

Abstract
Background
The hereditary long QT syndrome is characterized by prolonged ventricular repolarization that can be caused by mutations to the KCNQ1 gene, which encodes the α subunits of the cardiac potassium channel complex that carries the IKs current (the β subunits are encoded by KCNE1). In this study, we characterized a deleterious variant, KCNQ1-S277L, found in a patient who presented with sudden cardiac death in the presence of cocaine use.
Methods
The KCNQ1-S277L mutation was analyzed via whole-cell patch clamp, confocal imaging, surface biotinylation assays, and computer modeling.
Results
Homomeric mutant KCNQ1-S277L channels were unable to carry current, either alone or with KCNE1. When co-expressed in a 50/50 ratio with WT KCNQ1, current density was reduced in a dominant-negative manner, with the residual current predominantly wild type. There was no change in the activation rate and minimal changes to voltage-dependent activation for both KCNQ1 current and IKs current. Immunofluorescence confocal imaging revealed reduced surface expression of mutant KCNQ1-S277L, which was biochemically confirmed by surface biotinylation showing a 44% decrease in mutant surface expression. Expression of KCNQ1-S277L with human ether-a-go-go-related gene (HERG) did not significantly affect HERG protein or current density compared to KCNQ1-WT co-expression.
Conclusion
The KCNQ1-S277L mutation causes biophysical defects that result in dominant-negative reduction in KCNQ1 and IKs current density, and a trafficking defect that results in reduced surface expression, both without affecting HERG/IKr. KCNQ1-S277L mutation in the proband resulted in defective channels that compromised repolarization reserve, thereby enhancing the arrhythmic susceptibility to pharmacological blockage of IKr current.
Keywords: KCNQ1, IKs, long QT syndrome, sudden death, genetic phenotype, disease mechanism
Introduction
Long QT syndrome (LQTS) is a cardiac disorder characterized by a prolonged QT interval on surface electrocardiogram (ECG), reflecting an abnormally delayed repolarization phase of the cardiac action potential. LQTS patients are susceptible to potentially lethal arrhythmias, in particular the Torsade de Pointes-type polymorphic ventricular tachycardia1 that can progress to syncope, ventricular fibrillation, and sudden death. LQTS can be due to congenital mutations, or can be acquired as a result of therapeutic drug use or structural heart disease.2
Two delayed rectifier K+ currents are largely responsible for myocardial repolarization: the slowly activating current IKs, carried by a channel complex composed of KCNQ1 with its regulatory subunit KCNE1,3,4 and the rapidly activating IKr, carried by the human ether-a-go-go-related gene (HERG) subunits complexed with either KCNE1 or KCNE2.5,6 There are presently 13 proposed LQTS loci, but congenital mutations in KCNQ1 account for approximately 45% of inherited LQTS in patients with an identified gene mutation.7 Arrhythmias associated with LQT1 are most often triggered by exercise-related increases in heart rate.8
The voltage-gated KCNQ1/KCNE1 channel complex that carries the IKs current has α-subunits encoded by KCNQ1 and β-subunits encoded by KCNE1. Each α-subunit contains intracellular N- and C-terminal tails, and six membrane-spanning segments (S1 through S6) that tetramerize with four-fold symmetry around the K+ ion conduction pathway, which is comprised of S5, S6, and the intervening loop that confers ion selectivity. LQT1 mutations are found throughout the length of the protein, and most commonly cause changes in the biophysical properties of the channel, affecting voltage sensing, gating, or permeation. Some mutations, however, cause defects in channel trafficking or tetramerization. This is in contrast to the HERG channel, where the majority of mutations cause assembly and trafficking defects.9 The two channels also differ in the distribution of arrhythmia-associated mutations. For KCNQ1, the majority of LQT mutations are found in the pore region (29% S5–S6), the C-terminus (32%), and the intracellular linkers (20%); whereas for HERG, they are more widely distributed: N-terminal Per-Arnt-Sim (PAS)-domain (27%), pore region (23%), the extracellular linkers (23%), and C-terminus (15%).10
In the present study, we functionally characterized the LQT1-associated mutation KCNQ1-S277L, which was initially clinically identified in a Chinese family.11,12 We present a case of a 26-year-old woman with a prolonged QT interval on surface ECG who succumbed to sudden cardiac death, and was subsequently found to have the S277L mutation. Leucine 277 is located within the conduction pathway-lining S5 segment, close to the ion selectivity loop. Our findings show a unique combination of a biophysical defect coupled with a trafficking defect that together result in reduction of cardiac action potential repolarization. We also discuss the clinical implications of this type of LQTS mutation and enhanced susceptibility to proarrhythmic agents.
Methods
Genetic Analysis
Genomic DNA was extracted from dried blood on a 3 × 3-mm piece of bloodstain card (Whatman, Piscataway, NJ, USA) using a MagAttract DNA Mini M48 Kit (Qiagen, Valencia, CA, USA) and Qiagen’s BioRobot M48 following manufacturer’s instruction. All the coding regions and intron-exon boundaries of five long-QT genes, KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2, were amplified by the polymerase chain reaction. All sequence specific primers were designed by online programs ExonPrimer or Primer3 and purchased from Integrated DNA Technologies. For high throughput sequencing, the primers were tagged with universal primer sequences M13 on the 5′ end of forward primers and FKS on the 5′ end of reverse primers. The KCNQ1-S277L mutation is located on exon 6; the primers used were as follows: KCNQ1_6FW+M13 5′-TGT AAA ACG ACG GCC AGT CTG GGA CTC GCT GCC TTA G-3′; KCNQ1_6R+FKS 5′-TCG AGG TCG ACG GTA TCG ATA CAC CAG TGC CCA GAT GTC-3′. For polymerase chain reaction (PCR), two types of master mixtures were used: Master Mix A (1.5 mM MgCl2, 100 μM dNTPs, and 0.625 U AmpliTaq Gold with 1X PCR Buffer II [Applied Biosystems, Carlsbad, CA, USA]) for the majority of amplicons and Master Mix B (3 mM MgCl2, 200 μM dATP, 200 μM dCTP, 200 μM dTTP, 100 μM dGTP, 100 μM 7-deaza-dGTP [USB], 0.6 M betaine, 5% DMSO, and 1.25 U AmpliTaq Gold with 1X PCR Buffer II), for amplicons with high GC content. Approximately 5 ng of genomic DNA were used for each amplicon in a 25-μL reaction volume. The reaction also included fluorescence labeled primers 6-FAM-M13 and NED-FKS (0.1 μM each for Master Mix A, and 0.04 and 0.08 μM, respectively, for Master Mix B) to visualize the PCR products for sizing on an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems). The PCR program was as follows: 95°C × 5 min, followed by 35 cycles of denaturing at 95°C × 40 s, annealing at 60°C × 60 s, and extension at 72°C × 60 s, followed by additional extension at 72°C × 5 min. Amplicons were treated with ExoSAP-IT (USB, Cleveland, OH, USA) to remove remaining primers and dNTPs and, following sizing, subjected to bidirectional cycle sequencing using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) as per manufacturer’s instructions. Following the cycle sequencing reaction samples were precipitated with 250-mM sodium acetate and 10-mM ethylenediaminetetraacetic acid (EDTA) in 70% ethanol, washed with 70% ethanol and resuspended in 10-μL Hi-Di formamide (Applied Biosystems). Sequencing was performed on an ABI Prism 3130xl Genetic Analyzer. Sequencing data analysis and editing were performed using Sequencher 4.2 (Gene Codes Corporation, Ann Arbor, MI, USA). Each chromatograph must meet rigorous quality standards for correct base-calling. Sequence variants are numbered based on cDNA reference sequences in National Center for Biotechnology Information (NCBI).
Plasmids and Cell Culture
Construction and validation of Myc-KCNQ1 and 3X-FLAG-KCNE1 expression plasmids has been previously described.13,14 KCNQ1-S277L mutation was introduced by QuikChange® Site-Directed mutagenesis (Stratagene Corp., La Jolla, CA, USA). The following primer was used for mutagenesis: 5′-GGC CTC ATC TTC TCC TTG TAC TTT GTG TAC CTG-3′. The sequence of the mutated cDNA was verified by automated sequencing.
For electrophysiology experiments, Chinese Hamster Ovary (CHO) cells (American Type Culture Collection, Manassas, VA, USA) were maintained in Ham’s F-12 (Mediatech Inc., Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and Penicillin/Streptomycin (Mediatech Inc.) at 37°C and 5% CO2. Transient transfections were performed using Fugene 6 (Roche), with green fluorescent protein (GFP) plasmid to allow identification of transfected cells by fluorescence. Electrophysiology studies were performed 48 hours after transfection. Creation of CHO cells stably transfected with HERG has been previously described.6 The stably transfected cells were cultured and transfected in the same manner as the CHO cells.
For biochemistry and immunofluorescence assays, Human Embryonic Kidney 293 (HEK-293) cells (American Type Culture Collection) were cultured in RPMI 1640 (Mediatech Inc.) supplemented with 10% FBS (HyClone) and Penicillin/Streptomycin (Mediatech Inc.) at 37°C and 5% CO2. Phenol red-free RPMI 1640, additionally supplemented with L-glutamine, was used for immunofluorescence imaging. Transient transfections were performed using Fugene 6 (Roche), and experiments were performed 48 hours after transfection. Creation of HEK-293 cells stably transfected with HERG has been previously described.15 The stably transfected cells were cultured and transfected in the same manner as the HEK-293 cells.
Electrophysiology
CHO cells were used for electrophysiological experiments because they lack endogenous potassium currents. Transfected CHO cells were grown on sterile glass cover slips and placed in an acrylic/polystyrene perfusion chamber (Warner Instruments, Hamden, CT, USA) mounted in an inverted microscope outfitted with fluorescence optics and patch pipette micromanipulators. Extracellular solution consisted of NaCl 150 mM, CaCl2 1.8 mM, KCl 4 mM, MgCl2 1 mM, glucose 5 mM, and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer 10 mM (pH 7.4) at room temperature (20–22°C). Intracellular pipette solution contained KCl 126 mM, K-ATP 4 mM, MgSO4 1 mM, EGTA 5 mM, CaCl2 0.5 mM, and HEPES buffer 25 mM (pH 7.2) at room temperature (20–22°C). For experiments involving the HERG channel, CHO cells stably transfected with HERG were used. Intracellular pipette solution was the same as above except for the use of Mg-ATP instead of K-ATP. Chromanol 293B was diluted from a 10-mM stock dissolved in DMSO to a final concentration of 100 μM in the extracellular solution in order to block any KCNQ1 currents.
The whole-cell configuration of the patch clamp technique was used to measure potassium currents.16 A MultiClamp 700B patch-clamp amplifier was used and protocols were controlled via PC using pCLAMP10 acquisition and analysis software (Axon Instruments, Sunnyvale, CA, USA). Patch-clamp pipettes were manufactured and tips polished to obtain a resistance of 2–3 megaOhm in the test solutions. The pipette offset potential in these solutions was corrected to zero just prior to seal formation. The junction potential for these solutions was calculated between 3 and 4 mV (by pClamp analysis software) and was not corrected for analyses. Whole cell capacitance (generally 10–25pF) was compensated electronically through the amplifier. Whole-cell series resistance of 6–12 MΩ was compensated to 75%–90% using amplifier circuitry such that the voltage errors for currents of 2nA were always less than 6 mV. A standard holding potential was −80 mV, and figure insets show applied voltage protocols. Data were filtered using an 8-pole Bessel filter at 1 kHz.
SDS-PAGE and Western Blotting
HEK-293 cells were used for these experiments because of their higher transfection efficiency and expression levels versus CHO cells. Forty-eight hours posttransfection, HEK-293 cells were lysed with a cell lysis buffer containing 150 mM NaCl, 1% NP-40, 0.4% deoxycholic acid, 5 mM EDTA, 25 mM Tris-HCL at pH 7.5 with complete protease inhibitor cocktail (Roche) for 15 minutes. Cell lysates were centrifuged at 13,000 rotations per minute at 4°C for 10 minutes, and the supernatants were mixed with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. Proteins were separated by 4%–20% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA) by semi-dry blotting unit (FisherBiotech, Thermo Fisher Scientific, Pittsburgh, PA, USA). The nitrocellulose membranes were blocked with Tris-buffered saline containing 0.5% Tween-20 (TBS-T) and 5% non-fat dry milk for 30 minutes at room temperature, and then incubated with goat-anti-KCNQ1 antibody (1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and mouse-anti-tubulin antibody (1:10,000, Sigma-Aldrich, St. Louis, MO, USA) or mouse-anti-sodium/potassium ATPase antibody (1:1000, Affinity Bioreagents, Rockford, IL, USA) for 2 hours at room temperature. The membrane was washed with TBS-T, and then incubated in the corresponding infrared-fluorescence IRDye® 800 conjugated donkey anti-goat and IRDye® 700DX conjugated donkey anti-mouse secondary antibodies (1:10,000, Rockland Immunochemicals Inc., Gilbertsville, PA, USA) for 30 minutes at room temperature in the dark followed by washing with TBS-T. The membranes were then scanned to visualize the signal at 680 nm or 780 nm by the Odyssey detection system (Li-Cor Biosciences, Lincoln, NE, USA) and densitometry of the protein bands was performed using Adobe Photoshop software (Adobe, San Jose, CA, USA). For experiments involving the HERG channel, the stably transfected HEK-293 cells were used, with transient transfection of wild-type or mutant KCNQ1. Cells were lysed 72 hours posttransfection and processed as stated above. To detect HERG protein, rabbit-anti-HERG antibody (1:200, Santa Cruz Biotechnology) was used, with IRDye® 700DX conjugated donkey anti-rabbit secondary antibody (1:10,000, Rockland Immunochemicals Inc.).
Immunofluorescence
HEK-293 cells were used for these experiments because their subcellular architecture is better suited for microscopic analysis with immunofluorescence, versus CHO cells. HEK-293 cells were transiently transfected as above. Twenty-four hours after transfection, cells were plated onto 35-mm glass bottom culture dishes (MatTek Corporation, Ashland, MA, USA). Forty-eight hours after transfection, cells were fixed in 3.7% formaldehyde in phosphate-buffered saline (PBS) for 20 minutes at room temperature, followed by permeabilization with 0.2% triton-X100 in PBS at room temperature for 10 minutes and then blocking with 5% BSA in 0.2% triton-X100 in PBS at room temperature for 30 minutes. The cells were then incubated with goat anti-KCNQ1 (1:200, Santa Cruz Biotechnology) and mouse anti-Cadherin (1:100, Abcam, Cambridge, MA, USA) antibodies in 5% BSA, 0.2% triton-X100 in PBS at 37°C for 1 hour. The cells were washed with 0.2% triton-X100 in PBS at room temperature for 5 minutes three times before incubation with Alexa Fluor 488 donkey anti-goat (Molecular Probes, Invitrogen Corp., Carlsbad, CA, USA) and Alexa Fluor 568 donkey anti-mouse (Molecular Probes) secondary antibodies (1:2000) in 5% BSA, 0.2% triton-X100 in PBS at 37°C for 1 hour in the dark. After incubation, the cells were washed with 0.2% triton-X100 in PBS at room temperature five times, mounted in Gel/Mount™ (Biømeda Corp., Foster City, CA, USA) with cover slips, and kept at 4°C in the dark until subjected to fluorescence microscopy examination. Images were collected with a Leica TCS SP2 AOBS confocal microscope (Leica Microsystems CMS GmbH, Mannheim, Germany) with 40X oil immersion optics. Laser lines at 488 nm and 561 nm for excitation of Alexa Fluor 488 and 568 dyes were provided by an Ar laser and a diode laser. Detection ranges were set to eliminate crosstalk between fluorophores. Fluorescent images were analyzed with the Image Correlation Analysis plug-in for ImageJ Software (http://rsbweb.nih.gov/ij/),17 with Pearson’s correlation quotient (ranging from 0 to 1) obtained as a quantification of KCNQ1 co-localization.
Cell Surface Analyses
Cell surface protein expression was determined by labeling with the membrane-impermeant biotinylation reagent, NHS-SS-biotin (Pierce, Rockford, IL, USA). Forty-eight hours posttransfection of HEK-293 cells, the cells were washed twice with PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS-CaMg), and incubated on ice in NHS-SS-biotin (1.5 mg/mL) in 20-mM HEPES, pH 9.0, 2-mM CaCl2, and 150-mM NaCl for 30 minutes on ice. After labeling, the cells were rinsed briefly with PBS-CaMg and incubated in 100-mM glycine in PBS-CaMg for 20 minutes on ice to quench unreacted NHS-SS-biotin. Cells were lysed in 100 μL of lysis buffer (50 mM Tris-HCl (pH 7.5), 1% Triton X-100, 1% SDS, 150-mM NaCl, 5-mM EDTA, complete protease inhibitor cocktail [Roche]) with gentle shaking on ice for 15 minutes. The cell lysates were then diluted by the addition of 900 μL of lysis buffer without SDS and then homogenized by QiaShredder (Qiagen). Protein concentration of each sample was determined by Micro BCA Assay (Pierce). Equal amounts of biotinylated proteins were precipitated from the supernatant solution with UltraLink® Immobilized Streptavidin (Pierce) and overnight incubation at 4°C with gentle agitation. The beads were washed three times with lysis buffer, twice with high-salt lysis buffer (lysis buffer with 500-mM NaCl and 0.1% Triton X-100), and once with 50-mM Tris-HCl (pH 7.5). The biotinylated surface proteins were eluted from the beads with SDS sample-loading buffer at 25°C for 30 minutes. The remaining supernatant represented the unlabeled proteins that had not yet been trafficked to the cell surface. Proteins were then subjected to SDS-PAGE and immunoblotting. Cadherin, a plasma membrane protein, was detected as the positive control for biotinylation of surface protein, whereas calnexin, an integral endoplasmic reticulum (ER) protein, was used as the negative control.
KCNQ1 Mutation Modeling
The structural model of KCNQ1 based on Kv1.2 crystal structure was used to examine the potential impact of the S277L mutation.18 Amino acid mutations were introduced into the protein database file (PDB) of the predicted open and closed states of KCNQ1 in PyMOL. Nearest contacts of the mutated residue were visualized and measured by UCSF Chimera package (Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco) and PyMOL. Additional analysis of mutational affects was performed using the PoPMuSiC Server (http://babylone.ulb.ac.be/popmusic/).19
Statistics
Values presented are means ± standard error. Analysis of variance was used for statistical analysis of the data, and P values < 0.05 were considered to be significant.
Results
Case Presentation
A 26-year-old Hispanic female was referred for cardiac evaluation after a prolonged QT interval was noted on a screening ECG obtained during a preoperative assessment for umbilical hernia repair. She reported having a syncopal episode preceded by dizziness at age 10, and another episode several weeks prior to her cardiac evaluation. She had a vague family history of cardiovascular events, including possible arrhythmia. Her medications at the time included a fentanyl patch and oxycodone/acetaminophen started recently for hernia pain relief.
Physical exam and laboratory tests were within normal limits, including potassium of 4.8 mEq/L and calcium of 9.0 mg/dL. ECG revealed normal sinus rhythm with a variable resting QTc duration (maximal recorded resting QTc = 494 ms (QT corrected by Bazett’s formula, Fig. 1). A stress exercise ECG began with a QTc = 427 ms at 2 minutes; 482 ms after 1 minute of recovery; and 517 ms after 2 minutes of recovery. No arrhythmias occurred during exercise. A 2-dimensional echocardiogram revealed a structurally normal heart with an ejection fraction of 70%. A subsequent 24-hour Holter did not show any evidence of tachyarrythmias and only very rare atrial premature complexes and ventricular premature complexes.
Figure 1.

Pedigree of the family affected by the KCNQ1-S277L mutation. Proband is indicated by the asterisk (*). Individuals are assigned as males (squares) or females (circles); affected (filled black), unaffected (white), not genotyped (white with question mark); possible arrhythmia or sudden cardiac death (gray). Triangle indicates stillborn infant of unknown sex. Leads II and V3 from 12-lead ECGs obtained at rest are shown for the proband and two affected offspring labeled with age and corrected QT interval (QTc).
Several months after the above evaluation, the patient presented to a local emergency room for a sprained elbow. A urine toxicology screen was performed, which was positive for benzodiazepines, opiates, methadone, and cocaine. Following the emergency visit, the patient was lost to follow-up.
The next presentation by this patient was sudden collapse at home. Ventricular fibrillation that could not be defibrillated was recorded by the emergency response team. Resuscitation efforts were continued en route to the hospital where attempts to convert ventricular fibrillation failed. Subsequent autopsy report failed to uncover structural myocardial pathology or coronary occlusions. Postmortem toxicology study revealed ethanol 0.01g%, cocaine (0.06 mg/L), and cocaine derivatives (ethylbenzoylecgonine 0.16 mg/L and benzoylecgonine 1.0 mg/L) in her serum.
The patient had two young children without any history of palpitations, dizziness, seizures, or syncope. Resting QTc intervals for the children were not prolonged (Fig. 1). Genetic analysis revealed that both children had the same mutation in KCNQ1 as the proband, and were subsequently started on 15-mg nadolol daily. A treadmill test was performed while off the β-blocker, with both children exhibiting prolonged QTc at 2 min 40 sec recovery (QTc > 440 ms is considered prolonged in childhood20). The son had a QTc baseline of 410 ms (69 beats per minute [BPM]), 470 ms (85 BPM) after 2 min 40 sec recovery, and 450 ms (78 BPM) after 10 minutes recovery. The daughter had a QTc baseline of 430 ms (90 BPM), 490 ms (90 BPM) after 2 min 40 sec recovery, and 430 ms (93 BPM) after 10 minutes recovery.
Genetic Analysis
All the coding regions and intron-exon boundaries of five Long-QT genes, KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2, were bidirectionally sequenced. From this set of candidate genes, the KCNQ1–830 C-to-T mutation was identified within exon 6. Mutational analysis of the children showed that both had also inherited the mutation. To date, neither child has exhibited a prolonged QTc at rest and neither have had any symptoms attributable to cardiac arrhythmia. This mutation is predicted to result in a change of serine to leucine at position 277 of the KCNQ1 protein. Based on a structural model of KCNQ1,18 this site is predicted to reside in the S5 transmembrane segment in close proximity to the selectivity filter pore loop-helix (Fig. 2).
Figure 2.
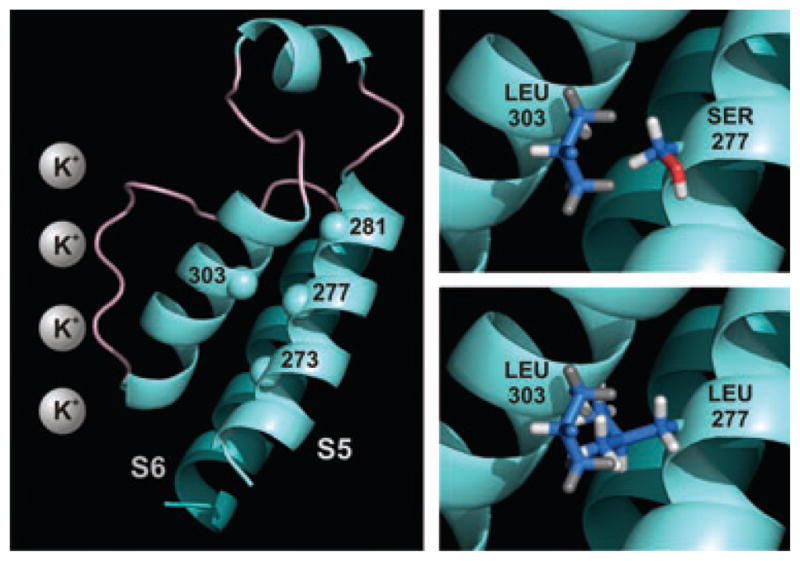
KCNQ1 residues 268–340 (left panel). Residue 277 in transmembrane domain S5 forms hydrogen bonds with leucine 273 and tyrosine 281. Grey spheres represent potassium ions in the selectivity filter. When residue 277 is mutated from serine to leucine, it clashes with alanine 302 and leucine 303 in the pore turret. Insets highlight the interactions between the native and mutated side chains of residue 277 with leucine 303.
Modeling
Nearest contacts, clashes, and hydrogen bonding of the native serine 277 residue and mutated leucine residue were visualized and measured by the UCSF Chimera package, using the structural model of KCNQ1 based on Kv1.2 crystal structure.18 In the open conformation, the closest contacts for the native serine residue were leucine 273 and tyrosine 281, directly below and above it on the alpha helix, with which it forms hydrogen bonds (Fig. 2). The native serine did not clash with any nearby residues (measured at a Vanderwaals radius overlap of 0.6 Å). When residue 277 is mutated to leucine, however, it formed closer contacts with residues 302, 303, and 306 (alanine, leucine, and glycine, respectively) than it did with 273 or 281. In addition, clashes with alanine 302 and leucine 303 were detected. The findings were similar in the closed conformation. Prediction of changes in protein stability showed that the serine to leucine mutation causes a destabilizing (ΔΔG = 0.42 kcal/mol) change in the folding free energy of KCNQ1 (PoPMuSiC Server).
Protein Expression and Electrophysiological Characterization
To examine the potential pathophysiological role of the KCNQ1-S277L mutation, we systematically examined the effects of the mutation on protein expression, potassium current conduction, and intracellular localization. Western blot analysis consistently showed that KCNQ1-S277L was expressed in both CHO and HEK-293 cells (CHO cell data not shown). However, expression levels were lower than that of wild-type KCNQ1 (Figs. 3A and B). In particular, the S277L tetramer and trimer bands were disproportionately fainter than the monomer band, when compared to KCNQ1-WT (Fig. 3C). Quantification of the fraction of each band to the total expression of protein under each condition (WT, S277, or 50/50 mixed) showed that the S277L and WT/S277L mixed conditions had a smaller fraction of tetramer and trimer and a larger fraction of dimer and monomer bands as compared to WT alone.
Figure 3.

Expression of KCNQ1-WT and S277L, alone or co-transfected in a 50/50 ratio into HEK-293 cells. (A) Samples were separated by 4%–15% gradient SDS-PAGE. Western blot analysis was performed with goat anti-KCNQ1 and mouse anti-tubulin antibodies. Left lane shows signal from cells transfected with empty vector. (B) Densitometry quantification of total protein levels, with WT expression normalized to 1.0. n = 3, **P < 0.01, ***P <0.001. (C) Densitometry quantification of mono- and oligomeric KCNQ1 bands comparing the ratio of S277L expression to WT expression, which was normalized to one. For each experiment, the densitometry of each band was expressed as a fraction of the total expression for each condition (KCNQ1-WT or -S277L), and then the ratio of mutant to wild type was graphed. n = 3.
Wild-type and mutant KCNQ1 were expressed in CHO cells, both without and with KCNE1, and whole-cell patch clamp recordings were made of KCNQ1 current or IKs current. KCNQ1-S277L produced no current (Fig. 4A). When wild-type KCNQ1 was co-transfected with mutant KCNQ1-S277L in a 50/50 ratio, KCNQ1 current was produced, though with significantly smaller current density (Figs. 4A and B). The mean current reduction was 81 ± 2%. There was a change in shape of the voltage dependence of activation curve for the mixed KCNQ1-WT and S277L currents (Fig. 4C), which may also indicate a mixed pool of channel complexes, each having from one to four subunits formed by KCNQ1-S277L, and each differing in its ability to carry K+ current. The half-max rise time, an indication of activation rate, was unchanged (Fig. 4D).
Figure 4.

Effect of the KCNQ1-S277L mutation on KCNQ1 current. (A) Whole-cell current traces of KCNQ1 (Q1) channels, with patch protocol in inset. WT and S277L channels were transiently expressed in CHO cells either alone, or in a 50/50 ratio. For the 50/50 expression, trace is enlarged for clarity. (B) Current density. n = 5, ***P < 0.001. (C) Normalized voltage-dependent activation curves. Curves were fitted using a Boltzmann function. Vh for KCNQ1-WT = −26.0 ± 1.4 mV, KCNQ1-WT/KCNQ1-S277L = −15.0 ± 4.1 mV. n = 5, P < 0.05. (D) Rates of activation measured as half-max rise time. n = 5, ***P < 0.001.
Co-expressing WT and mutant KCNQ1 with KCNE1 yielded similar results in regards to IKs current. KCNQ1-S277L co-expressed with KCNE1 produced no current (Fig. 5A). When also co-expressed with WT KCNQ1 in a 50/50 ratio, IKs-like current was produced with significantly smaller current density (Fig. 5A), with a 90±4% reduction in current density (Fig. 5B). The voltage dependence of activation curve and half-max rise time were both unchanged (Figs. 5C and D).
Figure 5.

Effect of the KCNQ1-S277L mutation on IKs current. (A) Whole-cell current traces of KCNQ1/KCNE1 channels. Patch protocol same as in Fig. 3. WT and S277L channels were transiently expressed in CHO cells either alone, or in a 50/50 ratio, and also with KCNE1. For the 50/50 expression, trace is enlarged for clarity. (B) Current density. n = 5, ***P < 0.001. (C) Normalized voltage-dependent activation curves. Curves were fitted using a Boltzmann function. Vh for KCNQ1-WT/KCNE1 = 9.8 ± 4.5 mV, KCNQ1-WT/KCNQ1-S277L/KCNE1 = 0.6 ± 12.0 mV. n = 5, P not significant. (D) Rates of activation, measured as half-max rise time. n = 5, ***P < 0.001.
Cellular Localization
To examine whether the S277L affects KCNQ1 trafficking and membrane localization, we performed immunofluorescence assays on HEK-293 cells transiently transfected with either WT or mutant KCNQ1. The cells were stained with commercial anti-KCNQ1 antibody and counter-stained with cadherin antibody as the control for a membrane-localized protein. Our imaging results show that KCNQ1-S277L is primarily distributed throughout the interior of the cell, with a smaller portion appearing to reach the surface membrane as compared to WT KCNQ1 (Fig. 6A). This was quantified by a Pearson’s correlation analysis of KCNQ1 and cadherin co-localization. For WT KCNQ1 the Pearson correlation is 0.843 ± 0.02 and for S277L, it is 0.573 ± 0.0, P < 0.001 with an n of six cells each.
Figure 6.
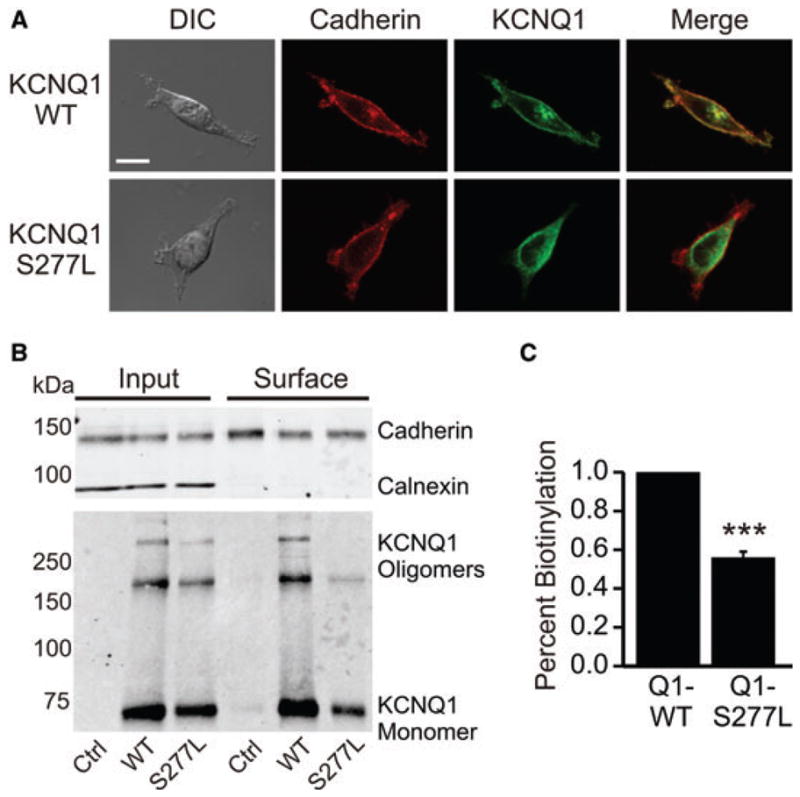
Surface expression of KCNQ1-S277L compared to WT. (A) Confocal immunofluorescence micrographs of HEK-293 cells transfected with ether KCNQ1-WT or KCNQ1-S277L and counter stained with anti-cadherin antibody to indicate the cell membrane. Pearson’s correlation of KCNQ1 and cadherin co-localization is 0.843 ± 0.02 for WT KCNQ1 and 0.573 ± 0.02 for S277L, P < 0.001. n = 6, scale bar = 10 μm, DIC = differential interference contrast. (B) Surface proteins were isolated by NHS-biotin labeling and precipitation from transiently transfected HEK cells, and samples were separated by 7.5% linear SDS-PAGE. Calnexin serves as negative control for surface labeling and cadherin as positive control for surface labeling in the upper panel. Western blot analysis was performed with goat anti-KCNQ1, rabbit anti-calnexin, and mouse anti-cadherin antibodies. (C) Densitometry analysis of the efficiency of surface presentation of KCNQ1 proteins expressed as the amount of surface KCNQ1 divided by the total cellular KCNQ1 normalized for loading, and also normalized for streptavidin pull-down of biotinylated protein (normalization calculated using the calnexin and cadherin controls). WT KCNQ1 is normalized to one. n = 3, ***P < 0.001.
We also performed surface biotinylation experiments to quantify surface expression of WT versus S277L KCNQ1. Using NHS-SS-Biotin to bind only proteins at the surface of the cell membrane, we were able to quantitatively show that KCNQ1-S277L surface expression is only 56% of WT KCNQ1 (Figs. 6B and C). The quantification is normalized to KCNQ1 expression, total protein expression, and degree of pull-down of protein-bound NHS-SS-Biotin by streptavidin beads.
Effects on HERG Expression and Current
Under normal physiologic conditions in the heart, both the KCNQ1/KCNE1 and HERG channels carry outward potassium currents to repolarize the cardiomyocyte. There is evidence for interactions between the two channels that influence their expression and current densities.21–23 When we transiently transfected KCNQ1-WT, KCNQ1-S277L, or Kv1.5 as a control into HEK-293 cells that stably express HERG, overall protein expression levels were unchanged compared to the HERG levels in cells that were transfected with empty vector (Fig. 7A). The percent of mature HERG protein (155 kDa glycosylated form found at the cell membrane) was slightly decreased over all three conditions compared to the control (and immature 135 kDa HERG reciprocally increased), but the data are not statistically significant over three averaged experiments (Fig. 7B). There is no difference between the percentages of mature or immature HERG expressed under the three transfection conditions. Electrophysiological data confirmed these findings: while overall HERG repolarization current density at −40 mV was decreased in cells co-transfected with either KCNQ1-WT or S277L, the HERG current density remained the same between the WT and mutant KCNQ1 co-transfections (Figs. 7C and D).
Figure 7.
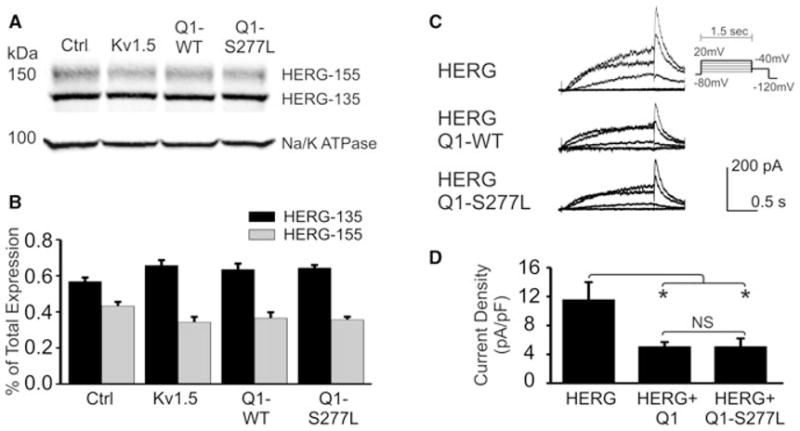
Effect of KCNQ1-WT and S277L on HERG expression and current density. (A) KCNQ1-WT (Q1-WT) and KCNQ1-S277L (Q1-S277L) were transiently transfected into HEK-293 cells that stably express HERG protein. Left lane is empty vector control, and second lane is transfected with Kv1.5. Samples were separated by 4%–15% gradient SDS-PAGE 72 hours after transfection. Western blot analysis was performed with rabbit anti-HERG and mouse anti-sodium/potassium ATPase antibodies. (B) Densitometry quantification of HERG mature (155 kDa) and immature (135 kDa) bands expressed as percent of total protein. n = 3, no statistical significance. (C) Whole cell current traces of HERG channels, from CHO cells stably expressing HERG protein and transfected with empty vector, KCNQ1-WT or S277L, patched 72 hours after transfection. Chromanol 293B was used in the bath solution to block KCNQ1 current. (D) Summary of repolarization current density measured at −40mV. n = 10, NS = no statistical significance.
Discussion
We identified a KCNQ1 variant, S277L, in a patient with noted prolonged QT interval on ECG and who presented with sudden cardiac death. The possible deleterious mutation was first reported in a Chinese family with two cases of sudden death (proband and proband’s mother) out of four members of the family with the mutation,11 with the trigger for arrhythmia being exercise or excitement.
In our studies, the S277L mutation causes complete loss of function of channels formed by homomeric KCNQ1-S277L α-subunits, with or without the KCNE1 β-subunit. Co-expression in a 50/50 ratio of wild-type KCNQ1 with S277L shows at least partially dominant-negative suppression of KCNQ1 and IKs current. For a fully dominant mutation, 94% current suppression would be expected, as predicated by the binomial distribution in a 50/50 mix of mutant and wild-type subunits that assemble in a tetramer (only channels with four wild-type subunits would carry current). KCNQ1-S277L suppresses wild-type current by 81%, leading us to conclude that current is being carried by a mixture of homomeric wild-type channels and a percentage of heteromeric wild-type/mutant channels. Another interpretation is that the S277L mutation impairs multimerization with wild-type subunits; therefore, more wild-type subunits are available to homotetramerize than is predicted by the binomial distribution. Indeed, our data do show possible impaired tetramerization of mutant channels, as seen in the reduction of KCNQ1 tetramer band on Western blot. Addition of KCNE1 appears to increase the dominant-negative effect of the S277L mutation, as IKs current is suppressed by 90%. For wild-type KCNQ1 channels, addition of KCNE1 stabilizes the closed state of the KCNQ1/KCNE1 channel complex, slowing channel activation and shifting the voltage dependence of activation to the right. Therefore, it is possible that with KCNE1, the conditions under which a heteromeric mutant channel can open is further impeded by structural constraints introduced by KCNE1.
Our functional data are in agreement with recent electrophysiological studies of the S277L mutation conducted in Xenopus oocytes, which showed decrease in current density of KCNQ1 and IKs current.24 However, our biochemical and imaging studies revealed an additional mechanism for current reduction. Confocal imaging showed reduced surface expression of mutant KCNQ1-S277L, which was confirmed by surface-biotinylation labeling of the wild type and mutant channels that showed a 44% decrease in surface expression of the mutant. Thus, a single amino acid change causes both a reduction in surface expression as well as biophysical functional impairment of the channels that do reach the cell membrane. Given the mutation’s proximity to the pore loop, it is not surprising that the ability to carry potassium current would be compromised. In addition, the experimental data showing a partial trafficking defect indicates that the mutation causes misfolding to the point of being recognized by the protein-folding quality control system. Some mutants are able to traffic to the membrane and carry current when complexed with wild-type subunits, indicating that the mutation does not perturb folding as much as other more dominant mutations that can suppress the trafficking of wild-type channels.25 The intermediate severity of the mutation allows us to detect both the protein-folding and biophysical mechanisms of current reduction.
Our analysis of a structural model of KCNQ1 reveals that residue 277 makes contact with residues 302 and 303, which are located in the α-helical portion of the pore turret that is connected to the selectivity filter. Mutation of residue 277 from serine to leucine increases the bulkiness of the side chain and causes an increased overlap of the van der Waals radius of these three amino acids. The mutated residue may push on the turret, compromising the selectivity filter’s ability to pass potassium ions. Arrhythmia-associated mutations of residues 302 and 303 have also been identified, but have yet to be functionally characterized. However, mutations in residue 305 and 306, which make additional contacts with residue 277 only when it is mutated to leucine, have been characterized: W305S and G306R both have dominant-negative loss-of-function effects on wild-type subunits.26,27 Thus, it appears that amino acid side chains in this structural region have low tolerance for changes in mass or polarity in terms of biophysical properties of ion conductance.
The structural model, made by Smith et al.,18 used the Kv1.2 crystal structure as a template. The alignment of the KCNQ1 sequence to the Kv1.2 sequence was most confident in the ion conduction region consisting of transmembrane domains S5 and S6 and the intervening pore loop, based on both sequence identity (38%) and secondary structure prediction. The modeling gives us the ability to speculate about a possible mechanism for the biophysical effects of the S277L mutation; however, the actual mechanism of current reduction by the mutation remains unknown since we are limited by the lack of a crystal structure for KCNQ1.
Out of the 21 amino acids spanning the putative transmembrane S5 domain (residues 262–282), 17 mutations on 13 residues have been identified, though few have been functionally characterized. Of those that have been characterized, the defects range from completely inactive channel with no trafficking defect, as seen with the Y281C mutation,28 to the G269S mutation that causes significantly reduced KCNQ1 current when expressed homozygously, yet produces near-wild-type-like current when expressed heterozygously with wild-type KCNQ1.29 One other S5 mutation has been characterized as having a dual effect on both the biophysical properties of the channel as well as causing a trafficking defect. F275S causes ER retention of the channel, and the channels that do successfully traffic to the membrane show a right shift in voltage-dependent activation, decreased activation and deactivation rates, and decreased current density of IKs.30 The S277L mutation is unique in that it forms a completely inactive channel, whether expressed alone or with KCNE1, in addition to causing a significant reduction in surface expression. Two other genetic variants at this site, S277P and S277W, have been reported in LQTS patients, but neither has been phenotypically characterized. Based on the functional and biochemical data for S277L and the structural modeling, we would predict moderate-to-severe deleterious effects from these other mutations.
Our data also indicate that the KCNQ1-S277L mutation does not have a deleterious effect on HERG expression or IKr current. Co-expression of KCNQ1 with HERG, whether wild type or mutant, does appear to decrease surface expression of HERG, as seen with the decrease in IKr current density, similar to the findings of the Koren group.23 However, the S277L mutation did not decrease IKr current density more than WT KCNQ1.
Despite the eventual outcome in the proband, the clinical phenotype of the KCNQ1-S277L mutation in this family appears rather mild. Besides one possible syncopal event as a child, there had been no other symptoms until just before death. Neither of the surviving children (both who harbor the KCNQ1-S277L mutation) has prolonged QTc or arrhythmias. The mild severity of the clinical phenotype is also in agreement with the recent report of the family from Germany where most of the mutation carriers have little or no signs or symptoms of LQTS.24 Several factors may contribute to a mild clinical phenotype despite significant reduction in IKs current in vitro, including variable penetrance of the mutation and epigenetic mechanisms that can affect the expression of alleles. Indeed, phenotypic variation between patients with the same mutation has been documented, even among members of the same family.31,32 That KCNQ1-S277L did not negatively affect HERG/IKr (contrary to other more severe mutations23) may also contribute to the rather mild clinical phenotype.
For our patient, an important factor to consider when trying to explain the contrast in severe cellular defect compared to the clinical phenotype is repolarization reserve, the concept that multiple overlapping potassium currents contribute to normal repolarization.33 Thus, reduced function of one K+ current does not necessarily result in a clinical phenotype until another one is also compromised. One study showed that in the absence of sympathetic stimulation, IKr is the predominant repolarizing current human ventricular myocytes, and that pharmacological block of IKs does not prolong action potential duration (APD) under basal conditions.34 Under sympathetic stimulation, however, if IKr is pharmacologically reduced, then additional IKs block did lengthen APD. Therefore, reduction in the repolarization reserve becomes significant if the second current is also affected. This concept may be particularly applicable to our patient, whose use of cocaine, ethanol, and prescription pain medications may have reduced her repolarization reserve, compounding the underlying genetic defect and thereby contributing to sudden cardiac death. The patient’s resting QTc interval of 494 ms was abnormally prolonged during a period when it appeared that she was taking various drugs that may have QT-prolonging properties. Her prescription medications fentanyl and oxycodone/acetaminophen have not been directly shown to prolong QT interval, though an association between increasing oxycodone dosage and longer QTc has been observed.35 Oxycodone and fentanyl reduce HERG current in vitro,35,36 and as do cocaine and cocaine metabolites,37,38 but none of these drugs affect the IKs current. The cocaine levels found in her blood on autopsy were in such low levels that her death cannot be directly attributed to cocaine use. In one study that compared the postmortem levels of cocaine and cocaine derivatives in cases where death was attributed either to cocaine or noncocaine related causes, postmortem levels cocaine-related deaths averaged 908 ng/mL and levels of the derivative benzoylecgonine averaged 3960 ng/mL, whereas in noncocaine deaths, cocaine levels averaged 146 ng/mL and benzoylecgonine 888 ng/mL.39 Postmortem toxicology on our patient showed 60 ng/mL cocaine and 1000 ng/mL benzoyleconine. Despite these relatively low levels, cocaine (along with the prescription oxycodone and fentanyl) may have blocked her IKr current, thus leaving her with a reduction of both repolarizing currents significant enough to trigger an arrhythmia that lead to sudden cardiac death. In addition, the toxicology report showed the presence of ethanol. In the presence of ethanol, cocaine is metabolized to cocaethylene, which may be an even more potent blocker of HERG than cocaine itself (1.5–3.7 fold lower IC50).40,41 Taken together, the patient had a deleterious genetic mutation of KCNQ1, and had been using drugs that pharmacologically block HERG. Therefore, loss of repolarization reserve may have been clinically significant for our patient.
Conclusion
The KCNQ1-S277L point mutation causes biophysical defects that result in dominant-negative reduction in KCNQ1 and IKs current density, and also a trafficking defect that results in reduced surface expression. In a patient with prolonged QT interval who subsequently presented with sudden death, the mutation yielded a defective channel that compromised her repolarization reserve, making repolarization almost entirely dependent on the action of the HERG channel carrying the IKr current. Cocaine usage therefore may have contributed to sudden cardiac death by significantly reducing the remaining repolarizing IKr current. Our companion article characterizes the HERG-G816V mutation, which reduces IKr current density via a trafficking error.42 Like the patient presented here, the relatively severe cellular phenotype of HERG-G816V did not correlate with a seemingly mild clinical phenotype as measured by patient’s QT interval; yet, the patient presented with sudden death in a presumed hypokalemic state. In both situations, patients with a pathogenic mutation in KCNQ1 or HERG were dependent on their repolarization reserve, and had a mild clinical phenotype until a second environmental factor diminished their remaining repolarizing current. Together, these reports highlight the continued need for detailed phenotypic analyses of genetic variants of unknown significance, particularly in atypical clinical cases.
Acknowledgments
Funding: There was no funding.
Financial Support: This work was supported in part by grants from the NIH/NHLBI (HL093440 and 1RC1HL100756).
Footnotes
Conflicts of Interest: None.
References
- 1.Anantharam A, Markowitz SM, Abbott GW. Pharmacogenetic considerations in diseases of cardiac ion channels. J Pharmacol Exp Ther. 2003;307:831–838. doi: 10.1124/jpet.103.054569. [DOI] [PubMed] [Google Scholar]
- 2.Roden DM. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc Res. 2001;50:224–231. doi: 10.1016/s0008-6363(00)00302-3. [DOI] [PubMed] [Google Scholar]
- 3.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 5.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 6.McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, Goldstein SA, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- 7.Saenen JB, Vrints CJ. Molecular aspects of the congenital and acquired Long QT Syndrome: Clinical implications. J Mol Cell Cardiol. 2008;44:633–646. doi: 10.1016/j.yjmcc.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 9.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, et al. Most LQT2 mutations reduce Kv11. 1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 10.Jackson HA, Accili EA. Evolutionary analyses of KCNQ1 and HERG voltage-gated potassium channel sequences reveal location-specific susceptibility and augmented chemical severities of arrhythmogenic mutations. BMC Evol Biol. 2008;8:188. doi: 10.1186/1471-2148-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Yang J, Hu D, Kang C, Li C, Zhang S, Li P, et al. KCNQ1 and KCNH2 mutations associated with long QT syndrome in a Chinese population. Hum Mutat. 2002;20:475–476. doi: 10.1002/humu.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Krumerman A, Gao X, Bian JS, Melman YF, Kagan A, McDonald TV. An LQT mutant minK alters KvLQT1 trafficking. Am J Physiol Cell Physiol. 2004;286:C1453–C1463. doi: 10.1152/ajpcell.00275.2003. [DOI] [PubMed] [Google Scholar]
- 14.Melman YF, Um SY, Krumerman A, Kagan A, McDonald TV. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Chen K, Sroubek J, Wu ZY, Thomas D, Bian JS, McDonald TV. Post-transcriptional control of human ether-a-go-go-related gene potassium channel protein by alpha-adrenergic receptor stimulation. Mol Pharmacol. 78:186–197. doi: 10.1124/mol.109.062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv: Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 17.Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with image. J Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 18.Smith JA, Vanoye CG, George AL, Jr, Meiler J, Sanders CR. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry. 2007;46:14141–14152. doi: 10.1021/bi701597s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwasigroch JM, Gilis D, Dehouck Y, Rooman M. PoPMuSiC, rationally designing point mutations in protein structures. Bioinformatics. 2002;18:1701–1702. doi: 10.1093/bioinformatics/18.12.1701. [DOI] [PubMed] [Google Scholar]
- 20.Garson A, Jr, Dick M, 2nd, Fournier A, Gillette PC, Hamilton R, Kugler JD, van Hare GF, 3rd, et al. The long QT syndrome in children. An international study of 287 patients. Circulation. 1993;87:1866–1872. doi: 10.1161/01.cir.87.6.1866. [DOI] [PubMed] [Google Scholar]
- 21.Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, et al. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrlich JR, Pourrier M, Weerapura M, Ethier N, Marmabachi AM, Hebert TE, Nattel S. KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J Biol Chem. 2004;279:1233–1241. doi: 10.1074/jbc.M309087200. [DOI] [PubMed] [Google Scholar]
- 23.Ren XQ, Liu GX, Organ-Darling LE, Zheng R, Roder K, Jindal HK, Centracchio J, et al. Pore mutants of HERG and KvLQT1 downregulate the reciprocal currents in stable cell lines. Am J Physiol Heart Circ Physiol. 2010;299:H1525–H1534. doi: 10.1152/ajpheart.00479.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aidery P, Kisselbach J, Schweizer PA, Becker R, Katus HA, Thomas D. Biophysical properties of mutant KCNQ1 S277L channels linked to hereditary long QT syndrome with phenotypic variability. Biochim Biophys Acta. 2011;1812:488–494. doi: 10.1016/j.bbadis.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Wilson AJ, Quinn KV, Graves FM, Bitner-Glindzicz M, Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1) Cardiovasc Res. 2005;67:476–486. doi: 10.1016/j.cardiores.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 26.Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J. 1997;16:5472–5479. doi: 10.1093/emboj/16.17.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Tristani-Firouzi M, Xu Q, Lin M, Keating MT, Sanguinetti MC. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:817–826. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi L, Priori SG, Napolitano C, Surewicz KA, Dennis AT, Memmi M, Schwartz PJ, et al. Mechanisms of I(Ks) suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol. 2000;279:H3003–H3011. doi: 10.1152/ajpheart.2000.279.6.H3003. [DOI] [PubMed] [Google Scholar]
- 29.Murray A, Potet F, Bellocq C, Baro I, Reardon W, Hughes HE, Jeffery S. Mutation in KCNQ1 that has both recessive and dominant characteristics. J Med Genet. 2002;39:681–685. doi: 10.1136/jmg.39.9.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Wang QF, Du R, Xu QM, Ke QM, Wang B, Chen XL, et al. Congenital long QT syndrome caused by the F275S KCNQ1 mutation: Mechanism of impaired channel function. Biochem Biophys Res Commun. 2009;380:127–131. doi: 10.1016/j.bbrc.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 31.Saenen JB, Paulussen AD, Jongbloed RJ, Marcelis CL, Gilissen RA, Aerssens J, Snyders DJ, et al. A single hERG mutation underlying a spectrum of acquired and congenital long QT syndrome phenotypes. J Mol Cell Cardiol. 2007;43:63–72. doi: 10.1016/j.yjmcc.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453. [DOI] [PubMed] [Google Scholar]
- 33.Roden DM. Taking the “idio” out of “idiosyncratic”: Predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 34.Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, et al. Restricting excessive cardiac action potential and QT prolongation: A vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- 35.Fanoe S, Jensen GB, Sjogren P, Korsgaard MP, Grunnet M. Oxycodone is associated with dose-dependent QTc prolongation in patients and low-affinity inhibiting of hERG activity in vitro. Br J Clin Pharmacol. 2009;67:172–179. doi: 10.1111/j.1365-2125.2008.03327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katchman AN, McGroary KA, Kilborn MJ, Kornick CA, Manfredi PL, Woosley RL, Ebert SN. Influence of opioid agonists on cardiac human ether-a-go-go-related gene K(+) currents. J Pharmacol Exp Therapeutics. 2002;303:688–694. doi: 10.1124/jpet.102.038240. [DOI] [PubMed] [Google Scholar]
- 37.Clarkson CW, Xu YQ, Chang C, Follmer CH. Analysis of the ionic basis for cocaine’s biphasic effect on action potential duration in guinea-pig ventricular myocytes. J Mol Cell Cardiol. 1996;28:667–678. doi: 10.1006/jmcc.1996.0062. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Rajamani S, Chen Y, Gong Q, Rong Y, Zhou Z, Ruoho A, et al. Cocaine blocks HERG, but not KvLQT1+minK, potassium channels. Mol Pharmacol. 2001;59:1069–1076. doi: 10.1124/mol.59.5.1069. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins AJ, Levine B, Titus J, Smialek JE. The interpretation of cocaine and benzoylecgonine concentrations in postmortem cases. Forensic Sci Int. 1999;101:17–25. doi: 10.1016/s0379-0738(99)00006-7. [DOI] [PubMed] [Google Scholar]
- 40.O’Leary ME. Inhibition of HERG potassium channels by cocaethylene: A metabolite of cocaine and ethanol. Cardiovasc Res. 2002;53:59–67. doi: 10.1016/s0008-6363(01)00458-8. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira S, Crumb WJ, Jr, Carlton CG, Clarkson CW. Effects of cocaine and its major metabolites on the HERG-encoded potassium channel. J Pharmacol Exp Therapeutics. 2001;299:220–226. [PubMed] [Google Scholar]
- 42.[please add the details of this article’s companion article (Krishnan, Y., et al., PACE-11-0278.R2) when available].