride dyslipidemia and differs from the traditional hypercholesterolemia of the lean patient with atherosclerosis. How these and other factors impact the vasculature remains incompletely understood.
The vascular endothelium is a major interface between arterial function and the physical and chemical components of blood flow. Proper function of the endothelium is necessary to preserve hemostasis, maintain vascular tone and limit the extent of vascular diseases such as atherosclerosis. Given its central role in vascular health, endothelial dysfunction has been the source of considerable research interest in diabetes and obesity. In the current review, we will examine the pathologic impact of obesity and diabetes on coronary function and the extent to which these two factors impact endothelial function.
Obesity, diabetes and coronary disease in the human population
Obesity in clinical terms has been defined by a series of indices, the most common of which is the Body Mass Index or BMI. Increasing BMI is associated with increasing cardiovascular risk when the value exceeds 25, especially in women [1]. In addition to the well-described increase in the incidence of diabetes, additional evidence suggests that components of the metabolic syndrome such as insulin resistance and dyslipidemia are equally important. Indeed, data from the NHANES III Study indicate that coronary disease is much more prevalent in individuals with metabolic syndrome whether diabetes is present or not (Figure 1) [2].
Figure 1.

Effects of obesity and diabetes on the prevalence of coronary heart disease in adults. From Alexander et al, 2003 [2]. Reproduced with permission.
The heart of the obese, diabetic individual faces a number of unique challenges. Most notable among these is an increase in cardiac output to service the larger body mass of the obese patient [3]. The increase in cardiac output is multi-factorial, reflecting an increase in venous return from the body’s larger blood volume [4] combined with a sympathoactivation that is prevalent in the obese population [5]. Elevations in cardiac output increases cardiac oxygen consumption [6] and thus the need for perfusion is increased.
In parallel to this increased demand, the deleterious effects of the obese state tend to drive down perfusion due to increasing incidence of atherosclerosis and microvascular disease [7]. Flow restriction via large artery stenosis and/or loss of microvascular vasomotor function contribute to reduced perfusion, and obesity and diabetes impact each of these in human populations. The paradigm of increasing demand combined with threats to perfusion underlies the central clinical dilemma in the cardiac health of obese and metabolically compromised patients.
Atherosclerosis
The effects of diabetes on atherosclerosis are well known and have been subject to extensive review [8, 9]. Data from the MRFIT study suggest that incidence of coronary atherosclerosis may be as high as 4-fold compared to non-diabetic patients [10] and the diabetic patient often presents with a progressive and complex form of atherosclerosis. It has been suggested that diabetics may be refractory to Glagov remodeling [11, 12], a vascular adaptation to the presence of a lesion that preserves lumen patency in the early stages of disease [13]. The loss of this effect contributes to premature encroachment on the lumen by the lesion and accelerates ischemic risk. The mechanisms by which diabetes limits Glagov remodeling are unclear and some controversy exists regarding diabetic risk [14]. Similar defects thwart revascularization therapy as diabetics are often more prone to restenosis of arterial bypass grafts and intrusion of lesion into coronary stents [15, 16]. Finally, diabetes may impair the formation of collateral arteries, undermining in the hearts intrinsic protections against chronic ischemia [17]. While precise links between coronary disease and frank diabetes remain to be fully elucidated, the diabetic milieu presents a toxic state of elevated lipids, disproportionate levels of triglycerides, high glucose, activation of inflammatory cells and oxidant stress [18, 19]. Indeed, when so many injurious factors are present, it may be virtually impossible to tease out specific sources of injury that contribute to atherosclerosis.
An important avenue of intervention may be a more aggressive management of the obese and pre-diabetic patient population. Mounting evidence suggests that, even in the absence of frank diabetes, metabolic dysfunction can be identified as a predictor of atherosclerosis [20, 21]. As early as 1970, Kayshap et al [22] determined that in “asymptomatic diabetics”, individuals without observable diabetes but evidence of glucose intolerance, those with ischemic heart disease demonstrated more severe metabolic alterations when challenged with oral glucose or tobutamide. The authors concluded that these deficits might lay the ground work for atherosclerosis even before the effects of frank diabetes were evident. Over the intervening decades, this concept has been refined from “exaggerated insulin production” [23] to the modern concept of insulin resistance or metabolic syndrome used today [24–26]. Consistent with the observations of the NHANES Study[2], obesity is also a positive and independent risk factor for pro-atherosclerotic conditions traditionally attributed to diabetes, such as rates of restenosis after stenting [15, 27]. Obese individuals commonly have an atherogenic lipid profile with disproportionate levels of plasma triglycerides and low HDL[28]. Keaney et al documented that oxidant stress, a major suspect in diabetic vasculopathy, correlated with increased body mass independently of diabetes [29]. Finally obesity is associated with a heightened state of inflammation with elevated levels of pro-inflammatory cytokines such as TNF-alpha and interleukin-6 [30]. Thus, many of the “usual suspects” in atherogenesis are present and potentially causal in the pre-diabetic patient as well as those with later stage disease.
Microvascular disease
While much of the functional impact of coronary disease is secondary to atherosclerotic stenosis, microvascular disease is also an important contributor [31–33]. Impaired vasomotor tone limits the ability of the heart to adapt to a stenosis by autoregulatory dilation [34] and in some cases, limits coronary reserve or produces ischemia even in the absence of significant plaque burden [35–37]. Moreover, diabetics have a greater incidence of silent ischemia [38], a precursor condition to infarction in which the microcirculation likely plays a greater role [39–41]
As with atherosclerosis, the impact of diabetes on the human coronary circulation is well-known and profound. Diabetic microvascular disease in the coronary circulation is characterized by a reduced vasodilator tone[37, 42], inappropriate vasoconstriction [43] and detrimental structural remodeling to stiffer vessels with smaller lumina [44]. The impairments in vasodilator function include decrements in endothelium-dependent dilation [43] as well smooth muscle dilation to hypoxia [45] or adenosine [46].
While deleterious effects of diabetes are well-documented, microvascular data from obese, pre- or non-diabetic humans are significantly less abundant. This stems in part from the relatively recent increase in the prevalence of obesity, even more recent recognition that obesity may itself be a risk factor for cardiovascular disease and the fact that most obese patients undergoing cardiac procedures (where material for study is largely derived) usually have more advanced disease than obesity alone.
Nevertheless, some basic observations indicate that obesity itself has impact on the coronary circulation. Recent work from Kiviniemi et al [47] indicates that in healthy young adult men, coronary flow reserve varied inversely with waist-to-hip ratio, suggesting that accumulation of visceral fat has a detrimental impact on the coronary vasculature (Figure 2). Furthermore, Dagres and colleagues found that insulin resistance was a predictor of reductions in coronary flow reserve in obese individuals [46]. Mechanistic studies from intact coronary arterioles remains limited, though some evidence of endothelial dysfunction has been reported [48].
Figure 2.
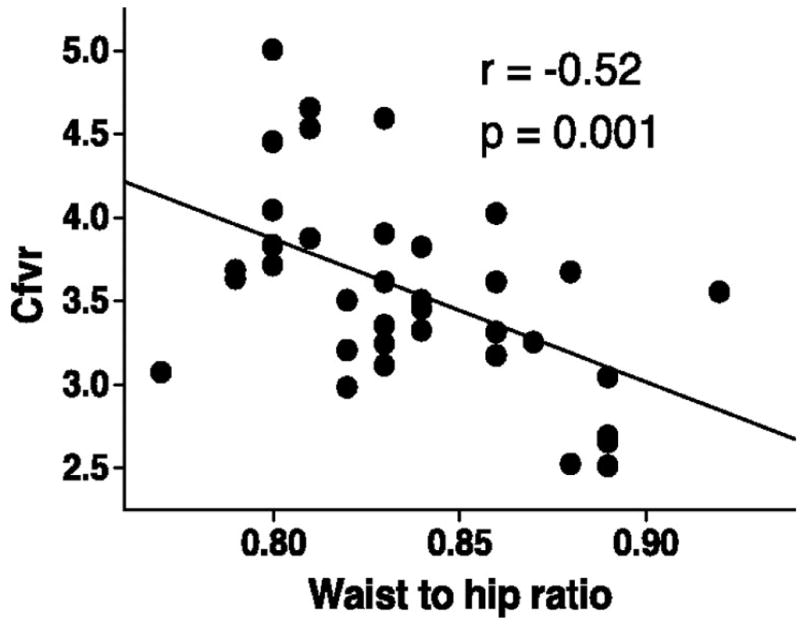
Effects of obesity on coronary flow velocity reserve in health young men. From Kiviniemi et al, 2008 [47]. Reproduced with permission.
Taken together, the above evidence makes a convincing case that obesity and diabetes have profound negative consequences for the coronary circulation. The current review will examine the effects of obesity and metabolic disease on endothelial and smooth muscle cells, the two predominant cell types in the vasculature. Insight into smooth muscle defects is offered in the sister article to this one by Berwick et al [49]. Insights into endothelial cell defects will be offered from a broad understanding of effects of obesity and diabetes on vascular endothelium in general with specific discussion of the coronary circulation following.
Obesity, diabetes and the vascular endothelium
Endothelial control of vascular tone
Represented by a monolayer of cells which lines the entire vascular system, the vascular endothelium constitutes the interface between the circulating blood and the vascular wall but also serves as an important autocrine and paracrine organ that regulates vascular function. The endothelium is a dynamic organ sensitive to mechanical forces exerted by the flowing blood (shear stress) and to chemical signals and secretes a wide range of vasoactive and trophic factors that regulate vascular tone, cell adhesion, platelet function, plasmatic coagulation, vascular smooth muscle cell proliferation, and vessel wall inflammation. The importance of the endothelium was first recognized for its effects on vascular tone more than 30 years ago when Furchgott and Zawadsky demonstrated elegantly that vascular relaxation depends on a vasodilator substance released by the endothelium [50]. The endothelium-derived relaxing factor, as they called it, was subsequently identified to be nitric oxide (NO). NO is released in response to an increase in shear stress but is also secreted in response to signaling molecules such as bradykinin, adenosine, vascular endothelial growth factor and insulin. NO is generated by the conversion of L-arginine to L-citruline by the endothelial NO synthase (eNOS or NOS3), in the presence of co-factors such as tetrahydrobiopterin (BH4) [51]. Once released, NO diffuses across the endothelial cells into the adjacent smooth muscle cells where it activates the soluble guanylate cyclase and induces cGMP-mediated vascular relaxation[52].
Although NO is the main vasodilator substance released by the endothelium, endothelium-dependent control of vascular tone also involves NO-independent mechanisms. Endothelial cells could lead to vascular relaxation through the secretion of endothelium-derived hyperpolarizing factors (EDHF) able to mediate hyperpolarization of smooth muscle cells. EDHF is released under endothelial stimulation by agonists such as bradykinin and acetylcholine [53]. EDHF increases potassium conductance and facilitates the propagation of hyperpolarization of smooth muscle cells to maintain the vasodilator tone. The nature of EDHF and its involvement in the control of vascular relaxation are partially understood and differ between vascular beds [54].
In the endothelium, the conversion of arachidonic acid (AA) by the cyclooxygenases leads to the formation of prostacyclins (PGI2, PGE2) and thromboxane A2 (TxA2) that respectively relax and constrict vascular smooth muscle cells. Cyclooxygenases are present in 2 isoforms in the endothelium, a constitutive form, COX-1 and an inducible cyclooxygenase, COX-2. Both isoforms secrete prostanoids and are involved in the endothelium-mediated constriction[55]. COX-derived prostacyclins (PGI2, PGE2) bind the IP receptor at the surface of the smooth muscle cells and activate adenylate cyclase which triggers vascular relaxation by the synthesis of cyclic adenosine monophosphate (cAMP). On the other hand, once generated, TxA2 activates thromboxane-prostanoid (TP) receptors present at the surface of the smooth muscle cells, a phenomenon that will lead to an increase in intracellular Ca2+ levels and trigger vasoconstriction [56].
Additionally, endothelial cells modulate vasomotion by secreting endothelin 1 (ET-1) [57, 58]. Mostly activated by inflammatory cells and interleukin secretion, ET-1 secretion is also downregulated by shear stress, NO and PGI2 [59]. Due to the presence of ET-1 receptor at both the endothelial and smooth muscle cells level, ET-1 presents the particularity to either relax or constrict blood vessels. Activation of the ETB receptor on the endothelium causes vasodilation via the release of NO and PGI2 [60, 61] while ET-1 binding on ETA receptors at the smooth muscle cell level increases intracellular Ca2+ and triggers vasoconstriction.
Finally, endothelial cells can control vasomotion is by facilitating the conversion of angiotensin I to angiotensin II [62]. Secreted at the endothelial surface, angiotensin II could either induce vasodilation through the activation AT2 receptor expressed in endothelial cells [63] or constriction via binding AT1 receptors at the surface of the smooth muscle cells.
Common mechanisms of dysfunction (superoxide and inflammation)
Endothelial dysfunction could be defined as an inadequate vasodilation and/or paradoxical vasoconstriction in response to stimuli that release nitric oxide (NO). This improper response of the endothelial cells could be explained in most cases by a decreased bioavailability of the vasodilator NO and/or in some cases by an increase in the production of constrictor factors by the endothelium. NO bioavailability represents the balance between the amounts of NO produced by eNOS and the amount of active NO scavenged by reactive oxygen species (ROS), particularly superoxide anion (•O2−).
An important consideration in the study of endothelial dysfunction is progression. While studying the effects of a gradual increase in insulin resistance in obese mice, we demonstrated that the endothelial function of obese and lean mice is progressively and gradually reduced with the addition of risk factors such as type 2 diabetes and aging[64]. This progressively impedes the ability of the vessels to respond functionally and structurally to chronic changes in blood flow and ischemic injury[65].
ROS have been shown to reduce NO bioavailability through several mechanisms. By reacting directly with NO, ROS form peroxynitrite (•ONOO−). This will have for consequence to directly reduce NO bioavailability but also to alter eNOS functioning and to lead to eNOS uncoupling. When uncoupled, instead of releasing NO, eNOS produces ROS [66, 67]. Depletion of the eNOS substrate L-arginine [68] or ROS-mediated oxidation of the eNOS cofactor, tetrahydrobiopterin (BH4) are the main mechanisms responsible for eNOS uncoupling [69]. ROS-mediated eNOS uncoupling is a major source of NO reduction, however, ROS can also directly affect NO release by inhibiting the dimethylarginine dimethylaminohydrolase (DDAH) enzyme that converts the endogenous eNOS inhibitor called asymmetric dimethylarginine (ADMA). Inhibition of DDAH causes ADMA accumulation and suppression of NO secretion [70].
While reduction in NO bioavailability plays a key role in the reduced ability of the vessels to relax in response to dilator stimuli, an exaggerated secretion of constrictor agents is also involved in this dysfunction. In the presence of high ROS levels, peroxynitrite inhibits the prostacyclin synthase reducing PGI2 release. This induces a shift in the arachidonic acid metabolism to the PGI2 precursor PGH2 and other TP receptor agonists such as TXA2 promoting a constrictive phenotype [71].
In mammalian cells potential enzymatic sources of ROS include the mitochondrial electron transport chain, xanthine oxidase, cyclooxygenase, lipoxygenase, NO synthase, heme oxygenase, peroxidases and NADH oxidases. However, in the vasculature, the NAD(P)H oxidases represent the primary source of ROS. In endothelial cells, NAD(P)H oxidase isoforms are expressed in the endoplasmic reticulum, and in the perinuclear membranes generating ROS as modulators of redox sensitive signaling pathways [72, 73]. NAD(P)H oxidase is functionally active in all cells within the vascular wall (endothelial, vascular smooth muscle cells, fibroblasts). NAD(P)H oxidase is a multi-complex catalyzer that possesses a membrane component formed by the association of two transmembrane proteins p22phox and gp91phox (termed NOX2 or its homologues NOX1,3-5) and a cytoplasmic component represented by the proteins p47phox, p67phox and the small proteins Rac-1 and Rac-2 that play a key role in NAD(P)H oxidase activation. Upon activation, p47phox is phosphorylated, and the cytosolic subunits form a complex that translocates to the membrane in which it associates with cytochrome b558 (heterodimeric flavoprotein formed by the association of p22phox and gp91phox), to assemble the active oxidase, which transfers electrons from the substrate to O2 forming •O2[74, 75]. Produced at the molecular level, ROS act as signaling molecules to influence diverse signal transduction pathways, such as oxidation of reactive cysteine residues. However, when generated in excess ROS causes the vascular dysfunction described above.
Risk factors driving endothelial function
At the interface between the circulating blood and the vascular wall, the endothelium is highly exposed and the first organ submitted to changes in physical forces and chemical signals, making it highly susceptible to alterations. Endothelial dysfunction represents the earliest abnormality in the development of vascular diseases and is associated with a number of traditional risks factors including diabetes mellitus, hypercholesterolemia, hypertension, insulin resistance, advanced age and obesity [76]. Obesity is one of the most relevant health issues of the last decades, and represents one of the highest risk factor for the development of endothelial dysfunction. The authors of the Framingham Heart Study established an independent relationship between body mass index and blunted brachial artery flow-mediated dilation [77] that was further confirmed by several studies highlighting the deleterious effects of abdominal fat deposition on the endothelial function [78–80]. Using animals models of obesity and type 2 diabetes, obesity, from its early stage to the development of frank type 2 diabetes induces a progressive and gradual decrease in endothelial function [65, 81–85].
Obesity is a multi-factorial and complex disease that is associated with metabolic (insulin resistance, hyperglycemia, hypercholesterolemia, dyslipidemia, hyperleptinemia), and hemodynamic dysfunction (hypertension) but also recognized as an inflammatory disease. While the endothelial dysfunction observed in obesity and type II diabetes could mainly be explained by a single factor represented by a decrease in NO availability, the origins of this dysfunction are multiple. However, they do not involve a decrease in eNOS expression. Indeed eNOS expression was either reported not to be affected by insulin resistance and obesity [65, 86] or to be increased in pathological states associated with oxidative stress [87, 88]. Reduced NO bioavailability due to excess ROS generation becomes then the major source of endothelial dysfunction in obesity and diabetes. The complexity and the multi-factorial aspect of metabolic disease explain the multiple sources involved in ROS generation.
A key player in excessive ROS generation associated with obesity and diabetes is NADPH oxidase [89]. Endothelial cells isolated form obese individuals have been reported to exhibit an increased NADPH oxidase expression [90]. However, obesity per se is probably not the factor triggering NADPH overexpression but insulin resistance and its consequent metabolic disturbances likely represent the main source. The arguments to support this postulate are provided by a recent study from Ali et al analyzing the vascular consequences of correcting insulin sensitivity in obese mice. Insulin sensitivity was restored in obese mice via the deletion of the molecular restrain of the insulin signaling pathway, the protein tyrosine phosphatase 1B. In this study, we demonstrated that despite obesity, insulin sensitive mice present a normal endothelial function and lower levels of oxidative stress and NADPH subunits expression in the microcirculation, compared to obese insulin resistant animals [81]. This clearly highlights the role of insulin resistance in the endothelial dysfunction associated with obesity and more precisely its importance in excessive ROS generation.
The metabolic disturbances associated with insulin resistance are certainly the main cause of increased oxidative stress observed with obesity. Insulin resistance is characterized by a decreased ability of insulin to promote glucose uptake in skeletal muscle and adipose tissue and to suppress hepatic glucose output which combined with impaired beta cells function increases circulating blood glucose [91]. As reported by several studies hyperglycemia has been shown to be involved in NADPH-derived ROS generation. Indeed, treatment of human [92] or mouse microvascular endothelial cells [87] with high glucose increases NADPH oxidase expression and levels of oxidative stress. Under hyperglycemic conditions advanced glycation end-products (AGE) are also formed and stimulate NADPH activity. Furthermore, AGE scavenging with soluble form receptor for AGE induced a partial restoration of the endothelial function of obese diabetic mice that was associated with a reduced NADPH oxidase expression [93]. According to these authors [94, 95] and others [96, 97] AGEs promote NADPH oxidase-derived ROS secretion through an inflammatory process involving the NF-κB and TNF-α signaling pathway, highlighting the role of inflammation in the vascular dysfunction associated with obesity.
By affecting mitochondrial metabolism, hyperglycemia generates an increase in mitochondrial-derived superoxide production increasing diacylglycerol (DAG) formation. DAG activates protein kinase C isoforms and more specifically PKC-β that is implicated in the regulation and activation of membrane associated NADPH oxidase regulatory subunits p22phox, p47phox and p67phox in the vessels of diabetic patients. In the latter patients, PKC inhibition has been reported to reduce superoxide generation [89].
With insulin resistance, DAG levels could also be increased independently of mitochondrial-generated ROS. Under hyperglycemic conditions, elevated glucose levels also increase the glycolytic pathway flux and leads to an elevation in the levels of intracellular glyceraldehyde-3-phosphate. This in turn can stimulate increases in the de novo synthesis of DAG and activate PKC [98–100]. Upon activation, PKCs inhibit the activity of the PI3 kinase/Akt signaling pathway thereby limiting the subsequent phosphorylation of eNOS and NO release in response to insulin. In addition PKC activation mediates the overexpression of adhesion molecules such as ICAM-1, VCAM-1 and E-selectin and enhances vascular contractility by increasing ET-1 release [101].
Finally, hyperglycemia is involved in the up-regulation of angiotensin 2 secretion associated with obesity and diabetes [102]. Angiotensin II promotes NADPH-induced ROS release [103] and ET-1 stimulation and/or release, which counteract NO activities and impair endothelial function.
While insulin resistance is characterized by high circulating glucose levels, it also involves high circulating lipid levels resulting from the reduced sensitivity of the adipose to the metabolic effects of insulin. Dyslipidemia and more specifically hypercholesterolemia was the first pathological condition associated with an impaired endothelium dependent relaxation. In hypercholesterolemic rabbits and monkeys vasorelaxation to acetylcholine was almost absent or changed into vasoconstriction [104, 105]. Similar observations were made in patients with coronary artery diseases [106, 107]. Once again ROS are the main player in the endothelial dysfunction. However contrary to high glucose levels that increase ROS through NADPH oxidase activation, hypertriglyceridemia-derived ROS appears to induce a xanthine oxidase-mediated ROS release in the endothelial cells [108, 109].
Increased ROS in response to high glucose and high lipids levels also activate NF-kB, which further stimulates production of other proinflammatory cytokines including TNF-α and IL-6 and C reactive protein [110, 111] that will further impair the endothelial function by further activating NADPH oxidase [112].
Taken together, these data suggest that obesity and insulin resistance produce an oxidant injury to the endothelium that reduces NO-mediated dilation (Figure 3). The exact metabolic components and the role of accessory processes such as inflammation remain to be determined.
Figure 3.

The impact of obesity and insulin resistance on endothelial NO production and action.
Unique characteristics of the coronary circulation
As discussed above, the plasma milieu of the diabetic and obese individual has pervasive effects on the vascular endothelium across virtually all beds studied to date, by largely similar mechanisms. Interestingly, studies from animal models find that while many diabetic models demonstrate endothelial injury in the coronary circulation, models of obesity are much less likely to have similar defects. Knudson et al [113] observed normal vasodilator responses to acetylcholine in obese, pre-diabetic dogs, Zhang et al [114] made similar observations in young db/db mice and parallel results have been obtained in young adult obese Zucker Rats [115–117].
As metabolic disease worsens toward diabetes, the impact on the coronary circulation is unambiguous. In the Ossabaw swine model of the metabolic syndrome, endothelial dilation is markedly reduced to bradykinin [118] and TRPV1 channel activation [119], effects that may be attributed to factors released from periadventitial fat. As shown in other beds [120, 121], the loss of endothelial dilation in the coronary circulation is progressive as shown in older Zucker rats [115] and db/db mice [114]. Moreover, the progressive injury may reflect the accumulation of differing insults rather than simple escalation of metabolic variables as the impact of cytokine blockade varies over the course of progression. TNF-α, for example, makes a larger contribution to the total impairment in 12 week old db/db mice vs. 24 week olds. This observation correlated with an ability of TNF-α antagonism to reduce superoxide in younger animals but the antagonism was much less potent in older animals, suggesting that other factors contribute to the oxidant injury driving endothelial dysfunction in obesity and metabolic disease [114].
The apparent limitation in endothelial injury in the coronary circulation begs the question: why? Perhaps the simplest answer is that unlike many organs in obese individuals, the heart must work significantly harder to maintain effective cardiac output to larger individuals. Increases in cardiac output correlate with increased myocardial oxygen consumption in obese but otherwise healthy adults [6]. The increased work would, at least initially, produce increased flow and increased shear stress which is well documented to influence coronary tone in vivo [122, 123]. Evidence for the concept is found in the Zucker rat model of obesity, in which metabolic injury is relatively minor at younger ages but cardiac output is significantly increased [116]. Vasodilation to acetylcholine is normal in the septal arteries from these animals or even augmented at supra-normal levels of stimulation. The concept of a shear-mediated preservation of endothelial function is further supported by studies of aortic vasodilation. An increase in cardiac output translates to an increase in aortic flow and shear stress and the earliest studies of aortic dilation in the Zucker rat demonstrated an increased dilator capacity to endothelial stimulation [124]. Taken together, it seems plausible that an increase in shear may offset the deleterious defects to endothelial function caused by metabolic changes early in obesity.
The mechanisms by which endothelial tone is protected or preserved are incompletely understood. Work from Katakam et al suggests an increase in NO generation, as evidenced by more potent buffering of endothelin-mediated vasoconstriction by NO [125]. In studies using a high-fat model of obesity (one of the mildest metabolic models), Jebelovszki et al observed an increased sensitivity to NO which was attributed to an increase in soluble guanylate cyclase activity [126]. Finally, it should be considered that eventual impairment in endothelial signaling might be supplemented by other dilators. Indeed, Szerafin et al determined in human coronary arteries that metabolic disease increases vasodilation to cyclooxygenase products, providing an alternative NO-independent mechanism to preserve vasodilator capacity and cardiac perfusion [127].
Summary
Taken together, the literature reviewed above indicates that diabetes and obesity have progressive deleterious effects on the endothelial lining. An emerging consensus indicates that the primary injury is oxidant-mediated corruption of nitric oxide signaling, with oxidation being driven in large part by a pro-inflammatory tissue environment. The primary risk factor associated with endothelial function appears to be primarily the insulin resistant state, possibly secondary to pro-inflammatory advanced glycation end products or changes in lipid status. The coronary circulation appears to be afforded a measure of protection early on in the progression of metabolic disease, most likely due to hemodynamic changes associated with increased cardiac output. Ultimately, the balance shifts in favor of metabolic disease, leaving the heart vulnerable to ischemic heart disease.
Highlights.
Obesity and diabetes are major risk factors for coronary endothelial dysfunction.
Insulin Resistance is the major risk factor for this endothelial dysfunction
Oxidation and inflammation are the primary mechanisms of endothelial dysfunction.
The coronary circulation may be more slowly injured due to effects of increased cardiac work.
Acknowledgments
The authors wish to acknowledge Michelle Davis of Michelle Davis Studios (http://www.mdavisstudios.com) for art for Figure 3.
Footnotes
Disclosures: I hereby confirm that any and all potential conflicts of interest have been fully and properly disclosed in the manuscript as outlined.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Poirier P, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113(6):898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 2.Alexander CM, et al. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes. 2003;52(5):1210–4. doi: 10.2337/diabetes.52.5.1210. [DOI] [PubMed] [Google Scholar]
- 3.Kardassis D, et al. Impact of body composition, fat distribution and sustained weight loss on cardiac function in obesity. Int J Cardiol. doi: 10.1016/j.ijcard.2011.02.036. [DOI] [PubMed] [Google Scholar]
- 4.Frohlich ED, Susic D. Mechanisms underlying obesity associated with systemic and renal hemodynamics in essential hypertension. Curr Hypertens Rep. 2008;10(2):151–5. doi: 10.1007/s11906-008-0028-8. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez GE, et al. Sympathetic neural activation in visceral obesity. Circulation. 2002;106(20):2533–6. doi: 10.1161/01.cir.0000041244.79165.25. [DOI] [PubMed] [Google Scholar]
- 6.Peterson LR, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109(18):2191–6. doi: 10.1161/01.CIR.0000127959.28627.F8. [DOI] [PubMed] [Google Scholar]
- 7.Barton M. Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch. 460(5):825–37. doi: 10.1007/s00424-010-0860-y. [DOI] [PubMed] [Google Scholar]
- 8.D’Souza A, et al. Pathogenesis and pathophysiology of accelerated atherosclerosis in the diabetic heart. Mol Cell Biochem. 2009;331(1–2):89–116. doi: 10.1007/s11010-009-0148-8. [DOI] [PubMed] [Google Scholar]
- 9.Sitia S, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 9(12):830–4. doi: 10.1016/j.autrev.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Stamler J, et al. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16(2):434–44. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 11.Dangas G, et al. Preintervention arterial remodeling as an independent predictor of target-lesion revascularization after nonstent coronary intervention: an analysis of 777 lesions with intravascular ultrasound imaging. Circulation. 1999;99(24):3149–54. doi: 10.1161/01.cir.99.24.3149. [DOI] [PubMed] [Google Scholar]
- 12.van der Feen C, et al. Angiographic distribution of lower extremity atherosclerosis in patients with and without diabetes. Diabet Med. 2002;19(5):366–70. doi: 10.1046/j.1464-5491.2002.00642.x. [DOI] [PubMed] [Google Scholar]
- 13.Glagov S, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371–5. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 14.Reddy HK, et al. Remodeling of coronary arteries in diabetic patients-an intravascular ultrasound study. Echocardiography. 2004;21(2):139–44. doi: 10.1111/j.0742-2822.2004.03014.x. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann R, et al. Impact of the metabolic syndrome on angiographic and clinical events after coronary intervention using bare-metal or sirolimus-eluting stents. Am J Cardiol. 2007;100(9):1347–52. doi: 10.1016/j.amjcard.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 16.Aronson D, Edelman ER. Revascularization for coronary artery disease in diabetes mellitus: angioplasty, stents and coronary artery bypass grafting. Rev Endocr Metab Disord. 11(1):75–86. doi: 10.1007/s11154-010-9135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Celik T, et al. Impaired coronary collateral vessel development in patients with proliferative diabetic retinopathy. Clin Cardiol. 2005;28(8):384–8. doi: 10.1002/clc.4960280808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fatehi-Hassanabad Z, Chan CB, Furman BL. Reactive oxygen species and endothelial function in diabetes. Eur J Pharmacol. 636(1–3):8–17. doi: 10.1016/j.ejphar.2010.03.048. [DOI] [PubMed] [Google Scholar]
- 19.Niswender K. Diabetes and obesity: therapeutic targeting and risk reduction - a complex interplay. Diabetes Obes Metab. 12(4):267–87. doi: 10.1111/j.1463-1326.2009.01175.x. [DOI] [PubMed] [Google Scholar]
- 20.Targher G. High-Sensitivity C-Reactive Protein, Obesity, and Subclinical Atherosclerosis: Implications of JUPITER From the MESA Study. Arterioscler Thromb Vasc Biol. 31(6):1251–2. doi: 10.1161/ATVBAHA.111.228320. [DOI] [PubMed] [Google Scholar]
- 21.Malik S, Wong ND. Metabolic syndrome, cardiovascular risk and screening for subclinical atherosclerosis. Expert Rev Cardiovasc Ther. 2009;7(3):273–80. doi: 10.1586/14779072.7.3.273. [DOI] [PubMed] [Google Scholar]
- 22.Kashyap ML, et al. Insulin and non-esterified fatty acid metabolism in asymptomatic diabetics and atherosclerotic subjects. Can Med Assoc J. 1970;102(11):1165–9. [PMC free article] [PubMed] [Google Scholar]
- 23.Stout RW. The role of insulin in atherosclerosis in diabetics and nondiabetics: a review. Diabetes. 1981;30(Suppl 2):54–57. doi: 10.2337/diab.30.2.s54. [DOI] [PubMed] [Google Scholar]
- 24.Baron AD. Insulin resistance and vascular function. J Diabetes Complications. 2002;16(1):92–102. doi: 10.1016/s1056-8727(01)00209-4. [DOI] [PubMed] [Google Scholar]
- 25.Keller KB, Lemberg L. Obesity and the metabolic syndrome. Am J Crit Care. 2003;12(2):167–70. [PubMed] [Google Scholar]
- 26.Meigs JB. Epidemiology of the insulin resistance syndrome. Curr Diab Rep. 2003;3(1):73–9. doi: 10.1007/s11892-003-0057-2. [DOI] [PubMed] [Google Scholar]
- 27.Nikolsky E, et al. Impact of obesity on revascularization and restenosis rates after bare-metal and drug-eluting stent implantation (from the TAXUS-IV trial) Am J Cardiol. 2005;95(6):709–15. doi: 10.1016/j.amjcard.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima K. Pharmacotherapy of mixed dyslipidemia in the metabolic syndrome. Curr Clin Pharmacol. 5(2):133–9. doi: 10.2174/157488410791110760. [DOI] [PubMed] [Google Scholar]
- 29.Keaney JF, Jr, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23(3):434–9. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 30.Espinola-Klein C, et al. Inflammatory markers and cardiovascular risk in the metabolic syndrome. Front Biosci. 16:1663–74. doi: 10.2741/3812. [DOI] [PubMed] [Google Scholar]
- 31.Celik T, et al. The clinical significance of microvascular impairment in patients with pure uncomplicated diabetes mellitus. Int J Cardiol. 2008;131(1):123–4. doi: 10.1016/j.ijcard.2007.07.008. author reply 128. [DOI] [PubMed] [Google Scholar]
- 32.Yodaiken RE. The relationship between diabetic capillaropathy and myocardial infarction: a hypothesis. Diabetes. 1976;25(2 SUPPL):928–30. [PubMed] [Google Scholar]
- 33.Liu Y, Gutterman DD. Vascular control in humans: focus on the coronary microcirculation. Basic Res Cardiol. 2009;104(3):211–27. doi: 10.1007/s00395-009-0775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kersten JR, Brooks LA, Dellsperger KC. Impaired microvascular response to graded coronary occlusion in diabetic and hyperglycemic dogs. Am J Physiol. 1995;268(4 Pt 2):H1667–74. doi: 10.1152/ajpheart.1995.268.4.H1667. [DOI] [PubMed] [Google Scholar]
- 35.Sari I, et al. Uncomplicated diabetes mellitus is equivalent for coronary artery disease: new support from novel angiographic myocardial perfusion-myocardial blush. Int J Cardiol. 2008;127(2):262–5. doi: 10.1016/j.ijcard.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Eliot RS, Baroldi G, Leone A. Necropsy studies in myocardial infarction with minimal or no coronary luminal reduction due to atherosclerosis. Circulation. 1974;49(6):1127–31. doi: 10.1161/01.cir.49.6.1127. [DOI] [PubMed] [Google Scholar]
- 37.Nahser PJ, Jr, et al. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. 1995;91(3):635–40. doi: 10.1161/01.cir.91.3.635. [DOI] [PubMed] [Google Scholar]
- 38.Koistinen MJ. Prevalence of asymptomatic myocardial ischaemia in diabetic subjects. Bmj. 1990;301(6743):92–5. doi: 10.1136/bmj.301.6743.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Picano E. The alternative “ischemic” cascade in coronary microvascular disease. Cardiologia. 1999;44(9):791–5. [PubMed] [Google Scholar]
- 40.L’Abbate A. Large and micro coronary vascular involvement in diabetes. Pharmacol Rep. 2005;57(Suppl):3–9. [PubMed] [Google Scholar]
- 41.Galderisi M, D’Errico A. Beta-blockers and coronary flow reserve: the importance of a vasodilatory action. Drugs. 2008;68(5):579–90. doi: 10.2165/00003495-200868050-00002. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Gutterman DD. The coronary circulation in diabetes: influence of reactive oxygen species on K+ channel-mediated vasodilation. Vascul Pharmacol. 2002;38(1):43–9. doi: 10.1016/s1537-1891(02)00125-8. [DOI] [PubMed] [Google Scholar]
- 43.Nitenberg A, et al. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42(7):1017–25. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- 44.Zoneraich S. Small-vessel disease, coronary artery vasodilator reserve, and diabetic cardiomyopathy. Chest. 1988;94(1):5–7. doi: 10.1378/chest.94.1.5. [DOI] [PubMed] [Google Scholar]
- 45.Miura H, et al. Diabetes mellitus impairs vasodilation to hypoxia in human coronary arterioles: reduced activity of ATP-sensitive potassium channels. Circ Res. 2003;92(2):151–8. doi: 10.1161/01.res.0000052671.53256.49. [DOI] [PubMed] [Google Scholar]
- 46.Dagres N, et al. Insulin sensitivity and coronary vasoreactivity: insulin sensitivity relates to adenosine-stimulated coronary flow response in human subjects. Clin Endocrinol (Oxf) 2004;61(6):724–31. doi: 10.1111/j.1365-2265.2004.02156.x. [DOI] [PubMed] [Google Scholar]
- 47.Kiviniemi TO, et al. Determinants of coronary flow velocity reserve in healthy young men. Am J Physiol Heart Circ Physiol. 2006;291(2):H564–9. doi: 10.1152/ajpheart.00915.2005. [DOI] [PubMed] [Google Scholar]
- 48.Fulop T, et al. Adaptation of vasomotor function of human coronary arterioles to the simultaneous presence of obesity and hypertension. Arterioscler Thromb Vasc Biol. 2007;27(11):2348–54. doi: 10.1161/ATVBAHA.107.147991. [DOI] [PubMed] [Google Scholar]
- 49.Berwick ZC, Dick GM, Tune JD. Heart of the matter: Coronary dysfunction in metabolic syndrome. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 51.Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333(6174):664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 52.Oppermann M, et al. Regulation of vascular guanylyl cyclase by endothelial nitric oxide-dependent posttranslational modification. Basic Res Cardiol. 106(4):539–49. doi: 10.1007/s00395-011-0160-5. [DOI] [PubMed] [Google Scholar]
- 53.Cohen RA, Vanhoutte PM. Endothelium-Dependent Hyperpolarization: Beyond Nitric Oxide and Cyclic GMP. Circulation. 1995;92(11):3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- 54.Shimokawa H, et al. The Importance of the Hyperpolarizing Mechanism Increases as the Vessel Size Decreases in Endothelium-Dependent Relaxations in Rat Mesenteric Circulation. Journal of Cardiovascular Pharmacology. 1996;28(5):703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 55.Ge T, et al. Endothelium-Dependent Contractions Are Associated With Both Augmented Expressionof Prostaglandin H Synthase-1 and Hypersensitivity to Prostaglandin H2in the SHR Aorta. Circ Res. 1995;76(6):1003–1010. doi: 10.1161/01.res.76.6.1003. [DOI] [PubMed] [Google Scholar]
- 56.Alexander RW, Griendling KK. Signal transduction in vascular smooth muscle. J Hypertens Suppl. 1996;14(5):S51–4. [PubMed] [Google Scholar]
- 57.Kinlay S, et al. Role of Endothelin-1 in the Active Constriction of Human Atherosclerotic Coronary Arteries. Circulation. 2001;104(10):1114–1118. doi: 10.1161/hc3501.095707. [DOI] [PubMed] [Google Scholar]
- 58.Yanagisawa M, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 59.Alonso D, Radomski MW. The Nitric Oxide-Endothelin-1 Connection. Heart Failure Reviews. 2003;8(1):107–115. doi: 10.1023/a:1022155206928. [DOI] [PubMed] [Google Scholar]
- 60.Lerman A, et al. Inhibition of endothelium-derived relaxing factor enhances endothelin- mediated vasoconstriction. Circulation. 1992;85(5):1894–1898. doi: 10.1161/01.cir.85.5.1894. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki S, et al. Effects of endothelin-1 on endothelial cells in the porcine coronary artery. Circ Res. 1991;69(5):1361–1368. doi: 10.1161/01.res.69.5.1361. [DOI] [PubMed] [Google Scholar]
- 62.Saye JA, Singer HA, Peach MJ. Role of endothelium in conversion of angiotensin I to angiotensin II in rabbit aorta. Hypertension. 1984;6(2):216–221. [PubMed] [Google Scholar]
- 63.Matrougui K, et al. Activation of AT2 Receptors by Endogenous Angiotensin II Is Involved in Flow-Induced Dilation in Rat Resistance Arteries. Hypertension. 1999;34(4):659–665. doi: 10.1161/01.hyp.34.4.659. [DOI] [PubMed] [Google Scholar]
- 64.Belin de Chantemele EJ, et al. Obesity-induced insulin resistance causes endothelial dysfunction without reducing the vascular response to hindlimb ischemia. Basic Res Cardiol. 2009;104(6):707–17. doi: 10.1007/s00395-009-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bouvet C, et al. Flow-Induced Remodeling in Resistance Arteries From Obese Zucker Rats Is Associated With Endothelial Dysfunction. Hypertension. 2007;50(1):248–254. doi: 10.1161/HYPERTENSIONAHA.107.088716. [DOI] [PubMed] [Google Scholar]
- 66.Laursen JB, et al. Endothelial Regulation of Vasomotion in ApoE-Deficient Mice: Implications for Interactions Between Peroxynitrite and Tetrahydrobiopterin. Circulation. 2001;103(9):1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 67.Skalak TC, Price RJ. The role of mechanical stresses in microvascular remodeling. Microcirculation. 1996;3(2):143–65. doi: 10.3109/10739689609148284. [DOI] [PubMed] [Google Scholar]
- 68.Imaizumi T, et al. Effects of L-arginine on forearm vessels and responses to acetylcholine. Hypertension. 1992;20(4):511–517. doi: 10.1161/01.hyp.20.4.511. [DOI] [PubMed] [Google Scholar]
- 69.Milstien S, Katusic Z. Oxidation of Tetrahydrobiopterin by Peroxynitrite: Implications for Vascular Endothelial Function. Biochemical and Biophysical Research Communications. 1999;263(3):681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 70.Lin KY, et al. Impaired Nitric Oxide Synthase Pathway in Diabetes Mellitus: Role of Asymmetric Dimethylarginine and Dimethylarginine Dimethylaminohydrolase. Circulation. 2002;106(8):987–992. doi: 10.1161/01.cir.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- 71.Zou MH, Shi C, Cohen RA. High Glucose via Peroxynitrite Causes Tyrosine Nitration and Inactivation of Prostacyclin Synthase That Is Associated With Thromboxane/Prostaglandin H2 Receptor†“Mediated Apoptosis and Adhesion Molecule Expression in Cultured Human Aortic Endothelial Cells. Diabetes. 2002;51(1):198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 72.Cave AC, et al. NADPH Oxidases in Cardiovascular Health and Disease. Antioxidants & Redox Signaling. 2006;8(5–6):691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 73.Fredenburgh LE, et al. Absence of Cyclooxygenase-2 Exacerbates Hypoxia-Induced Pulmonary Hypertension and Enhances Contractility of Vascular Smooth Muscle Cells. Circulation. 2008;117(16):2114–2122. doi: 10.1161/CIRCULATIONAHA.107.716241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Touyz RM, et al. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25(3):512–8. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- 75.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23(6):981–7. doi: 10.1161/01.ATV.0000069236.27911.68. [DOI] [PubMed] [Google Scholar]
- 76.Steinberg HO, et al. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97(11):2601–10. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benjamin EJ, et al. Clinical Correlates and Heritability of Flow-Mediated Dilation in the Community: The Framingham Heart Study. Circulation. 2004;109(5):613–619. doi: 10.1161/01.CIR.0000112565.60887.1E. [DOI] [PubMed] [Google Scholar]
- 78.Arcaro G, et al. Body fat distribution predicts the degree of endothelial dysfunction in uncomplicated obesity. Int J Obes Relat Metab Disord. 1999;23(9):936–42. doi: 10.1038/sj.ijo.0801022. [DOI] [PubMed] [Google Scholar]
- 79.Brook RD, et al. Usefulness of visceral obesity (waist/hip ratio) in predicting vascular endothelial function in healthy overweight adults. Am J Cardiol. 2001;88(11):1264–9. doi: 10.1016/s0002-9149(01)02088-4. [DOI] [PubMed] [Google Scholar]
- 80.Williams IL, et al. Effect of fat distribution on endothelial-dependent and endothelial-independent vasodilatation in healthy humans. Diabetes, Obesity and Metabolism. 2006;8(3):296–301. doi: 10.1111/j.1463-1326.2005.00505.x. [DOI] [PubMed] [Google Scholar]
- 81.Ali MI, et al. Deletion of protein tyrosine phosphatase 1b improves peripheral insulin resistance and vascular function in obese, leptin-resistant mice via reduced oxidant tone. Circ Res. 2009;105(10):1013–22. doi: 10.1161/CIRCRESAHA.109.206318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Belin de Chantemele EJ, et al. Cyclooxygenase-2 preserves flow-mediated remodelling in old obese Zucker rat mesenteric arteries. Cardiovascular Research. 2010;86(3):516–525. doi: 10.1093/cvr/cvp411. [DOI] [PubMed] [Google Scholar]
- 83.Frisbee JC. Impaired dilation of skeletal muscle microvessels to reduced oxygen tension in diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol. 2001;281(4):H1568–74. doi: 10.1152/ajpheart.2001.281.4.H1568. [DOI] [PubMed] [Google Scholar]
- 84.D’Angelo G, et al. Exaggerated cardiovascular stress responses and impaired beta-adrenergic-mediated pressor recovery in obese Zucker rats. Hypertension. 2006;48(6):1109–15. doi: 10.1161/01.HYP.0000247306.53547.d4. [DOI] [PubMed] [Google Scholar]
- 85.Vessieres E, et al. Cyclooxygenase-2 inhibition restored endothelium-mediated relaxation in old obese Zucker rat mesenteric arteries. Frontiers in Physiology. 2010:1. doi: 10.3389/fphys.2010.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fulton D, et al. Insulin resistance does not diminish eNOS expression, phosphorylation, or binding to HSP-90. Am J Physiol Heart Circ Physiol. 2004;287(6):H2384–93. doi: 10.1152/ajpheart.00280.2004. [DOI] [PubMed] [Google Scholar]
- 87.Ding H, Aljofan M, Triggle CR. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. Journal of Cellular Physiology. 2007;212(3):682–689. doi: 10.1002/jcp.21063. [DOI] [PubMed] [Google Scholar]
- 88.Li H, et al. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide. 2002;7(3):149–164. doi: 10.1016/s1089-8603(02)00111-8. [DOI] [PubMed] [Google Scholar]
- 89.Guzik TJ, et al. Mechanisms of Increased Vascular Superoxide Production in Human Diabetes Mellitus: Role of NAD(P)H Oxidase and Endothelial Nitric Oxide Synthase. Circulation. 2002;105(14):1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 90.Silver AE, et al. Overweight and Obese Humans Demonstrate Increased Vascular Endothelial NAD(P)H Oxidase-p47phox Expression and Evidence of Endothelial Oxidative Stress. Circulation. 2007;115(5):627–637. doi: 10.1161/CIRCULATIONAHA.106.657486. [DOI] [PubMed] [Google Scholar]
- 91.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 92.Quagliaro L, et al. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells. Diabetes. 2003;52(11):2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 93.Gao X, et al. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2008;295(2):H491–8. doi: 10.1152/ajpheart.00464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gao X, et al. Tumor Necrosis Factor-{alpha} Induces Endothelial Dysfunction in Leprdb Mice. Circulation. 2007;115(2):245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 95.Picchi A, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99(1):69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 96.Csiszar A, et al. Vasculoprotective Effects of Anti-Tumor Necrosis Factor-[alpha] Treatment in Aging. The American Journal of Pathology. 2007;170(1):388–398. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmidt AM, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. The Journal of Clinical Investigation. 1995;96(3):1395–1403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Inoguchi T, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 99.Inoguchi T, et al. Insulin’s effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol. 1994;267(3 Pt 1):E369–79. doi: 10.1152/ajpendo.1994.267.3.E369. [DOI] [PubMed] [Google Scholar]
- 100.Xia P, et al. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43(9):1122–9. doi: 10.2337/diab.43.9.1122. [DOI] [PubMed] [Google Scholar]
- 101.Sheetz MJ, King GL. Molecular Understanding of Hyperglycemia’s Adverse Effects for Diabetic Complications. JAMA: The Journal of the American Medical Association. 2002;288(20):2579–2588. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 102.Gabriely I, et al. Hyperglycemia modulates angiotensinogen gene expression. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2001;281(3):R795–R802. doi: 10.1152/ajpregu.2001.281.3.R795. [DOI] [PubMed] [Google Scholar]
- 103.Griendling KK, et al. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 104.Freiman PC, et al. Atherosclerosis impairs endothelium-dependent vascular relaxation to acetylcholine and thrombin in primates. Circ Res. 1986;58(6):783–9. doi: 10.1161/01.res.58.6.783. [DOI] [PubMed] [Google Scholar]
- 105.Jayakody RL, et al. Cholesterol feeding impairs endothelium-dependent relaxation of rabbit aorta. Can J Physiol Pharmacol. 1985;63(9):1206–9. doi: 10.1139/y85-199. [DOI] [PubMed] [Google Scholar]
- 106.Golino P, et al. Divergent Effects of Serotonin on Coronary-Artery Dimensions and Blood Flow in Patients with Coronary Atherosclerosis and Control Patients. New England Journal of Medicine. 1991;324(10):641–648. doi: 10.1056/NEJM199103073241001. [DOI] [PubMed] [Google Scholar]
- 107.Ludmer PL, et al. Paradoxical Vasoconstriction Induced by Acetylcholine in Atherosclerotic Coronary Arteries. New England Journal of Medicine. 1986;315(17):1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 108.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. The Journal of Clinical Investigation. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White CR, et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences. 1996;93(16):8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ungvari Z, et al. Increased Superoxide Production in Coronary Arteries in Hyperhomocysteinemia: Role of Tumor Necrosis Factor-{alpha}, NAD(P)H Oxidase, and Inducible Nitric Oxide Synthase. Arterioscler Thromb Vasc Biol. 2003;23(3):418–424. doi: 10.1161/01.ATV.0000061735.85377.40. [DOI] [PubMed] [Google Scholar]
- 113.Knudson JD, et al. Leptin resistance extends to the coronary vasculature in prediabetic dogs and provides a protective adaptation against endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289(3):H1038–46. doi: 10.1152/ajpheart.00244.2005. [DOI] [PubMed] [Google Scholar]
- 114.Zhang C, et al. Maturation-induces endothelial dysfunction via vascular inflammation in diabetic mice. Basic Res Cardiol. 2008;103(5):407–16. doi: 10.1007/s00395-008-0725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oltman CL, et al. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2006;291(4):H1780–7. doi: 10.1152/ajpheart.01297.2005. [DOI] [PubMed] [Google Scholar]
- 116.Prakash R, Mintz JD, Stepp DW. Impact of obesity on coronary microvascular function in the Zucker rat. Microcirculation. 2006;13(5):389–96. doi: 10.1080/10739680600745919. [DOI] [PubMed] [Google Scholar]
- 117.Katakam PV, et al. Impaired insulin-induced vasodilation in small coronary arteries of Zucker obese rats is mediated by reactive oxygen species. Am J Physiol Heart Circ Physiol. 2005;288(2):H854–60. doi: 10.1152/ajpheart.00715.2004. [DOI] [PubMed] [Google Scholar]
- 118.Payne GA, et al. Periadventitial adipose tissue impairs coronary endothelial function via PKC-beta-dependent phosphorylation of nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297(1):H460–5. doi: 10.1152/ajpheart.00116.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bratz IN, et al. Impaired capsaicin-induced relaxation of coronary arteries in a porcine model of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2008;294(6):H2489–96. doi: 10.1152/ajpheart.01191.2007. [DOI] [PubMed] [Google Scholar]
- 120.Sanchez D, Miguel M, Aleixandre A. Arterial blood pressure and aortic responses in obese, age-grouped Zucker rats. Methods Find Exp Clin Pharmacol. 32(6):421–6. doi: 10.1358/mf.2010.32.6.1444767. [DOI] [PubMed] [Google Scholar]
- 121.Belin de Chantemèle E, et al. Obesity induced-insulin resistance causes endothelial dysfunction without reducing the vascular response to hindlimb ischemia. Basic Research in Cardiology. 2009;104(6):707–717. doi: 10.1007/s00395-009-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stepp DW, Nishikawa Y, Chilian WM. Regulation of shear stress in the canine coronary microcirculation. Circulation. 1999;100(14):1555–61. doi: 10.1161/01.cir.100.14.1555. [DOI] [PubMed] [Google Scholar]
- 123.Stepp DW, et al. Nitric oxide limits coronary vasoconstriction by a shear stress- dependent mechanism. Am J Physiol Heart Circ Physiol. 2001;281(2):H796–803. doi: 10.1152/ajpheart.2001.281.2.H796. [DOI] [PubMed] [Google Scholar]
- 124.Auguet M, Delaflotte S, Braquet P. Increased influence of endothelium in obese Zucker rat aorta. J Pharm Pharmacol. 1989;41(12):861–4. doi: 10.1111/j.2042-7158.1989.tb06389.x. [DOI] [PubMed] [Google Scholar]
- 125.Katakam PV, et al. Impaired endothelin-induced vasoconstriction in coronary arteries of Zucker obese rats is associated with uncoupling of [Ca2+]i signaling. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R145–53. doi: 10.1152/ajpregu.00405.2005. [DOI] [PubMed] [Google Scholar]
- 126.Jebelovszki E, et al. High-fat diet-induced obesity leads to increased NO sensitivity of rat coronary arterioles: role of soluble guanylate cyclase activation. Am J Physiol Heart Circ Physiol. 2008;294(6):H2558–64. doi: 10.1152/ajpheart.01198.2007. [DOI] [PubMed] [Google Scholar]
- 127.Szerafin T, et al. Increased Cyclooxygenase-2 Expression and Prostaglandin-Mediated Dilation in Coronary Arterioles of Patients With Diabetes Mellitus. Circ Res. 2006;99(5):e12–317. doi: 10.1161/01.RES.0000241051.83067.62. [DOI] [PubMed] [Google Scholar]