

Abstract
In recent years, genome-sequencing projects of pathogens and humans have revolutionized microbial drug target identification. Of the several known genomic strategies, subtractive genomics has been successfully utilized for identifying microbial drug targets. The present work demonstrates a novel genomics approach in which codon adaptation index (CAI), a measure used to predict the translational efficiency of a gene based on synonymous codon usage, is coupled with subtractive genomics approach for mining potential drug targets. The strategy adopted is demonstrated using respiratory pathogens, namely, Streptococcus pneumoniae and Haemophilus influenzae as examples. Our approach identified 8 potent target genes (Streptococcus pneumoniae–2, H. influenzae–6), which are functionally significant and also play key role in host-pathogen interactions. This approach facilitates swift identification of potential drug targets, thereby enabling the search for new inhibitors. These results underscore the utility of CAI for enhanced in silico drug target identification.
Electronic supplementary material
The online version of this article (doi:10.1007/s11568-011-9152-7) contains supplementary material, which is available to authorized users.
Keywords: Subtractive genomics, Bacterial pathogens, Codon usage, CAI value, Drug targets
Introduction
The astonishing success of genomics has delivered an ever-increasing flow of sequence data. The sequence data serve as the raw material for in silico target discovery (Read et al. 2001). The strategies for drug design and development are progressively shifting from the genetic approach to the genomic approach (Galperin and Koonin 1999). Genomics and bioinformatics provide new opportunities for finding optimal targets. With the phenomenal growth of microbial sequence databases, it has now become possible to use in silico comparisons among genomes to identify potential targets at the beginning of the drug discovery process. To date, more than 500 complete microbial genomes and human genome sequence data have been published and are publicly available (for an updated list of published genomes, vide http://www.genomesonline.org/).
Genomics can be applied to assess the suitability of potential targets using two criteria: “essentiality” and “selectivity”. Essential gene products are considered to constitute the foundation of the organism’s life, and are therefore likely to be common to all cells (Mushegian and Koonin 1996; Kobayashi et al. 2003; Itaya 1995). Galperin and Koonin (1999) suggested searching for drug targets among previously characterized proteins that are specific and essential for a particular pathogen. Recently, Zhang et al. (2004) compiled a list of all currently available essential genes into the Database of Essential Genes (DEG) comprising of several bacterial members.
The possibilities of mining targets through genomics protocols have been increasing. An interesting approach named “differential genome display” or “subtractive genomics” has been proposed for the prediction of potential drug targets (Huynen et al. 1997; 1998). This strategy depends on the fact that genomes of parasitic microorganisms are generally much smaller and encode fewer proteins than the genomes of free-living organisms. The genes that are present in the genome of a parasitic microbe, but devoid in the genome of a taxonomically related free-living microbe, are likely to be important for its pathogenicity. Also, this target should not exhibit any well-conserved homolog in the human host. Hence, they may be considered as candidate drug targets (Sakharkar et al. 2004; Dutta et al. 2006). Subtractive genomics has been successfully utilized by several authors to locate novel drug targets in Pseudomonas aeruginosa (Sakharkar et al. 2004; Perumal et al. 2007), Helicobacter pylori (Dutta et al. 2006), Mycobacterium tuberculosis (Anishetty et al. 2005), Neisseria species (Sarangi et al. 2009; Barh and Kumar 2009), Burkholderia pseudomallei (Chong et al. 2006), Salmonella typhi (Rathi et al. 2009), Aeromonas hydrophila (Sharma et al. 2008), and Clostridium perfringens (Chhabra et al. 2010). The pursuit has been effectively complemented with the compilation of the Database of Essential Genes (DEG) for several pathogenic microorganisms (Zhang et al. 2004).
Codon usage analysis aids in identifying the selectivity and expression levels of the genes (Bennetzen and Hall 1982; Grosjean and Fiers 1982; Ikemura 1985; Ikemura and Ozeki 1982; Nichols et al. 1980). The codon adaptation index (CAI) (Sharp and Li 1987) is an extensively used measure of codon bias in prokaryotes and eukaryotes (Akashi 1994; Frohlich and Wells 1994). It summarizes the adaptation of codon usage in the set of genes known to be highly expressed. The frequency of codon usage in the highly expressed genes is used to define the relative fitness values for each synonymous codon. These values are calculated based on Relative Synonymous Codon Usage (RSCU) rather than raw codon usage and are therefore essentially independent of amino acid composition. Thus, CAI is defined as the geometric mean of relative adaptiveness values.
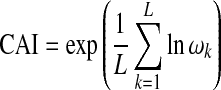 |
where ωk is the relative adaptedness of the kth codon, and L is the number of synonymous codons in the gene.
The CAI values of genes in a genome range between 0 and 1. Higher CAI value usually suggests that the gene of interest is likely to be highly expressed.
Streptococcus pneumoniae or pneumococcus is a gram-positive, alpha-hemolytic, capsulated, lanceolate diplococcus bacterium. This significant human pathogen was recognized as a major cause of pneumonia in the late nineteenth century. In children, they are the most prevalent bacterial agent in community-acquired pneumonia, otitis media, and bacterial meningitis. They also cause sinusitis, bronchitis, cellulitis, endocarditis, pericarditis, and bacteremia conditions (Ryan and Ray 2004). The genome of this organism was sequenced by the year 2001. It is a closed, circular DNA structure containing 2 million basepairs with a core set of 1553 essential genes, plus 154 genes in its virulome, which contribute to virulence, and 176 genes that maintain a non-invasive phenotype. It contains 39.7% of GC (Guanine, Cytosine) content (GenBank Acc. No. AE005672) (Tettelin et al. 2001).
Haemophilus influenzae, first described by Richard Pfeiffer (1892) during influenza pandemic, is a small, non-motile, non-sporing, oxidase positive, pleomorphic, and gram-negative bacilli. This bacterium was formerly called Pfeiffer’s bacillus or Bacillus influenza and is exclusive human pathogens, most strains being opportunistic. They are characterized by their requirement of one or both of two accessory growth factors (X and V) present in blood. It became the first free-living organism whose small circular genome was completely sequenced (Ryan and Ray 2004). The sequencing project, completed and published in science in 1995, was conducted at The Institute for Genomic Research (TIGR). The genome is made up of 1.8 Mbp of DNA and contains 1740 genes with a GC content of 38.1%. (GenBank Ac. No.L42023) (Fleischmann et al. 1995).
The present work aims to utilize subtractive genomics approach pertaining to the identification of potent drug targets in the two respiratory pathogens, Streptococcus pneumoniae and Haemophilus influenzae. An attenuated subtractive genomics approach was carried out by incorporating codon adaptation index (CAI) to increase the strength of prediction accuracy.
Materials and methods
Data collection
Databases primarily DEG, Kyoto Encyclopedia of Genes and Genomes (KEGG) (Ogata et al. 1999) and tools such as ACUA (Umashankar et al. 2007), CD-HIT, and BLAST were employed in this study. The essential gene datasets from DEG (version 3.0) of the two respiratory bacterial pathogens viz. Streptococcus pneumoniae TIGR4 (110 genes) and Haemophilus influenzae RdkW20 (638 genes) were utilized.
CAI calculation
The essential genesets obtained were subjected to CAI value (Sharp and Li 1987) calculation using ACUA. The complete respective microbial essential genesets were used as reference set, instead of highly expressed genes for CAI value calculation. This selection was based on the studies conducted by Rocha and Danchin (2004), wherein they suggest that essential genes are highly conserved and also tend to be more expressed than average. Non-synonymous codons and stop codons were excluded during calculation. Moreover, bacterial essential genes also follow a similar pattern of codon frequencies as it has been used for machine learning studies to classify essential genes in bacteria (Plaimas et al. 2010).
Mining significant genes from essential geneset
Wu et al. (2005) study on Streptomyces coelicolor and Streptomyces avermitilis demonstrated significance of CAI value and its correlation with experimental proteomic data. Based on this study, CAI value was chosen as a measure for mining significant essential genes. Hence, the genes with CAI value greater than or equal to 0.75 were segregated from the essential gene datasets studied.
Selection of taxonomically related non-pathogenic bacteria
The following organisms were selected as equivalent non-pathogenic species of the respiratory bacterial pathogens under study:
Streptococcus thermophilus CNRZ1066
Streptococcus thermophilus (Streptococcus salivarius subsp. thermophilus) is a gram-positive, facultative anaerobic, cytochrome-, oxidase- and catalase-negative, non-motile, non-spore forming, and homofermentative bacterium. This alpha-hemolytic species of the viridans group is classified as a thermophilic lactic acid bacterium (LAB) and is the second most important industrial dairy starter. This CNRZ1066 strain was isolated from yoghurt in France. It is “Generally Recognized As Safe” (GRAS) species, and more than 1021 live cells are being ingested annually by the human population (Bolotin et al. 2004; Hols et al. 2005). The completely sequenced genome of this organism has 1.7 Mbp (GenBank Acc. No. CP000024) and contains 39.1% of GC content coding 1915 proteins (Bolotin et al. 2004).
Mannheimia succiniciproducens MBEL55E
Mannheimia succiniciproducens MBEL55E is a non-sporing, mesophilic gram-negative coccobacillary bacterium. It has been isolated from a bovine rumen. It has ability to produce large amounts of succinic acid from several sources. This capnophilic organism is related to H. influenzae in its morphological characters, and it is non-pathogenic to humans (Hong et al. 2004). It has been identified to contain single chromosome (length of 2,314,078 bp) with no plasmid (GenBank Acc. No.AE016827). With 42.5% GC content, it also has 2384 protein-coding genes present in it (Hong et al. 2004).
Selections of taxonomically related non-pathogenic organisms were made based on morphological and physiological equivalence to pathogens, as well as also on the availability of the complete genome sequence.
Screening of pathogen-specific sequences
Each significant gene identified based on the CAI value was manually annotated to IDENTIFY the protein it encodes along with the metabolic pathway information available from KEGG. The significant datasets obtained were subjected to normalization using CD-HIT software to remove duplicate protein sequences or exclude paralogs as well as exclude proteins of less than 100 amino acid residues as described by Dutta et al. (2006). The normalized dataset sequences were subjected to BLASTP with E-value cutoff of 10−3 for screening significant essential protein sets versus respective free-living organism’s proteomes to identify the pathogen-specific sequences.
Selection of non-human homologs and potential target identification
The obtained gene sequences were subjected to BLASTP search against human proteome to identify proteins with no human homology. The E-value cutoff of 10−3 was adopted based on the study by Sakharkar et al. (2004), in which this cutoff was used to screen human non-homolog proteins of Pseudomonas aeruginosa. As a stringent measure, the proteins with “no hits found” as predicted by BLASTP were only chosen as potential drug targets and other sequences were excluded. The potentialities of the targets were further validated using supporting literature. Moreover, the functional significance of hypothetical proteins obtained as targets was assessed using INTERPROSCAN. The complete flowchart of the methodology adopted is shown in Fig. 1.
Fig. 1.

Codon usage analysis and drug target identification workflow
Results
Data collection and CAI value calculation
A total of 110 genes of Streptococcus pneumoniae TIGR4 strain were acquired from DEG database. Similarly, 638 genes of Haemophilus influenzae Rd KW20 were also obtained from DEG. All the genes collected are known to be essential for the cell’s survival (as per DEG). The collected gene sequences were segregated organism-wise and were subjected to codon adaptation index (CAI) calculation using ACUA. The CAI value of each gene was calculated using respective microorganism’s essential genes codon usage table as reference. Non-synonymous codons and stop codons were excluded during calculation. The overall range of CAI values shows significant variations in both the organisms studied: H. influenzae (CAI range = 0.547–0.884), S. pneumoniae (CAI range = 0.641–0.818).
Mining significant genes from essential geneset
CAI value cutoff was chosen as a measure for mining significant genes from the Essential Geneset based on the study by Wu et al. (2005). They demonstrated significance of CAI value and its correlation with experimental proteomic data. CAI summarizes the adaptation of codon usage in the set of genes known to be highly expressed. Thus, the genes with CAI value greater than or equal to 0.75 were segregated from the essential gene datasets under study. The identified genes were isolated from the respective microbial datasets and were tabulated with its corresponding functions after manual curation using KEGG database reference. Of the 638 essential genes in H. influenzae, only 97 genes were found to have CAI value above 0.75 (Fig. 2a). Similarly, of the 110 essential genes of Streptococcus pneumoniae, only 35 genes were shown to have CAI value greater than 0.75 (Fig. 2b) and were shortlisted as significant genes (Supplementary Table 1 and Supplementary Table 2).
Fig. 2.

Frequency of significant genes with CAI value above 0.75 in Haemophilus influenzae (a) and Streptococcus pneumoniae (b), CAI frequency of complete datasets: H. influenzae (c) and Streptococcus pneumoiae, Number in boxes over bar indicates the frequency. A total of 97 out of 638 and 35 out of 110 were identified as significant genes in H. influenzae and S. pneumoniae, respectively
All the corresponding protein sequences coded by the essential genes were retrieved by BATCH ENTREZ and were stored as organism specific, individual protein datasets. These datasets were further normalized using CD-HIT software to obtain a non-redundant data or exclude paralogs as well as to exclude proteins of less than 100 amino acid residues.
Selection of taxonomically related non-pathogenic bacteria
Selection of taxonomically related non-pathogenic organisms was made based on morphological and physiological equivalence to pathogens, as well as on the availability of the complete genome sequence. Hence, Streptococcus thermophilus CNRZ1066 and Mannheimia succiniciproducens MBEL55E were selected as non-pathogenic bacterial strains taxonomically related to Streptococcus pneumoniae TIGR4 and Haemophilus influenzaeRdkW20, respectively.
The complete genome sequence of the Streptococcus thermophilus strain LMG13811 (1889 genes) was also available during this study. However, Streptococcus thermophilus CNRZ1066 (1915 genes) strain was preferred due to its higher number of genes in the genome. On the other hand, the whole genome sequence of Mannheimia succiniciproducens MBEL55E was only available while exploring for taxonomically related non-pathogenic strain of H. influenzae. Hence, on this basis, the non-pathogenic bacteria were selected.
Pathogen-specific sequences were identified by performing BLASTP of normalized protein datasets versus corresponding non-pathogen datasets.
Pathogen-specific sequences screening and potential target identification
The obtained pathogen-specific sequences were further subjected to BLASTP search against human proteome to identify genes with no human homology. The resulting genes and their corresponding protein products were considered as potential drug targets. The potential efficacy of the targets was validated referring supporting literature. Furthermore, the functional significance of hypothetical proteins obtained as targets was assessed using INTERPROSCAN. The results reveal two significant genes in case of S. pneumoniae, namely, alcohol dehydrogenase (zinc-containing) and 2-C-methyl-D-erythritol 4-phosphate Cytidylyltransferase (Table 1). Whereas, in case of Haemophilus influenzae, six significant genes were identified, out of which five were hypothetical proteins, namely, HI0045, HI1627, HI1728, HI1053, HI1427 and other being the cell filamentation protein (Table 1).
Table 1.
List of final human non-homolog targets with corresponding interproscan annotation, functional significance, and host-pathogen interactions
| Accession. No | Gene product | Organism | Interproscan prediction | Functional significance | Host-pathogen interactions |
|---|---|---|---|---|---|
| NP_345734.1 | Alcohol dehydrogenase, Zinc-containing | S. pneumoniae | Not required | Modulation of ZN+ | Maintenance of the intracellular homeostasis of Zn+ ions is important for virulence and host interaction in S. pneumoniae (Kloosterman et al. 2007). |
| NP_345735.1 | 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase | S. pneumoniae | Not required | Isoprenoid biosynthesis, essential for cell viability and virulence | Isoprenoids are localized in membrane of S. pneumoniae and membrane components and essential for exhibiting virulence in host (Wilding et al. 2000) |
| NP_438218.1 | Hypothetical protein HI0045 | H. influenzae | Conserved hypothetical protein, YTFJ | Un- characterized conserved protein | Uncharacterized conserved protein |
| NP_439769.1 | Hypothetical protein HI1627 | H. influenzae | Endoribonuclease/chorism_mut-like, YJGF/chorismate_mutase-like | Modulation of fimbria expression genes, PUR and ILE, inhibit translation and even serve as mammalian tumor associated ANTIGEN | The airway colonization of H. influenzae in host is facilitated by fimbria (Read et al. 1992; Van Alphen and van Ham 1994) |
| NP_439869.1 | Hypothetical protein HI1728 | H. influenzae | Natural resistance-associated macrophage protein, function: transport, component: membrane | Metal ion transporters | Metal ion transport plays a key role in pathogenesis in the host (Agranoff and Krishna 1998) |
| NP_439212.1 | Hypothetical protein HI1053 | H. influenzae | Carboxymuconol-actone decarboxylase | Aromatic compound degradation | Unknown |
| NP_439576.1 | Hypothetical protein HI1427 | H. influenzae | Unintegrated, putative periplasmic-binding protein CBIK | Periplasmic intermediate | Periplasmic proteins mediate non-pilus adhesins attachment to human epithelial cells, an essential step during colonization of H. influenzae (St Geme and Grass 1998) |
| NP_439140.1 | Cell filamentation protein | H. influenzae | Not required | Cell division and chromosome partitioning | Filamentation proteins also play an important role in non-beta lactamase mediated beta-lactam resistance in H. influenzae despite serotype, origin of isolation, or geographic distribution (Clairoux et al. 1992) |
Discussion
Genome sequencing and bioinformatics have been driving the discovery and development of novel class of antimicrobial compounds and could enable medical science to keep pace with the increasing resistance of bacteria (Read et al. (2001)). Additionally, they have given valuable insights into the molecular mechanisms underlying several diseases. The optimization and modulation of existing bioinformatics strategies are likely to increase the pace and accuracy of drug target identification. Hence, the present study was formulated to evolve a novel strategy, which demonstrates the utilization of codon usage genomics coupled with subtractive genomics pertaining to potent drug target identification.
CAI is a statistical method used to predict gene expression levels and to analyze codon usage bias, which refers to differences in the frequency of occurrence of synonymous codons in coding DNA. This variation in codon bias can also be interpreted as evidence of selective pressure on the usage of synonymous codons. (Ermolaeva 2001; Lynn et al. 2002; Paul et al. 2008). CAI can be used as an effective tool to screen potential drug targets as it is directly related to selective pressure and gene expression. During CAI calculation, the codon usage tables used as reference set get normalized in the process of geometric mean calculation and exclude bias arising due to sample size. Hence, in this study, CAI was chosen as a parameter to narrow down potential drug targets, and the observed differences of CAI value in this study among the genes are genuine and are true representative of selective pressure in a gene-specific and species-specific manner. (Fig. 2a, c, b, d). Moreover, Wu et al. (2005) have already implemented the CAI value cutoff of 0.75 and 0.78 in case of Streptomyces coelicolor and Streptomyces avermitilis to predict the highly expressed genes and housekeeping genes. Furthermore, this cutoff was also found to correlate with 2D gel experimental results and was almost same for both the species. Thus, this cutoff was chosen for the study. Moreover, it is also rational to assume a stringent cutoff of 0.75, since this would avoid bias over choosing genes with less selective pressure and also lead to identification of most potential targets. Hence, the same was adopted and was proven to be plausible in choosing significant genes.
Most available antibiotics target microbe’s essential cellular processes. Essential gene products of microbial cells are promising targets for antibacterial drugs (Zhang et al. 2004). Targeting an essential gene necessary for bacterial cell survival may provide an efficient way to control infection. In this study, the essential genes of the two respiratory bacterial pathogens: Streptococcus pneumoniae and Haemophilus influenzae were screened to identify significant drug targets based on the codon usage genomics coupled with subtractive genomics approach.
In the initial step of codon usage genomics, the overall range of CAI values in the entire datasets showed significant variations: H. influenzae (CAI = 0.547–0.884), S. pneumoniae (CAI = 0.641–0.818), exposing the probable selective pressure among essential genes in an organism-specific manner. In the next step, the significant genes were identified and isolated from the respective microbial essential gene datasets based on their CAI value (> = 0.75).
Identified significant genesets were tabulated with its corresponding functions after manual curation by KEGG database reference (Supplementary Table 1 and Supplementary Table 2). The results reveal that H. influenzae and S. pneumoniae contained 97 and 35 significant genes, respectively. These significant genes probably are highly expressed due to selective pressure by means of high synonymous codon usage and subsequent translational selection. Hence, the possibility of these genes being essential for the survival of the cell is higher. The data obtained in this study also indicate that the genes identified as significant and were found to code for processes such as aminoacid synthesis, energy metabolism, fatty acid synthesis, nucleotide synthesis, which are obviously significant and play a crucial role in cellular processes: central dogma, energy metabolism, macromolecular transport, membrane formation, pathogenicity, and cell viability. Thus, this finding infers the predictive accuracy of the methodology adopted. Furthermore, deciphering the functional status of the unknown protein genes, hypothetical proteins, predicted coding region, and unclassified proteins reported as significant genes in this study by experimental methods would reveal about the level of essentiality. Moreover, these genes could also prove to be good targets as it formed the part of dataset retrieved while mining significant genes from essential genesets.
Subtractive genomics analysis of taxonomically related microorganisms (pathogen vs. non-pathogens) yields a wide range of targets with varied selective pressure, whereas in case of closely related species it is more likely to be narrow range of targets comprising mostly of virulent factors, excluding the genes which are crucial for pathogen’s survival. Moreover, many of the well-documented subtractive genomics studies have successfully implemented the strategy of using taxonomically related microorganisms for drug target identification, therefore the same was implemented in this study and is proven to be effective (Sakharkar et al. 2004; Anishetty et al. 2005).
A potent microbial drug target should not share any well-conserved homolog in the host. Thus, a stringent E-value cutoff of 10−3 was adopted based on the study by Sakharkar et al. (2004), wherein this cutoff was implemented to screen human non-homolog proteins in Pseudomonas aeruginosa. Hence, the proteins with “no hits found” as predicted by BLASTP were only chosen as potential drug targets in this study. Moreover, the same procedure was already proven to be potent by Anishetty et al. (2005) in mining non-pathogenic non-homologous sequences.
In the subtractive genomics step, the results show a total of eight significant genes (S. pnuemoniae–two, H.influenzae–six) that were non-homologous- to non-pathogenic-related bacteria as well as human genes (Table 1). Alcohol dehydrogenase (zinc-containing) (Acc. No. NP_345734.1) of S. pneumoniae is involved in the modulation of zinc ions. In 2007, Kloosterman et al. have reported that this enzyme is important for bacterial virulence and their host interaction, as it is necessary for maintenance of the intracellular homeostasis of zinc ions. The other potential target identified as follows: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (Acc. No. NP_345735.1) is involved in isoprenoid biosynthesis of S. pneumoniae. Isoprenoids are localized in membrane and membrane components of S. pneumoniae and essential for exhibiting virulence in host (Wilding et al. 2000). Both the identified targets are essential for cell viability and virulence.
In H. influenzae, six significant genes were identified as potential targets. Five were hypothetical proteins namely HI0045, HI1627, HI1728, HI1053, and HI1427 and a cell filamentation protein (Table 1). Cell filamentation protein (Acc. No. NP_439140.1) is involved in cell division and chromosome partitioning. Clairoux et al. (1992) have previously reported that filamentation proteins play an important role in non-beta lactamase-mediated beta-lactam resistance in H. influenzae despite serotype, origin of isolation, or geographic distribution. Hypothetical proteins obtained as targets were subjected to Interproscan functional annotation. Hypothetical protein HI1627 (Acc. no. NP_439769.1) was predicted to be an endoribonuclease/chorism_mut-like, yjgf/chorismate_mutase-like protein. This protein is supposed to have role in the modulation of fimbria expression genes, PUR and ILE, and inhibit their translation. The airway colonization of H. influenzae in host is facilitated by fimbria (Read et al. 1992; Van Alphen and van Ham 1994). This hypothetical protein may even serve as mammalian tumor-associated antigen.
Interproscan of the hypothetical protein HI1728 (Acc. No. NP_439869.1) predicts it to be a natural resistance-associated macrophage protein associated with bacterial membrane. Functionally, they are metal ion transporters. Metal ion transport plays a key role in pathogenesis in the host (Agranoff and Krishna 1998). The identified hypothetical protein HI1427 (Acc. No. NP_439576.1) of H. influenzae is unintegrated, putative periplasmic-binding protein CBIK. It is predicted to be a functional significant periplasmic intermediate molecule. Literature study indicates that periplasmic proteins mediate non-pilus adhesins attachment to human epithelial cells and is an essential step during colonization of H. influenzae (St Geme and Grass 1998).
Out of 8 finalized targets, 6 were found to be highly potential targets with well-proven role in host-pathogen interactions and functional significance. Proper annotations could not be performed for the other 2 hypothetical proteins due to limited literature availability. Furthermore, these hypothetical proteins are essential genes as per DEG, so research on these genes using modern molecular biology shall pave way to better understand their potentiality as efficient drug targets.
Subtractive genomics approach has been utilized by several scientists to identify potential drug targets in many bacteria (Sakharkar et al. 2004; Perumal et al. 2007; Dutta et al. 2006; Anishetty et al. 2005; Sarangi et al. 2009; Barh and Kumar 2009; Chong et al. 2006; Rathi et al. 2009; Sharma et al. 2008; Chhabra et al. 2010). However, the present work has employed a different novel approach, which the previous workers have not attempted. Here, the significant essential genes were selected based on CAI values before performing subtractive genomics, and the results obtained also indicate that this strategy would enhance the predictive accuracy of current approaches. Thus, from the above-discussed results, it could be concluded that the approach used in this study could prove to be a powerful tool toward identification of significant essential genes in bacteria, with reasonable accuracy.
Electronic supplementary material
References
- Agranoff DD, Krishna S. Metal ion homeostasis and intracellular parasitism. Mol Microbiol. 1998;28:403–412. doi: 10.1046/j.1365-2958.1998.00790.x. [DOI] [PubMed] [Google Scholar]
- Akashi H. Synonymous codon usage in Drosophila melanogaster natural selection and translational accuracy. Genetics. 1994;136:927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishetty S, Pulimia M, Pennathur G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 2005;29:368–378. doi: 10.1016/j.compbiolchem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Barh D, Kumar A. In silico identification of candidate drug and vaccine targets from various pathways in Neisseria gonorrhoeae. In Silico Biol. 2009;9:0019. [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD. Codon selection in yeast. J Biol Chem. 1982;257:3026–3031. [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Renault P, Sorokin A, Ehrlich SD, Kulakauskas S, Lapidus A, Goltsman E, Mazur M, Pusch GD, Fonstein M, Overbeek R, Kyprides N, Purnelle B, Prozzi D, Ngui K, Masuy D, Hancy F, Burteau S, Boutry M, Delcour J, Goffeau A, Hols P. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat Biotechnol. 2004;22:1554–1558. doi: 10.1038/nbt1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra G, Sharma P, Anant A, Deshmukh S, Kaushik H, Gopal K, Srivastava N, Sharma N, Garg LC. Identification and modeling of a drug target for Clostridium perfringens SM101. Bioinformation. 2010;4:278–289. doi: 10.6026/97320630004278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CE, Lim BS, Nathan S, Mohamed R. In silico analysis of Burkholderia pseudomallei genome sequence for potential drug targets. In Silico Biol. 2006;6:0031. [PubMed] [Google Scholar]
- Clairoux N, Picard M, Brochu A, Rousseau N, Gourde P, Beauchamp D, Parr TR, Jr, Bergeron MG, Malouin F. Molecular basis of the non-beta-lactamase-mediated resistance to beta-lactam antibiotics in strains of Haemophilus influenzae isolated in Canada. Antimicrob Agents Chemother. 1992;36:1504–1513. doi: 10.1128/aac.36.7.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Singh SK, Ghosh P, Mukherjee R, Mitter S, Bandyopadhyay D. In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori. In Silico Biol. 2006;6:43–47. [PubMed] [Google Scholar]
- Ermolaeva MD. Synonymous codon usage in bacteria. Curr Issues Mol Biol. 2001;3:91–97. [PubMed] [Google Scholar]
- Fleischmann R, Adams M, White O, Clayton R, Kirkness E, Kerlavage A, Bult C, Tomb J, Dougherty B, Merrick J. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Frohlich DR, Wells MA. Codon usage patterns among genes for Lepidopteran hemolymph proteins. J Mol Evol. 1994;38:476–481. doi: 10.1007/BF00178847. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Searching for drug targets in microbial genomes. Curr Opin Biotechnol. 1999;10:571–578. doi: 10.1016/S0958-1669(99)00035-X. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Fiers W. Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene. 1982;18:199–209. doi: 10.1016/0378-1119(82)90157-3. [DOI] [PubMed] [Google Scholar]
- St Geme JW, Grass S. Secretion of the Haemophilus influenzae HMW1 and HMW2 adhesins involves a periplasmic intermediate and requires the HMWB and HMWC proteins. Mol Microbiol. 1998;27:617–630. doi: 10.1046/j.1365-2958.1998.00711.x. [DOI] [PubMed] [Google Scholar]
- Hols P, Hancy F, Fontaine L, Grossiord B, Prozzi D, Leblond-Bourget N, Decaris B, Bolotin A, Delorme C, Ehrlich SD, Guédon E, Monnet V, Renault P, Kleerebezem M. New insights in the molecular biology and physiology of Streptococcus thermophilus revealed by comparative genomics. FEMS Microbiol. Rev. 2005;29:435–463. doi: 10.1016/j.femsre.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Hong SH, Kim JS, Lee SY, In YH, Choi SS, Rih JK, Kim CH, Jeong H, Hur CG, Kim JJ. The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nat Biotechnol. 2004;22:1275–1281. doi: 10.1038/nbt1010. [DOI] [PubMed] [Google Scholar]
- Huynen M, Dandekar T, Bork P. Differential genome analysis applied to the species-specific features of Helicobacter pylori. FEBS Lett. 1998;426:1–5. doi: 10.1016/S0014-5793(98)00276-2. [DOI] [PubMed] [Google Scholar]
- Huynen M, Diaz-Lazcoz Y, Bork P. Differential genome display. Trends Genet. 1997;13:389–390. doi: 10.1016/S0168-9525(97)01255-9. [DOI] [PubMed] [Google Scholar]
- Ikemura T, Ozeki H. Codon usage and transfer RNA contents: organism specific codon choice patterns in reference to the isoacceptor contents. Cold Spring Harbor Symposium Quantitative Biology. 1982;47:1087–1097. doi: 10.1101/sqb.1983.047.01.123. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and transfer-RNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Itaya M (1995) An estimation of minimal genome size required for life. FEBS Lett 362:257–260 (PMID: 7729508) [DOI] [PubMed]
- Kloosterman TG, van der Kooi-Pol MM, Bijlsma JJ, Kuipers OP (2007) The novel transcriptional regulator SczA mediates protection against Zn2+stress by activation of the Zn2+-resistance gene czcD in Streptococcus pneumoniae. Mol Microbiol 65:1049–1063 (PMID: 17640279) [DOI] [PubMed]
- Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P (2003) Essential Bacillus subtilis genes. Proc Natl Acad Sci USA 100:4678–4683 (PMID: 12682299) [DOI] [PMC free article] [PubMed]
- Lynn DJ, Singer GA, Hickey DA (2002) Synonymous codon usage is subject to selection in thermophilic bacteria. Nucleic Acids Res 30:4272–4277 (PMID 12364606) [DOI] [PMC free article] [PubMed]
- Mushegian AR, Koonin EV (1996) A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA 93:10268–10273 (PMID: 8816789) [DOI] [PMC free article] [PubMed]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27:29–34 (PMID:9847135) [DOI] [PMC free article] [PubMed]
- Nichols BP, Miozzari GF, van Cleemput M, Bennett GN, Yanofsky C (1980) Nucleotide sequence of the trpG regions of Escherichia coli, Shigella dysenteriae, Salmonella typhimurium and Serratia marcescens. J Mol Biol 142:503–517 (PMID: 7007652) [DOI] [PubMed]
- Paul S, Bag SK, Das S, Harvill ET, Dutta C (2008) Molecular signature of hypersaline adaptation: insights from genome and proteome composition of halophilic prokaryotes. Genome Biol 9:R70 (PMID 18397532) [DOI] [PMC free article] [PubMed]
- Perumal D, Lim CS, Sakharkar KR, Sakharkar MK (2007) Differential genome analyses of metabolic enzymes in Pseudomonas aeruginosa for drug target identification. In Silico Biol 7:0032 (PMID:18391237) [PubMed]
- Plaimas K, Eils R, König R (2010) Identifying essential genes in bacterial metabolic networks with machine learning methods, BMC Sys Biol 4:56 (PMID: 20438628) [DOI] [PMC free article] [PubMed]
- Rathi B, Sarangi AN, Trivedi N (2009) Genome subtraction for novel target definition in Salmonella typhi. Bioinformation 4:143–150 (PMID: 20198190) [DOI] [PMC free article] [PubMed]
- Read TD, Gill SR, Tettelin H, Dougherty BA (2001) Finding drug targets in microbial genomes. Drug Discov Today 6:887–892 (PMID: 11522517) [DOI] [PubMed]
- Read RC, Rutman AA, Jeffery PK, Lund VJ, Brain AP, Moxon ER, Cole PJ, Wilson R (1992) Interaction of capsulate Haemophilus influenzae with human airway mucosa in vitro. Infect Immun 60:3244–3252 (PMID: 1353481) [DOI] [PMC free article] [PubMed]
- Rocha EP, Danchin A (2004) An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol 21:108–116 (PMID: 14595100) [DOI] [PubMed]
- Ryan KJ, Ray CG (2004) Sherris medical microbiology. An introduction to infectious diseases. 4th edn. McGraw Hill Publications
- Sakharkar KR, Sakharkar MK, Chow VT (2004) A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. In Silico Biol 4:355–360 (PMID:15724285) [PubMed]
- Sarangi AN, Aggarwal R, Rahman Q, Trivedi N. Subtractive genomics approach for in silico identification and characterization of novel drug targets in Neisseria meningitidis serogroup B. J Comput Sci Syst Biol. 2009;2:255–258. doi: 10.4172/jcsb.1000038. [DOI] [Google Scholar]
- Sharma V, Gupta P, Dixit A (2008) In silico identification of putative drug targets from different metabolic pathways of Aeromonas hydrophila. In Silico Biol 8:0026 (PMID:19032165) [PubMed]
- Sharp PM, Li WH. The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001;293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- Umashankar V, Arunkumar V, Dorairaj S (2007) ACUA: a software tool for automated codon usage analysis. Bioinformation 2:62–63 (PMID: 18188422) [DOI] [PMC free article] [PubMed]
- Alphen L, Ham SM. Adherence and invasion of Haemophilus influenzae. Rev Med Microbiol. 1994;5:245–255. [Google Scholar]
- Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN (2000) Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in Gram-Positive Cocci. J Bact 8:4319–4327 (PMID: 10894743) [DOI] [PMC free article] [PubMed]
- Wu G, Culley DE, Zhang W (2005) Predicted highly expressed genes in the genomes of Streptomyces coelicolor and Streptomyces avermitilis and the implications for their metabolism. Microbiol 151:2175–2187 (PMID: 16000708) [DOI] [PubMed]
- Zhang R, Ou H-Y, Zhang C-T (2004) DEG: a database of essential genes. Nucleic Acids Res 32: D271–D272 (PMID: 14681410) [DOI] [PMC free article] [PubMed]
Websites
- http://www.bioinsilico.com/acua/
- http://www.ebi.ac.uk/InterProScan/
- http://www.genome.ad.jp/kegg/
- http://bioinformatics.org/cd-hit/
- http://www.ncbi.nlm.nih.gov/entrez/batchentrez.cgi
- http://tubic.tju.edu.cn/deg/
- http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi
- http://www.bioinsilico.com/acua/
- http://www.ebi.ac.uk/Tools/InterProScan/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.