

Abstract
In the present work, the factors that determine EPR lineshapes from spin labels at the protein-hydrocarbon interface of a β-barrel membrane protein are examined. The EPR spectra from hydrocarbon facing sites in the outer membrane protein A (OmpA) are highly dependent upon the detergent or lipid into which OmpA is reconstituted. In general, lineshapes at these sites are correlated with the solvent accessibility in the supporting amphiphile. A notable exception is CHAPS, which yields rigid limit EPR lineshapes for labels at every position along a transmembrane β-strand in OmpA. EPR lineshapes from the surface of OmpA are not strongly influenced by steric interference with neighboring side chains, but are modulated by solutes that should interact with hydrophobic surfaces. These results suggest that differences in EPR spectra in different supporting environments are not the result of differences in protein dynamics, but are a result of different configurations or rotameric states that are assumed by the label. This conclusion is supported by distance measurements across the OmpA β-barrel, which indicate that labels yielding more motionally restricted lineshapes interact more closely with the protein surface. These results have implications for the use of spin-label derived distance constraints in protein structure determination and demonstrate that spin labels on membrane proteins provide a highly sensitive probe for the environment surrounding a membrane protein.
Introduction
Site-directed spin labeling (SDSL) has become an important tool to provide information on protein structure and dynamics;1–5 moreover, because of the relatively fast time-scale of EPR spectroscopy, SDSL is also particularly well-suited to observe and quantitate protein conformational exchange.6–8 In the case of membrane proteins, SDSL is a particularly valuable approach because measurements can be made in a membrane environment and are not limited by protein molecular weight. As a result of the large electron gyromagnetic ratio, nitroxide-labeled side chains are widely used to generate long-range distance constraints from either paramagnetic enhancements of nuclear relaxation detected by NMR spectroscopy,9,10 or from electron-electron dipolar interactions measured using EPR based methods such as double electron-electron resonance (DEER).11 These long-range distances have proven to be powerful tools for protein structure determination and structure refinement.
There are a number of approaches that can be used to incorporate spin labels into proteins; however, the side chain R1 (see Figure 1a) is the most widely used because of the ease of generating site-specific cysteines and the specificity of the methanethiosulfonate reaction. The factors that determine the EPR lineshapes from R1 at exposed helical surface sites on soluble proteins are well-understood. At these sites, R1 does not interact strongly with neighboring side chains.12 Instead, the EPR spectra at helix surface sites are largely determined by rotations about χ4 and χ5 linking the nitroxide ring to the protein backbone13 and by local protein backbone motion on the ns time-scale.14 Previous work has identified the label rotamers that are most commonly encountered at aqueous exposed sites,15–18 and these findings are useful when assigning long-range distances to nitroxides on protein surfaces during simulated annealing or structure refinement.
Figure 1.
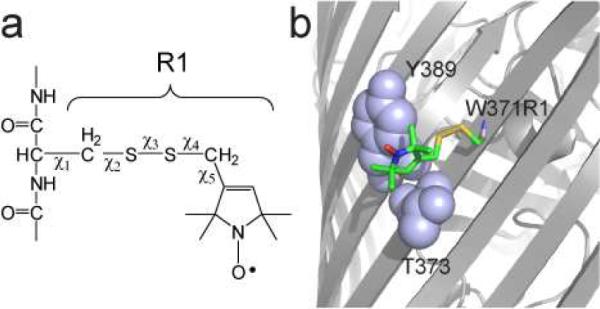
a) Model for the spin-labeled side chain R1 obtained by derivatization with an MTSL spin label. Five rotatable bonds link the nitroxide to the protein backbone, but motions that average the nitroxide magnetic interactions are often dominated by motion about χ4 and χ5. b) Structure of the R1 side chain at position 371 on the surface of the outer membrane transporter BtuB. The label is immobilized by interactions that are made in a pocket formed by Y389 and T373 (PDB ID: 3RGN).
The factors affecting spin labels on membrane proteins are not as well-characterized as labels on soluble proteins. A recent study on the membrane transporter LeuT indicated that at surface helical sites facing the membrane hydrocarbon, the R1 label tended to interact with the protein surface,19 although the rotamers assumed by R1 for χ1-χ3 were similar to those for soluble proteins at surface helical sites. Labels at hydrocarbon facing sites on the β-barrel membrane transport protein BtuB20 also interact with the protein backbone, although they are observed to assume different rotamers than labels at helical sites. For example, a spin label placed onto site 371 on the outer membrane transport protein BtuB gives rise to a room temperature EPR spectrum that is near the rigid limit on the X-band time scale (ca. 30 to 50 ns or longer).21 This is consistent with a model obtained by crystallography, which indicates that the spin label interacts with a pocket on the protein surface formed by the side chains Y389 and T373 (see Figure 1b).
In the present work, the spin-labeled side chain, R1, is engineered into several sites on the Escherichia coli outer membrane protein OmpA. The EPR spectra from spin-labeled sites on OmpA are shown to vary dramatically as a function of the environment into which OmpA is reconstituted, and changes in EPR lineshapes obtained in different detergents or lipids are generally correlated with aqueous accessibility at the labeled site. Mutagenesis and the effects of solutes suggest that the differences in EPR spectra as a function of environment are determined largely by differences in the interactions that the label makes with the protein surface. Measurements of distances across the OmpA barrel using pulse EPR are consistent with this result, and indicate that interspin distances can be modulated by environment even when the protein is rigid. These results show that the EPR spectra of R1 are a highly sensitive probe of the solvation environment at the protein-hydrocarbon interface.
Experimental Methods
Mutagenesis, protein expression, labeling, refolding, and reconstitution
The wild-type pET1113 plasmid for the transmembrane (TM) region, residues 1–177, of OmpA10 was generously provided by Prof. Lukas Tamm (University of Virginia). The TM domain of wild-type OmpA has no native cysteines, and single or double cysteine mutants of the OmpA TM domain were engineered using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) without further modification of the plasmid. OmpA was overexpressed in BL21(DE3)pLysS cells (Stratagene) under control of the T7 promoter by induction with isopropyl β-D-1-thiogalactopyranoside (IPTG; Roche Applied Science, Indianapolis, IN). Denatured protein was extracted and purified in 8 M urea as previously described.22
Mutant OmpA was spin labeled with S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSSL; Toronto Research Chemicals Inc., North York, Ontario) as described previously,10 except that dithiothreitol (DTT) was not added before addition of the spin label. This was done in order to avoid generating spin-labeled DTT. After passing the spin labeled protein through a desalting PD-10 column (GE Healthcare, Piscataway, NJ), the sample was dialyzed against 1 L of 8 M urea buffer in order to remove any residual spin label or spin labeled β-mercaptoethanol. The final concentration of the sample was about 15–50 mg/ml.
All detergents used were obtained from Anatrace (Santa Clara, CA), and refolding of the TM OmpA in each of the selected detergents was performed in a similar way as described previously.22 Briefly, approximately 1 mg of spin labeled protein was added to 5 ml of refolding buffer (20 mM sodium borate, 150 mM NaCl, 1 mM EDTA, pH 10) containing detergent at a total detergent concentration at least three times its critical micelle concentration (cmc), and with the total micelle concentration [(total detergent concentration – cmc)/aggregation number] being at least 20 times larger than the protein concentration. After an overnight incubation at 37 °C and buffer exchange (10 mM monopotassium phosphate, 50 mM NaCl, pH 6.3) samples were concentrated to about 4–5 mg/ml (~200–260 μM) for EPR experiments. Final detergent concentrations varied between 100 to 500 mM.
All lipids were obtained from Avanti Polar Lipids (Alabaster, AL). The TM OmpA region was refolded into palmitoyloleoylphosphatidylcholine (POPC)/palmitoyloleoylphosphatidylglycerol (POPG) (92.5:7.5) small unilamellar vesicles (SUVs).23 For EPR experiments, proteoliposomes were concentrated by ultracentrifugation for two hours at 150,000g and resuspended in ~20 μl potassium phosphate buffer. The final protein to lipid molar ratio was approximately 1:500. Correct refolding in detergent and lipid was monitored by circular dichroism (CD) spectroscopy and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Samples containing dioxane were prepared by adding the appropriate amount of dioxane to refolded TM OmpA in detergent in order to obtain a final dioxane concentration of either 10% or 20% (v/v). Samples were then incubated at room temperature for at least an hour before performing EPR experiments. After addition of dioxane, the protein remained folded as determined by SDS-PAGE.
CW EPR spectroscopy
Room temperature X-band EPR spectroscopy was performed on a Bruker EMX spectrometer with an ER4123D dielectric resonator (Bruker Biospin, Billerica, MA) or a modified Varian E-line 102 Century series spectrometer with a loop gap resonator (Medical Advances, Milwaukee, WI). All X-band spectra were taken using 2 mW incident microwave power, 1 G field modulation and a scan range of 100 G. Sample volumes were 5 μL and were loaded into capillaries (0.60 mm i.d. ×0.84 mm o.d.; Vitrocom, Mountain Lakes, NJ). Spectra were baseline corrected and normalized using LabVIEW software provided by Christian Altenbach (UCLA). A protein concentration of approximately 250 mM was used in all EPR experiments.
Continuous-wave progressive power saturation experiments were performed on the Bruker EMX spectrometer using gas-permeable TPX capillaries (Molecular Specialties, Milwaukee, WI) and carried out under three conditions: saturated with N2, in the presence of 20 mM Ni(II)EDDA, and saturated with air (20% O2). From the amplitudes as a function of microwave power, P1/2 and P1/2 values were determined as described previously,24 and then used to calculate a dimensionless collision parameter, Π, for Ni(II)EDDA or O2.25 The membrane depth parameter Φ was calculated according to: Φ=ln(ΔP1/2O2/ΔP1/2NiEDDA).
DEER measurements
Pulse-EPR measurements were performed on 25–30 μL of sample loaded into quartz capillaries (1.5 mm i.d. × 1.8 mm o.d. Vitrocom, Mountain Lakes, NJ). Samples for double electron-electron resonance (DEER) were flash frozen in isopropanol cooled with dry ice, and the data were recorded at 80 K on a Bruker Elexsys-E580 spectrometer fitted with an ER4118X-MS2 split ring resonator. Data were acquired using the four-pulse DEER sequence26 with a 16-ns π/2 and two 32-ns π observe pulses separated by a 32-ns π pump pulse. The dipolar evolution times were typically 0.8–1.7 s. The pump frequency was set to the center maximum of the nitroxide spectrum, and the observe frequency was set to the low field maximum. The phase-corrected dipolar evolution data were processed assuming a three-dimensional background, Fourier transformed, and the distance distributions were obtained with a Gaussian fit or Tikhonov regularization using the DeerAnalysis2009 package.27
Saturation Recovery EPR
Saturation recovery was performed on an X-band ELEXSYS E-580 spectrometer equipped with an MS-2 split-ring resonator (Bruker Biospin, Billerica, MA), using a Stanford Research Instruments amplifier SR445A (Sunnyvale, California) in place of the builtin Bruker video amplifier. The experiment was performed as described previously,20 using a 500 ns saturating pump pulse applied to the center of the mI=0 hyperfine line, and 2 mW CW observe power at the same frequency. Each measurement was independently repeated 3 times and the average T1 relaxation times and standard deviations are reported.
Simulation of EPR spectra using MOMD
EPR spectra were simulated using the Microscopic Order Macroscopic Disorder (MOMD) model28 implemented in the Multicomponent program developed by Dr. Christian Altenbach (University of California Los Angeles). The fitting was carried out as described previously,20 where the R tensor (diffusion tensor in Cartesian representation) was allowed to vary for each motional component independently. The Euler angles and R tensors were varied iteratively until a satisfactory fit was obtained. Subsequently, Lorentzian (and if necessary, Gaussian) broadening was included to obtain the final fit. In some cases, the A-tensor values were allowed to vary by up to 0.6 G. The quality of fit was assessed using the reduced χ2 between the experimental and theoretical spectra, as well as visually evaluating the match between prominent spectral features.
Modeling the spin label rotamer configurations
Inter-spin distances obtained by DEER measurements on doubly spin labeled TM OmpA were used as inputs for standard Xplor-NIH restrained simulated annealing and energy minimization protocols.29,30 In our annealing, the distance between the Sδ atom and the CHα in both R1 side chains was restricted to be ~3 Å. This stabilizing interaction has been observed in crystal structures of spin labeled proteins.16,19 Starting structures for simulated annealing were obtained by mutating in silico the spin labeled positions in the high-resolution NMR structure of TM OmpA (PDB ID: 1G90) to Cys and building the remainder of the R1 side chain. During the simulation backbone atoms were kept fixed while all side chains were allowed to vary. One hundred structures were generated in this way for each of the distances measured by DEER, and the ten with the lowest energy were visualized and analyzed with the program PyMOL (DeLano Scientific LLC).
Results
OmpA can be refolded into most detergent micelles
A structure of the transmembrane region (TM) of OmpA is shown in Figure 2a, and indicates the sites on β-strand 5 and on extracellular loop 3 that were chosen for attachment of the spin-labeled side chain R1. An SDS-PAGE shift assay31,32 was used to determine whether OmpA and these labeled mutants properly folded into each of the membrane mimetic systems used here, and circular dichroism (CD) spectroscopy was used to verify that the correct secondary structure of OmpA was obtained in each of these refolding environments (data not shown).32,33 The urea-denatured TM region of OmpA properly refolded into all the alkylmaltosides tested (which ranged from 8 to 14 carbons in chain length), but did not refold into alkylphosphocholines longer than 12 carbons in length. In addition, the TM region of OmpA refolded into CHAPS (3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate) and small unilamellar vesicles (SUVs).
Figure 2.

a) Model for OmpA (PDB ID 1G90), showing the sites on β-strand 5 and in loop 3 that were labeled with R1. X-band EPR spectra from OmpA for b) five outward facing sites along strand 5 and c) two sites on loop 3 in four different micelle or membrane environments: DPC (black trace), DoDM (red trace), SUVs (blue trace) and CHAPS (green trace). All spectra are 100 Gauss scans.
EPR spectra from outward facing sites on OmpA are highly dependent upon the supporting amphiphile
EPR spectra were obtained for spin labels placed along β-strand 5 in OmpA, which is expected to span the membrane hydrocarbon. These spectra, which are shown in Figure 2b, were recorded from the protein refolded or reconstituted into micelles of dodecylphosphocholine (DPC), dodecylmaltoside (DoDM), CHAPS and SUVs formed from POPC/POPG. At each site, the EPR lineshapes show considerable variation as a function of the micelle or lipid environment. Typically, the most mobile lineshapes are seen in DPC, and are dominated by a motional component having a correlation time in the range of 2 to 4 ns. Some of the spectra (for example from L97R1, T95R1 and L91R1) reveal two motional components (a mobile and immobile component). Compared to DPC, OmpA in DoDM or SUVs yield less mobile spectra, where the immobile component is enhanced relative to the mobile component. At every position along strand 5, OmpA in CHAPS produced EPR spectra that were near the rigid or slow motional limit on the X-band time scale (greater than 30 to 50 ns correlation time).
There are several possible origins for these dramatic differences in lineshape. The R1 side chain is known to be modulated by differences in backbone dynamics on the ns time-scale,34 and the differences might represent alterations in protein backbone dynamics within these different micellar and membrane environments. In the present case, this appears unlikely for several reasons. First, the differences in lineshape (particularly between CHAPS and DPC) are dramatic, and would require that OmpA assume dramatically different dynamic states in these two detergents. For a protein that is generally thought to be quite rigid,22,35–37 dramatically different amplitudes of backbone motion in these environments would not be expected. Second, as seen in Figure 2c, spectra from the extracellular loop 3 show very little variation as a function of the micelle or detergent environment, with DPC being the only detergent in which a significant difference appears. If the effects of detergent seen from labels in strand 5 were the result of protein dynamics, these changes would need to be localized to the membrane spanning portion of the protein.
It should be noted that the effective molecular size of the protein-detergent micelles is approximately 50kDa or larger, and as a result, the rotational diffusion rate of the protein-detergent complex should not significantly average the EPR lineshapes. We tested this assumption by altering the solution viscosity with the addition of 15% Ficoll 400 to the protein-detergent mixture. The addition of Ficoll did not significantly alter the EPR spectra from OmpA.
Another possible source of the lineshape differences seen in Figure 2b are differences in the rotameric states of R1. Different local environments, created by different membrane mimetic or lipid systems might modulate weak interactions that the spin label makes with the protein surface, altering motional modes of the spin label and the resulting EPR spectra. This is consistent with the finding that differences among EPR spectra are largely seen for labels within the membrane or micelle environment. In detergents such as CHAPS, labels along strand 5 may be making much stronger interactions with the protein backbone or with neighboring side chains.
EPR lineshapes are correlated with solvent accessibility, except for CHAPS
Progressive power saturation of the EPR spectrum in the presence of a paramagnetic reagent provides a measure of the collision frequency of the paramagnetic species with the spin label. In addition, the combined use of hydrophilic and hydrophobic paramagnetic reagents, such as Ni(II)EDDA and O2, can provide a depth parameter, Φ, that can be used to estimate the depth of a spin label within the hydrocarbon phase25.
Shown in Figure 3a are depth parameters obtained for positions along strand 5 in three different environments. Larger positive values are associated with a less polar environment (reduced Ni(II) and/or enhanced O2 collisions with the spin label). In each environment, power saturation of T95R1 produces the most positive value, indicating that this site is the most deeply buried in the micelle or bilayer. When different micelles or lipid are compared, labels attached to OmpA in SUVs experience the most hydrophobic environment, with both DoDM and DPC (in that order) yielding more polar values. This increase in Φ is primarily due to higher Ni(II)EDDA accessibility in DoDM and DPC. An examination of the spectra in Figure 2b indicates that the normalized amplitudes (which provide a relative indication of label motion) generally follow this trend. DPC gives the most mobile spectra followed by DoDM and SUVs. For these detergents (and for the series of maltosides shown below), motional averaging of the spin label is correlated with the depth parameter and with the accessibility of these reagents. It should be noted that to obtain a hydrocarbon depth from the depth parameter, an empirical calibration of Φ is required. This parameter has not been calibrated for the detergent micelles used here.
Figure 3.

a) Depth parameters (Φ) obtained by power saturation (see Methods) measured along β-strand 5 of OmpA in DPC or DoDM micelles, and in POPC/POPG bilayers. b) Collision parameters for O2 and Ni(II)EDDA and the depth parameter, Φ, for sites along β-strand 5 measured in CHAPS micelles. Error bars represent the uncertainty associated with the fit to P1/2 values.
The EPR lineshapes obtained for OmpA in CHAPS are different than those in other detergents or lipid (Fig. 2b). The collision parameters and depth parameters in CHAPS (Fig. 3b) are also very different, and do not follow the expected pattern. The spin label at position 95 has a negative depth parameter and this site is the most accessible to the aqueous reagent Ni(II)EDDA. This value for Φ is comparable to that for interfacial sites in bilayers. Moreover, there is no collision gradient in CHAPS such as that typically seen in a straight chain hydrocarbon micelle or bilayer environment. We do not believe that the differences seen in CHAPS can be attributed to OmpA aggregation (see Discussion). These results suggest that the protein-CHAPS interface is very different than that of the straight chain amphiphiles, and that there are significant packing defects between the protein and CHAPS. As discussed below, this inefficient packing in CHAPS may be the source of the highly immobile spectra observed in Figure 2b in this detergent.
Detergent chain length modulates EPR spectra from OmpA
Shown in Figure 4a are EPR spectra obtained for L91R1 in a series of maltosides having chain lengths that vary from 8 to 14 carbons. As indicated above, the normalized amplitudes of the EPR spectra indicate the relative mobility of R1. In the maltosides, the amplitudes decrease as chain length is increased; indicating that motion of the side chain slows in amphiphiles of longer chain length. The spectra from L91R1, like many of the spectra from strand 5, appear to be composed of both mobile and immobile motional components. These spectra, which were simulated using the MOMD model (see Methods), required two motional components for a reasonable fit (red traces Figure 4a). The parameters from these simulations are shown in Table 1. These simulations indicate that the population of more immobile spins increases as the chain length is increased. In addition, there are systematic changes in correlation times as chain length is varied.
Figure 4.

a) X-band EPR spectra from L91R1 obtained in four different alkyl maltosides having chain lengths of 8, 10, 12 and 14. Also shown (red traces) are simulated spectra using the MOMD model (see Methods). Parameters used in the simulation are given in Table 1. All spectra are 100 Gauss scans. b) Collision parameters for O2 (grey bars) and Ni(II)EDDA (red bars) and the depth parameter, Φ, (blue bars) for L91R1 in maltosides of different chain length. c) Saturation recovery (black trace) for OmpA L91R1 reconstituted into dodecylmaltoside. The recovery can be well fit by a single exponential (red trace) having a relaxation time of 1.27 ± 0.03 μs. The residual is shown in blue. The first 100 points corresponding to the instrumental defense pulse have been omitted.
Table 1.
Parameters used to simulate spectra for L91R1 in maltosides†
| Detergent | Component | % | τ(x) | τ(y) | τ(z) | α | β | γ |
|---|---|---|---|---|---|---|---|---|
| 8-maltoside | 1 | 19 | 4.4 | - | - | 0 | 0 | 23 |
| 2 | 81 | - | 16 | 1.6 | 33 | 52 | 30 | |
| 10-maltoside | 1 | 22 | 4.6 | - | - | 0 | 0 | 25 |
| 2 | 78 | - | 11 | 2.0 | 29 | 54 | 31 | |
| 12-maltoside | 1 | 28 | 5.5 | - | - | 0 | 0 | 19 |
| 2 | 72 | - | 10 | 3.4 | 26 | 62 | 35 | |
| 14-maltoside | 1 | 34 | 6.1 | - | - | 0 | 0 | 15 |
| 2 | 66 | - | 13 | 4.2 | 24 | 69 | 39 | |
| 8-malt+dioxane | ||||||||
| 2 | 100 | - | 13 | 1.4 | 36 | 51 | 24 | |
| 10-malt+dioxane | ||||||||
| 2 | 100 | - | 23 | 1.2 | 33 | 50 | 26 | |
| 12-malt+dioxane | ||||||||
| 2 | 100 | - | 13 | 1.4 | 33 | 50 | 26 | |
| 14-malt+dioxane | 1 | 19 | 4.1 | - | - | 0 | 0 | 56 |
| 2 | 81 | - | - | 0.82 | 17 | 58 | 33 |
Correlation times, τ, are in ns. 1 is the immobile component, 2 is the mobile component. Samples with dioxane contain 20% v/v dioxane in addition to detergent. Values for the magnetic tensors used in MOMD: gxx = 2.0085, gyy = 2.0059, gzz = 2.0021; Axx = 6.0, Ayy = 5.8, Azz = 35.0. The A tensor values were allowed to vary by ±0.8 Gauss if necessary to obtain the best fit. It should be noted that an isotropic rotational diffusion failed to reproduce the high-field region of the spectra.
Figure 4b shows the collision parameters for O2, Ni(II)EDDA and the depth parameter, Φ, for L91R1 in maltosides of increasing chain length. The collision parameters are proportional to the collision frequency of the paramagnetic reagent with the protein attached spin label. As the chain length of the detergent increases, the collision frequency for Ni(II)EDDA drops and the apparent aqueous accessibility decreases (the depth parameter increases). Although not unexpected, the result is consistent with the data presented above, and indicates that differences in the EPR spectrum of L91R1 with hydrocarbon chain length (Fig. 4a) are correlated with the aqueous accessibility of the label.
EPR spectra are not strongly influenced by hydrogen bonding and non-hydrogen bonding neighbors
In a soluble β-sheet protein, cellular retinol-binding protein, the EPR lineshapes from R1 are strongly influenced by nearest hydrogen bonded (HB) and non-hydrogen bonded (NHB) neighbors.38 We tested the effect of mutating HB or NHB neighbors to alanine, on EPR spectra from R1 reconstituted into SUVs at several sites in strand 5 of OmpA. In general, mutating these residues produced relatively little effect on the EPR lineshapes.
Shown in Fig. 5a is a model of R1 at position 91 in OmpA. At this position, the labeled side chain may make contact with Y129 (the NHB). As expected, the mutation Y129A produces a slight increase in label mobility for OmpA in POPC/POPG bilayers (Fig. 5d). At position 93, the mutations Y85A (HB) or V127A (NHB) produce almost no change in the EPR spectrum; even though a model for I93R1 (Fig. 5b) indicates that these side chains might strongly interact with the label. At position 97 (Fig. 5c), the mutation F123A actually shifts the label to a less mobile state, even though steric restraints from this side chain have been removed. These data indicate that neighboring side chains do not have a strong influence on the resulting EPR lineshapes. These results are consistent with those found previously for labels on LeuT19 and BtuB20 where the label has a strong tendency to make interactions with the protein surface.
Figure 5.
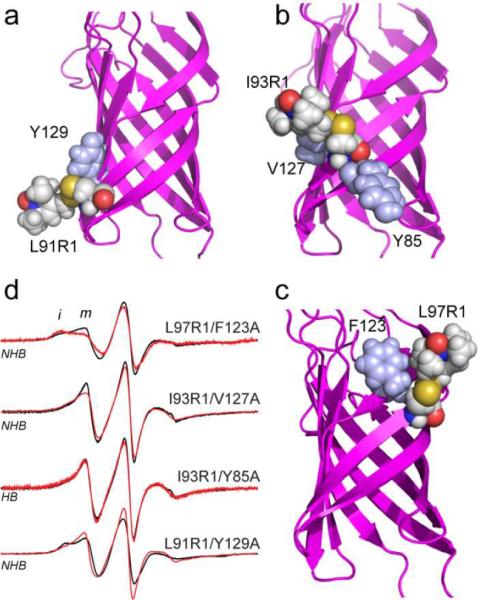
Models in a), b) and c) for L91R1, I93R1 and L97R1, respectively, showing the hydrogen-bonded and non hydrogen-bonded neighbors that were mutated to alanine. d) X-band EPR spectra obtained from labeled OmpA in POPC/POPG bilayers for the indicated spin label with the wild-type neighboring side chain (black trace) or with the indicate neighbor mutated to alanine.
Solutes modulate EPR lineshapes from the OmpA barrel
To test the idea that the motion of R1 may be influenced by interactions that are made with the protein surface, the solute dioxane was added to these samples. Dioxane has been used previously to probe weak interactions between R1 and protein surfaces17 and is expected to compete with R1 for binding sites on the protein surface. Shown in Figure 6 are three examples of R1 labeled sites in dodecylmaltoside to which dioxane was added: L91R1, L97R1 and L97R1 in the presence of the F123A mutation. In each case, the spectra are the result of two motional components, and the addition of dioxane increases the label mobility and increases the population of the more mobile component. The parameters required to simulate the spectra for L91R1 in the presence of 20 % v/v dioxane are shown in Table 1. In the case of maltosides having a chain length of 8, 10 or 12 carbons, the spectra in the presence of dioxane may be simulated with a single motional component. For the L97R1/F123A mutant, the change resulting from the substitution of alanine for phenylalanine (See Figure 5d) is reversed by dioxane addition. Changes are seen upon dioxane addition to other detergent systems and are consistent with the idea that this solute competes with the spin label for binding sites on the protein surface.
Figure 6.
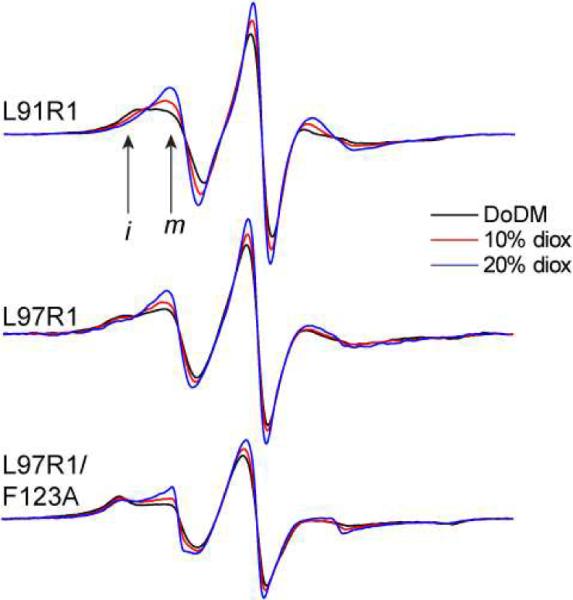
X-band EPR spectra for L91R1, L97R1 and L97R1/F123A in DoDM micelles with 0% (black trace), 10% (red trace) and 20% (blue trace) v/v dioxane addition. All spectra are normalized and increasing amplitudes reflect an increase in R1 side chain motion.
Saturation recovery indicates that hydrocarbon environment modulates label rotameric states
The rigidity of the OmpA barrel and the effect of solute suggest that differences in EPR lineshape in different detergents and lipids might result from changes in label rotamer populations. Another possibility is that the two components in the EPR spectra arise from two or more protein states that are in conformational exchange. To distinguish between these possibilities, the spin lattice relaxation rates for L91R1 were measured using saturation recovery. Shown in Figure 4c is the saturation recovery curve for L91R1 in the presence of dodecylmaltoside, which is a two component spectrum. This recovery curve is well-fit by a single exponential relaxation time of 1265 ± 35 ns. As explained in more detail elsewhere,39 the fact that a single exponential is obtained indicates that the conversion rate between the two label dynamic states is fast relative to the T1 timescale. That is, the exchange rates between motional modes, kex, is much greater than: , where T1a and T1b represent the relaxation rates of the two states. The spectrum of L91R1 in dodecylmaltoside with dioxane added yields a single component EPR spectrum (Figure 6), and a saturation recovery measurement on this sample also yields a single exponential recovery with a relaxation time of 797 ± 3 ns. The MOMD fits indicate that the more mobile component in the dodecylmaltoside spectrum is similar to the single mode of motion seen in the dodecylmaltoside spectrum with dioxane. If we assume that the T1 relaxation times of these modes are similar, the value of T1 from the dodecylmaltoside spectrum in dioxane can be taken to be T1A and we can solve for T1 of the slow component, T1B, by using the MOMD populations of each component (the measured saturation recovery signal should be a linear combination of T1A and T1B, (e.g. (0.72)(797ns) + (0.28)T1B = 1265ns)). Using this approach we estimate that the exchange rate between states can be no slower than 425 kHz or 2.35 μs. Since protein conformational exchange generally occurs on a time scale no slower than tens of μs,40 the two motional states of the label must represent different label rotamers.
Distance measurements indicate that rotameric states of R1 are modulated by different detergents
The data presented above provide strong evidence that the differences in EPR lineshape in different detergent or lipid environment are not the result of differences in OmpA backbone dynamics or conformational exchange, but result from changes in the rotameric states of R1. The spectra for OmpA reconstituted into CHAPS are the most immobile, and are predicted to result from R1 side chains that are strongly interacting with the protein surface. To test the idea that the least mobile lineshapes result from labels that are interacting more closely with the protein surface, distance measurements were made using double electron-electron resonance (DEER) for a pair of labels across the OmpA barrel.
A pair of spin labels (A11R1/T95R1) was engineered into the OmpA barrel (Figure 7a) and the protein was then reconstituted into CHAPS, DoDM, DPC or octylmaltoside (OM). Pulse EPR experiments were performed to measure distances across the OmpA barrel between these two spin labels. Shown in Figure 7b and 7c are the baseline-corrected DEER signals and distance distributions, respectively, obtained from the DEER signals. The mean distances and distributions for this spin pair are shown in Table 2. OmpA in these detergent systems produced very good DEER signals, which should have an accuracy of at least ± 0.5 Å. We were unable to obtain comparable DEER data in SUVs, due to the shorter phase memory times in lipid bilayers.
Figure 7.

a) Model of OmpA with two labeled side chains A11R1 and T95R1 constructed as described in the text. The separation between R1 nitrogen atoms is 26.2 Angstroms. b) DEER signals (black traces) obtained for the A11R1/T95R1 OmpA in four different environments: CHAPS, DoDM, DPC and OM. The red traces represent fits to the data using either a single Gaussian (used for CHAPS) or Tikhonov regularization implemented in DeerAnalysis 2009 (see methods). c) Distance distributions obtained in the indicated detergent environment, which represent the fits shown in b).
Table 2.
DEER derived distances from OmpA 11R1/95R1 in different micelle environments†
| Detergent | Distance (Å) | Distribution (Å) |
|---|---|---|
| CHAPS | 26.6 | 2.7 |
| dodecylphosphocholine | 28.3 | 4.1 |
| dodecylmaltoside | 27.7 | 5.0 |
| dodecylmaltoside (+dioxane) | 31.0 | 7.1 |
| octylmaltoside | 28.7 | 3.9 |
| octylmaltoside (+dioxane) | 30.3 | 5.4 |
Distances were determined from background corrected DEER signals using either a Gaussian fit or Tikhonov regularization implement in DeerAnalysis 200927. The distribution represents the standard deviation in the width of the distance distribution (this distribution does not contain the minor components seen in Fig. 7c). In the two maltoside samples, dioxane was added to a concentration of 20% (v/v). The uncertainty in the distance determination is ± 0.5 Å or better.
As seen in Table 2, the sample producing the shortest distances with the narrowest distance distribution is the sample reconstituted into CHAPS. The detergents DoDM, DPC and OM produced distances that were up to 2 Å longer, with a greater distance distribution. Table 2 also shows the effect of addition of dioxane on OmpA reconstituted into the two maltosides (OM or DoDM). In both cases, the distance and distributions are increased relative to those in detergent alone and are approximately 3 to 4 Å longer than those in CHAPS. It is unlikely that the TM core of OmpA changes shape to this extent, and the most reasonable explanation is that these detergents and solutes are altering the side chain configuration (or rotameric states) of R1 at the protein-detergent interface. The distances (and distributions) measured across the barrel are shortest for OmpA in the detergent that generates the most immobile EPR lineshapes (CHAPS), and the distances (and distributions) increase for OmpA in detergents (or combinations of detergent and dioxane) that produce most mobile EPR spectra.
It should be noted that the distances shown in Table 2 are consistent with the NMR structure of OmpA and rotamers that are expected for R1. The model shown in Figure 7a for 11R1/95R1 was generated by incorporating the two spin-labeled side chains into the NMR structure of OmpA, assigning the nitrogens in the two labels a distance of 26.2 Å, setting the Sδ – HCα distance to 3Å (as seen in many crystal structures of R1 in proteins16) and running simulated annealing and energy minimization routines in XPLOR (see Methods). For the labels in 7a, the result leaves the χ1 and χ2 angles of R1 in an {m,m} configuration, which is one of the common rotamers found for R1.18 In addition, the range of distances covered in Table 2 can be obtained by rotations of χ4 and χ5.
Discussion
The origins of EPR lineshapes from soluble proteins labeled with the side chain R1 have been well-characterized; however, less is known regarding the factors that affect EPR lineshapes from R1 on membrane proteins. At sites facing the membrane hydrocarbon, recent work on helical19 or β-barrel20 proteins indicates that interactions of R1 with the protein surface are important in defining the EPR lineshapes. The data presented here are consistent with this work and indicate that the local solvation environment at the protein-hydrocarbon interface has a profound effect on the interactions that the label makes with the protein surface.
As shown above, EPR spectra from strand 5 in OmpA exhibit dramatic changes in different micelle and membrane environments (see Figure 2). With the exception of CHAPS, the differences between spectra are correlated with a membrane depth parameter that should provide a measure of the aqueous accessibility of the label. The R1 side chain is known to make weak interactions with the protein surface,17 and it is likely that different solvation environments (for example, an aqueous phase versus bilayer phase) will modulate these interactions. In a hydrocarbon environment, the dielectric constant is lower and electrostatic interactions will be enhanced. If hydrocarbon-label interactions are not as favorable as water-label interactions, weak interactions of the label with the protein surface will be more important in a hydrocarbon phase.
CHAPS is a zwitterionic derivative of cholic acid with a hydrophobic core formed from a rigid polycyclic ring structure. In micelles formed from CHAPS, EPR spectra from labels on strand 5 are all near the rigid limit (Figure 2b) indicating that R1 at the surface of OmpA experiences a different environment in CHAPS than in other hydrocarbon amphiphiles. The fact that the core of CHAPS is rigid could lead to packing defects around OmpA, which may account for the increased collisions of R1 with Ni(II)EDDA and the lack of a collision gradient for Ni(II)EDDA and O2 within the CHAPS micelle (Figure 3b). We speculate that R1 does not interact favorably with CHAPS because good van der Waals contacts cannot be made with this detergent. Instead, R1 interacts strongly with the protein surface. Thus, EPR spectra from labels at the protein-membrane interface appear to be very sensitive to the interactions being made by lipid or amphiphile with the protein surface.
We considered the possibility that OmpA aggregation in CHAPS might account for the broad lineshapes for labels placed along strand 5 and the unusual saturation behavior in this detergent. However, several observations suggest that aggregation cannot account for these data. First, aggregation sufficient to immobilize R1 at every position on strand 5 would lead to very high local spin concentrations, which would make DEER measurements difficult. Moreover, phase memory times (which dampen the amplitude of a spin echo) are sensitive to local spin concentration.11 The phase memory times were measured and found to be similar in each detergent system and consistent with monomeric protein. Finally, collision parameters obtained in CHAPS indicate that the labeled sites are accessible to O2 and Ni(II)EDDA, which would not be the case it these labels became immobilized as a result of protein aggregation.
Site-directed spin labeling has proven to be a powerful approach to examine the association of soluble proteins or protein domains with membrane surfaces. EPR spectra from labeled sites on soluble proteins or domains indicate that R1 motion changes significantly and can even become immobilized upon membrane insertion. For example, dramatic changes in EPR lineshape are seen for R1 attached to the membrane binding loops of C2 domains41,42 or to the membrane interacting loops of the annexins.43 Although spectra from sites that become buried within the bilayer provide an assay for membrane insertion, the source of these changes has not been clear. The work presented here suggests that changes in EPR lineshape that accompany membrane insertion may not be due to label interactions with lipid or lipid headgroups. Instead, these changes may be due to weak interactions between the spin-labeled side chain and the protein surface that are enhanced in a hydrocarbon environment.
Previously, dramatic differences in the EPR spectra for R1 attached to hydrocarbon facing sites on the β-barrel membrane transporter BtuB were observed as a function of the environment into which BtuB was reconstituted. Outward facing spin-labeled sites on the second and third transmembrane β-strands of BtuB produce significantly more mobile EPR spectra when octylglucoside is added to POPC bilayers to form mixed micelles.44 The increases in label dynamics were attributed to an enhancement of protein backbone dynamics when the protein was placed into a mixed micelle environment. Although a contribution from protein dynamics cannot be ruled out, the data obtained here suggest that the hydrocarbon environment can modify the energetics of label rotamers at the protein-hydrocarbon interface. As a result, the differences in lineshape previously observed might result from a change in the rotamers sampled by labels at the hydrocarbon interface of BtuB.
The data shown in Figure 7 indicate that the environment at the protein-membrane interface modulates the inter-spin distances across the protein by as much as 4 Angstroms. Thus, in membrane proteins, environment can be expected to modulate long-range distances from spin-labeled side chains determined using pulse EPR methods, such as DEER, or high-resolution NMR methods, such as paramagnetic enhancements of nuclear relaxation. In addition, the configuration (or rotamers) of R1 will likely be different at hydrocarbon facing sites than they are at aqueous sites. In general, the spin-labeled side chain R1 has a limited number of rotatable bonds,13 and a limited set of rotamers is typically found for χ1 or χ2 of R1.16 As a result, distance measurements using R1 yield a narrower distribution of spin label positions than one might expect from a label that is linked by 5 bonds to the protein backbone.45 Nonetheless, such distributions can complicate the analysis of distances obtained from spin labels. In hydrophobic environments, R1 will tend to make interactions with the protein surface, and the label can be expected to select states where the motion of the nitroxide ring is limited by these interactions. This may further restrict the distance distribution and rotameric states assumed by the label.
Conclusions
OmpA has been used as a model for understanding the origins of EPR lineshapes from spin-labeled β-barrel membrane proteins. The motion of spin labels at the protein-hydrocarbon interface is shown to be strongly influenced by the supporting detergent or lipid environment, and the data presented here provide strong evidence that differences in label dynamics are not due to changes in protein dynamics or protein conformation, but are a result of changes in label rotameric states. This work suggests that spin labels will provide a useful probe of the packing environment at the protein-hydrocarbon interface and that rotamer selection for labels within the membrane hydrocarbon will be different than that seen on the exposed surfaces of soluble proteins. These findings will also facilitate the modeling of long-range distances obtained from pulse EPR measurements or paramagnetic enhancements of nuclear relaxation, and indicate that more hydrophobic environments, such as bilayers and long-chain detergents, will provide narrower distance distributions due to immobilizing interactions of the nitroxide ring with the protein surface.
Acknowledgements
LabVIEW programs for the processing and simulation of EPR spectra were kindly provided by Dr. Christian Altenbach (UCLA).
This work was supported by a grant from the National Institutes of Health, NIGMS, GM 035215.
References
- (1).Hubbell WL, McHaourab HS, Altenbach C, Lietzow MA. Structure. 1996;4:779. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- (2).Hubbell WL, Gross A, Langen R, Lietzow MA. Curr Opin Struct Biol. 1998;8:649. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- (3).Hubbell WL, Cafiso DS, Altenbach C. Nat Struct Biol. 2000;7:735. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- (4).Fanucci GE, Cafiso DS. Curr Opin Struct Biol. 2006;16:644. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- (5).Klug CS, Feix JB. Methods Cell Biol. 2008;84:617. doi: 10.1016/S0091-679X(07)84020-9. [DOI] [PubMed] [Google Scholar]
- (6).Lukasik SM, Ho KW, Cafiso DS. J Mol Biol. 2007;370:807. doi: 10.1016/j.jmb.2007.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Flores Jimenez RH, Do Cao MA, Kim M, Cafiso DS. Protein Sci. 19:269. doi: 10.1002/pro.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lopez CJ, Fleissner MR, Guo Z, Kusnetzow AK, Hubbell WL. Protein Sci. 2009;18:1637. doi: 10.1002/pro.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Clore GM, Tang C, Iwahara J. Curr Opin Struct Biol. 2007;17:603. doi: 10.1016/j.sbi.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liang B, Bushweller JH, Tamm LK. J Am Chem Soc. 2006;128:4389. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jeschke G, Polyhach Y. Phys Chem Chem Phys. 2007;9:1895. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- (12).McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Biochemistry. 1996;35:7692. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- (13).Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Biochemistry. 2001;40:3828. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- (14).Columbus L, Hubbell WL. Biochemistry. 2004;43:7273. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- (15).Langen R, Oh KJ, Cascio D, Hubbell WL. Biochemistry. 2000;39:8396. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- (16).Guo Z, Cascio D, Hideg K, Hubbell WL. Protein Sci. 2008;17:228. doi: 10.1110/ps.073174008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Guo Z, Cascio D, Hideg K, Kalai T, Hubbell WL. Protein Sci. 2007;16:1069. doi: 10.1110/ps.062739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fleissner MR, Cascio D, Hubbell WL. Protein Sci. 2009;18:893. doi: 10.1002/pro.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kroncke BM, Horanyi PS, Columbus L. Biochemistry. 2010;49:10045. doi: 10.1021/bi101148w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Freed DM, Khan AK, Horanyi PS, Cafiso DS. Biochemistry. 2011 doi: 10.1021/bi200971x. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ellena JF, Lackowicz P, Mongomery H, Cafiso DS. Biophys J. 2011;100:1280. doi: 10.1016/j.bpj.2011.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nat Struct Biol. 2001;8:334. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- (23).Hong H, Park S, Jimenez RH, Rinehart D, Tamm LK. J Am Chem Soc. 2007;129:8320. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- (24).Oh KJ, Altenbach C, Collier RJ, Hubbell WL. Methods Mol Biol. 2000;145:147. doi: 10.1385/1-59259-052-7:147. [DOI] [PubMed] [Google Scholar]
- (25).Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. Proc Natl Acad Sci U S A. 1994;91:1667. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. J Magn Reson. 2000;142:331. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- (27).Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. Appl Magn Reson. 2006;30:473. [Google Scholar]
- (28).Budil DE, Lee S, Saxena S, Freed JH. J Magn Reson Ser A. 1996;120:155. [Google Scholar]
- (29).Schwieters CD, Kuszewski JJ, Clore GM. Prog Nucl Mag Res Sp. 2006;48:47. [Google Scholar]
- (30).Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. Journal of Magnetic Resonance. 2003;160:65. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- (31).Dornmair K, Kiefer H, Jahnig F. J Biol Chem. 1990;265:18907. [PubMed] [Google Scholar]
- (32).Surrey T, Jahnig F. Proc Natl Acad Sci U S A. 1992;89:7457. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kleinschmidt JH, Wiener MC, Tamm LK. Protein Sci. 1999;8:2065. doi: 10.1110/ps.8.10.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Columbus L, Hubbell WL. Trends Biochem Sci. 2002;27:288. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- (35).Bond PJ, Sansom MS. J Mol Biol. 2003;329:1035. doi: 10.1016/s0022-2836(03)00408-x. [DOI] [PubMed] [Google Scholar]
- (36).Liang B, Arora A, Tamm LK. Biochim Biophys Acta. 2010;1798:68. doi: 10.1016/j.bbamem.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pautsch A, Schulz GE. J Mol Biol. 2000;298:273. doi: 10.1006/jmbi.2000.3671. [DOI] [PubMed] [Google Scholar]
- (38).Lietzow MA, Hubbell WL. Biochemistry. 2004;43:3137. doi: 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- (39).Bridges MD, Hideg K, Hubbell WL. Appl Magn Reson. 2010;37:363. doi: 10.1007/s00723-009-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Henzler-Wildman K, Kern D. Nature. 2007;450:964. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- (41).Frazier AA, Roller CR, Havelka JJ, Hinderliter A, Cafiso DS. Biochemistry. 2003;42:96. doi: 10.1021/bi0268145. [DOI] [PubMed] [Google Scholar]
- (42).Malmberg NJ, Van Buskirk DR, Falke JJ. Biochemistry. 2003;42:13227. doi: 10.1021/bi035119+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Isas JM, Kim YE, Jao CC, Hegde PB, Haigler HT, Langen R. Biochemistry. 2005;44:16435. doi: 10.1021/bi051751m. [DOI] [PubMed] [Google Scholar]
- (44).Fanucci GE, Cadieux N, Piedmont CA, Kadner RJ, Cafiso DS. Biochemistry. 2002;41:11543. doi: 10.1021/bi0259397. [DOI] [PubMed] [Google Scholar]
- (45).Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. Proc Natl Acad Sci U S A. 2008;105:7439. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]