

Abstract
The integration of nucleobase, amino acid, and glycoside into a single molecule results in a novel class of supramolecular hydrogelators, which not only exhibit biocompatibility and biostability, but also facilitate the entry of nucleic acids into cytosol and nuclei of cells. This work illustrates a simple way to generate an unprecedented molecular architecture from the basic biological building blocks for the development of sophisticated soft nanomaterials, including supramolecular hydrogels.
Introduction
This article reports a novel class of hydrogelators, which consist only of life's three fundamental building blocks—nucleobase, amino acid, and glycoside, to self-assemble in water for generating multifunctional, biocompatible, and biostable supramolecular nanofibers/hydrogels. Due to their morphological similarity to extracellular matrices (ECM) in tissues, hydrogels,1,2,3 which consist of crosslinked matrices and large amount of water, have emerged as an important class of biomaterials under intensive developments. Although both natural (e.g., collagen, gelatin, hyaluronic acid, and alginate) and synthetic polymers (e.g., poly(D-L-lactide-co-glycolide), poly(N-isopropyl acrylic amide), and poly(ethylene oxide)) have been serving as the matrices of hydrogels for biomedical applications (e.g., tissue engineering and drug delivery),1 each of them still has its own limitation: the separation and purification of natural polymers are nontrivial matter; the synthetic polymers are largely passive (despite the functionalization).4 Therefore, it is necessary and beneficial to explore alternative matrices for developing hydrogels that mimic the ECM both morphologically and functionally.
Among various alternative approaches, nanofibers of self-assembled peptides as the matrices of supramolecular hydrogels have exhibited considerable promises by serving as scaffolds to guide the differentiation of neuron progenitor cells,5 as media for cell culture,6 and as carriers for drug releases.7 Like the modified peptides, derivatives of glycosides are able to self-assemble to form nanofibers to result in supramolecular gels8 or hydrogels,9 which has led to the development of semi-wet peptide/protein arrays as biosensors and intelligent soft materials. Recently, nanostructures of deoxynucleic acid (DNA)10 have been demonstrated as the matrices of hydrogels, albeit that the hydrogels of DNA alone fail to promote cell growth.11 These results not only attest that molecular self-assembly is an ubiquitous process, but also imply that it is possible to combine the basic building blocks (i.e., nucleobase, amino acid, and glycoside) of the three major biomacromolecules (i.e., nucleic acid, proteins, and glycans) for exploring new molecular architectures to construct nanostructures that serve as the matrices of supramolecular hydrogels. Moreover, the existence of glycoproteins12 and nucleopeptides13 for a variety biological functions in nature and the recent demonstration of hydrogelators of nucleopetides14 support the notion that the integration of nucleobase, amino acid, and glycoside into a molecule to form the nanostructured matrices of supramolecular hydrogels will be an effective approach to impart hydrogels with both supramolecular orders and multiple functions.
Based on the above rationales, we simply connect a nucleobase (e.g., thymine), an amino acid (e.g., phenylalanine), and a glycoside (e.g., D-glucosamine) via covalent bonds and obtain 1T. Compound 1T forms molecular nanofibers to result in a supramolecular hydrogel at pH 7.0 and concentration of 3.0 wt%. The replacement of thymine with other nucleobases (e.g., adenine, cytosine, or guanine) and/or the introduction of diphenylalanine in 1T also results a series of novel hydrogelators (1A, 1G, 2T, 2C, 2A, and 2G) that self-assemble in water to form molecular nanofiber/hydrogels at 3.0 wt% and proper pH. Besides that these hydrogelators hardly inhibit the growth of mammalian cells, the inclusion of glycoside in the hydrogelators significantly enhances their resistance to proteases. Moreover, after the self-assembly, the nanofibers exhibit significant interbase interaction with nucleic acids. The hydrogelators are able to facilitate oligonucleic acids entering cells and the nuclei of cells. Thus, this work illustrates a simple way to generate unprecedented molecular architecture from the basic biological building blocks for developing sophisticated soft nanomaterials that promise a wide range of applications.
Results and Discussion
Scheme 1 shows the molecular design of two types of hydrogelators (1 and 2). 1 consists of a nucleobase (e.g., thymine, cytosine, adenine, or guanine), a phenylalanine, and a D-glucosamine; 2 consists of a nucleobase, a diphenylalanine,15 and a D-glucosamine. In both 1 and 2, the nucleobase and the D-glucosamine connect to the N-terminal and C-terminal, respectively, of the amino acid(s). Scheme 2 outlines typical synthetic routes for making these hydrogelators, exemplified by the cases of 1T, 2T, 1A, and 2A. The thymine acetic acid (3), being activated by N-hydroxysuccinimide (NHS), reacts with L-Phe to afford 4. After undergoing the same NHS activation, 4 couples with D-glucosamine to give the hydrogelator 1T. The addition of second phenylalanine to 4 affords 5, which couples with D-glucosamine to yield the hydrogelator 2T. The synthesis of other hydrogelators (i.e., 2C, 1A, 2A, 1G and 2G) and compound 1C starts from the protected nucleobases (i.e., (N4-bis-Boc-cytosine-1-yl)-acetic acid, (N6-bis-Boc-adenine-9-yl)-acetic acid, and (N2-bis-Boc-guanine-9-yl)-acetic acid). As exemplified by the process for making the hydrogelators consisting of adenine, following the procedures of making nucleobase acetic acid reported by Nieddu,16 we first synthesize bis(tert-butyloxycarbonyl) (bis-Boc) protected adenine, (N6-bis-Boc-adenine-9-yl)-acetic acid (6). After being activated by NHS, 6 reacts with L-Phe to afford 7, which undergoes the same NHS activation and D-glucosamine coupling to give the product 8. Subsequent removal of the Boc-protecting groups by the addition of trifluoroacetic acid (TFA) gives the hydrogelator 1A in 42% total yield. The addition of the second phenylalanine to the compound 7 gives 9, which reacts with D-glucosamine to afford intermediate 10. After the Boc groups being removed, 10 turns into hydrogelator 2A. This five-step synthesis affords 2A in 37% total yield. Based on the same strategy, we obtain 1C, 2C, 1G, and 2G in 45%, 39%, 41%, and 43% total yields, respectively.
Scheme 1. The structures of the hydrogelators (except 1C) consist only of nucleobase, amino acid, and glycoside.
Scheme 2. The typical synthetic route of hydrogelators of 1 and 2.

We find that all compounds, except 1C, behave as hydrogelators and self-assemble in water to form hydrogels. This result indicates that the covalent connection of nucleobase, amino acid, and glycoside presents a valid, simple approach to construct supramolecular hydrogelators. Since 1C, 2C, 1A, 2A, 1G, and 2G have amine groups on the nucleobases, we dissolve these compounds at low pH (via the protonation of their amine group(s)) and trigger hydrogelation by increasing the pH. Without any amine group for protonation, 1T and 2T dissolve completely in water at 3.0 wt% and pH 10.0 upon gentle heating. The change of the pH values of the solutions of 1T and 2T from 10.0 to 7.0 and 8.5, respectively, results in transparent hydrogels. 1C, however, remains as a solution at the same condition. 1A forms an opaque hydrogel at pH 5.0; 1G produces a semi-transparent hydrogel at pH 4.0. Hydrogelators 2T, 2C, 2A and 2G all self-assemble in water to form semi-transparent hydrogels at the concentration of 3.0 wt% and pH around 8.5, 7.5, 5.0 and 4.0, respectively. The different optical appearances of the hydrogels and the pHs for hydrogelation suggest the subtle difference of the self-assembly of these hydrogelators. Unlike 2T, 2A, 2C, and 2G, naphthalene-diphenylalanine-glucosamine fails to form hydrogel properly,17 suggesting a fundamental difference between the naphthalene unit and the nucleobases.
As revealed by transmission electron microscopy (TEM), each hydrogelator leads to a characteristic morphology of the nanostructures (Figure 2) in the corresponding hydrogels. For example, the nanofibers of 1T are thin and straight with a diameter of 12 nm; the nanofibers of 2T (15 nm in diameter) appears to bend easily and to crosslink relatively efficiently, thus forming a fine network. The TEM of the solution of 1C only shows featureless aggregates. The hydrogel of 2C consists of nanofibers (25 nm wide) that crosslink into a network. The nanofibers of 2C also form bundles, which likely contributes to the highest storage modulus among these hydrogels (Figure 3B). Both short nanofibers (14 nm in width and 200 nm in length) and nanoparticles (average diameter of 18 nm) present as the solid phase in the hydrogel of 1A. The hydrogel of 2A, similarly, consists of nanofibers (10 nm in width and 120 nm in length) and nanoparticles (average diameter of 21 nm), which tend to physically crosslink to afford the network. The hydrogel of 1G appears containing thin nanofibers (9 nm in width) and aggregated nanoparticles whose diameters are about 27 nm. In addition to form nanoparticles with 20 nm of average diameters, hydrogelator 2G mainly self-assembles in water to form long thin nanofibers with the width of 13 nm, and the nanofibers in 2G entangle with each other to form a dense nanofiber network.
Figure 2.
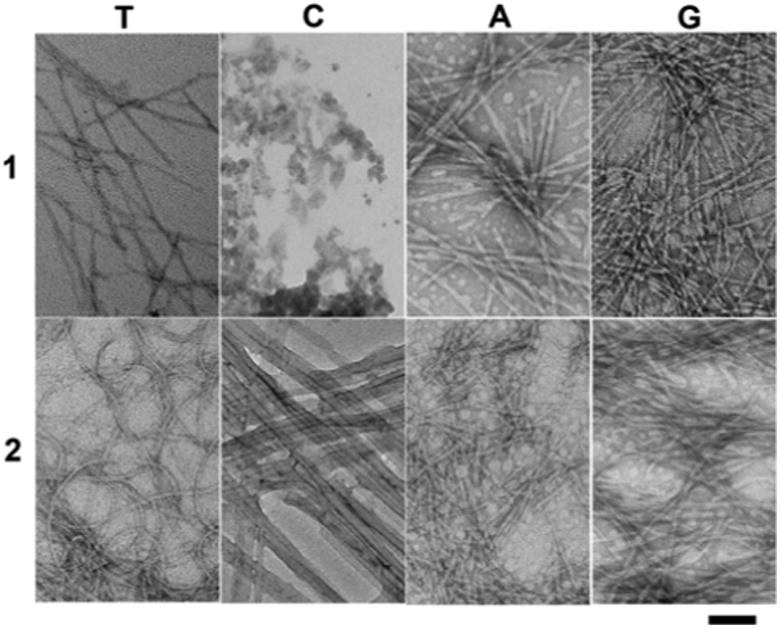
Transmission electron micrograph (TEM) of the negative stained18 hydrogels of 1T, 2T, 2C, 1A, 2A, 1G and 2G and solution 1C. Scale bar = 100 nm, and the concentration and pH value for each of them are same as in Figure 1.
Figure 3.
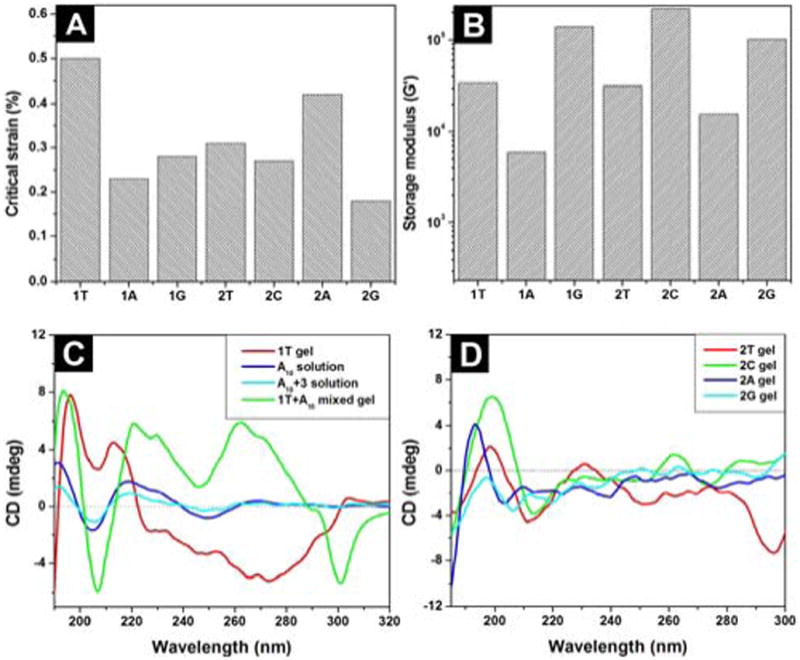
(A) The critical strain and (B) the dynamic storage moduli (G′) of the hydrogels of 1T, 2T, 2C, 1A, 2A, 1G and 2G. (C) The CD spectra of the hydrogel 1T, the solution of deoxyadenosine (A10), the mixture solution of thymine acetic acid with deoxyadenosine (A10) in 1:1 molecular ratio, and the hydrogel of 1T mixed with deoxyadenosine (A10) in 1:1 molecular ratio; (D) the CD spectra of hydrogels of 2T, 2C, 2A, and 2G. The concentrations and pH values are same as in Figure 1.
One characteristic property of hydrogels is their viscoelasticity, reflecting the mechanical properties to resist the deformation. We use rheometry to study the viscoelastic properties of the hydrogels. According to the results from the strain sweep (Figure S2), the hydrogel of 1T shows the critical strain value of 0.5 % (Figure 3A). The critical strain values of the hydrogels of 1A, 1G, 2T, 2C, 2A, and 2G are at 0.23, 0.28, 0.31, 0.27, 0.42, and 0.18 %, respectively, suggesting that the networks in these hydrogels lose the integrity relatively easily upon the application of an external force. The frequency sweep of the hydrogels shows that the dynamic storage moduli (G′) of the hydrogels (1T, 2T, 2C, 1A, 2A, 1G and 2G) dominate their dynamic loss moduli (G″) (Figure S2), indicating that all samples behave as viscoelastic materials. Among these hydrogels, the hydrogel of 2C exhibits the highest storage modulus (220 KPa). The hydrogels of 1G, 2G, 1T, 2T, and 2A possess relatively high storage moduli of 139, 101, 34, 32, and 15 KPa, respectively. The hydrogel of 1A exhibits the lowest storage modulus (6 KPa). Moreover, the addition of an oligomeric deoxyadenosine (A10) to the viscous solution of 1T (2.1 wt%, pH 7.0) affords a stable gel (Figure S5), accompanying by the increase of storage modulus (G′) from 4.4 × 102 Pa (of the solution of 1T) to 9.5 × 102 Pa (of the hydrogel of 1T plus A10), which suggests the interbase interaction between the self-assembly of 1T and A10 to favour molecular aggregation.14,19
As a useful tool to study the secondary structures of proteins, circular dichroism (CD) also provides insightful information about the self-assembled superstructures20 in the gel phase or the liquid crystal phases.21 We use CD to study the secondary structures of the self-assembled hydrogelators in the gel phase. As shown in Figure 3C, the hydrogel of 1T exhibits a peak near 195 nm and trough around 210 nm, suggesting that the backbones of the hydrogelators adopt β-sheet-like configurations in the self-assembled structures. The addition of A10 to the hydrogel of 1T results in distinctive changes in the CD spectra—the increase of the CD intensity at around of 194 nm and 207 nm (belonging to the β-sheet structure) and two new peaks at around 230 nm and 262 nm, suggesting the formation DNA-1T complex, which will enhance the base stacking of A10 and affects the superstructure of the self-assembly of 1T through interbase pairing and phosphate-sugar interactions.19,22,23 The CDs of hydrogels of 1A and 1G display a maximum around 201 nm and a minimum near 210 nm (Figure S3), slightly red-shifting from the CD signals of typical β-sheets, indicating that they share the common feature of a β-sheet structure but have a less ordered conformation or contain a mixture of β-sheet and random coil structures.24 This result also agrees with the TEM of the hydrogel of 1A or 1G. The solution of 1C exhibits the weakest CD signals (Figure S3), agreeing with that 1C fails to self-assemble in water to form a hydrogel. As shown in Figure 3D, hydrogels of 2T, 2C, 2A, 2G all exhibit a positive peak near 198, 198, 193, and 197 nm, and a negative peak around 211, 213, 202, and 206 nm, respectively, suggesting that the backbones of the hydrogelators adopt β-sheet-like configurations in the self- assembled structures. The CD of the hydrogel of 2T shows a negative broad band around 296 nm, which likely originates from the formation of a mesophase of 2T25 because it locates far from the chromophoric absorption region (ca. 268 nm) of 2T (Figure S2).
To verify the biocompatibility of the hydrogelators, we add hydrogelators 1 and 2 into the culture of mammalian cells and measure the proliferation of the cells. According to the results of MTT assay shown in Figures 4A and 4B, after being incubated with the 500 μM of the hydrogelator (1T, 1G, 2T, 2C, 2A, or 2G) for 72 hrs, the cell viability remain at 90%. Although the cell viability decreases slightly when they are incubated with 500 μM of 1C or 1A for 72 hrs, the value of IC50 is still > 500 μM. These results prove that hydrogelators 1 and 2 are biocompatible. In order to further examine the biocompatibility, we conduct a simple in vitro wound-healing assay26 with hydrogelator 2T. As shown in Figure 4D, the presence of hydrogelator 2T in cell culture has little inhibitory effect on the migration of cells, further confirming its biocompatibility.
Figure 4.
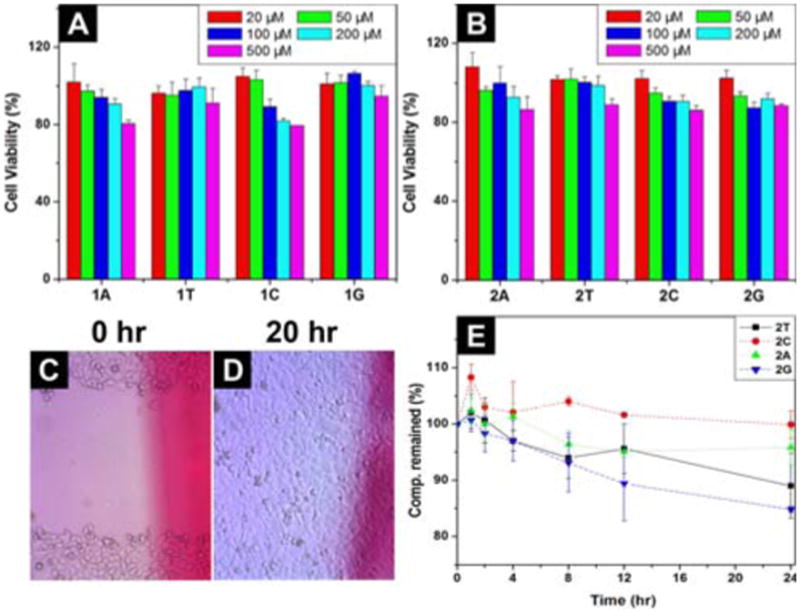
72 hr cell viability test of (A) hydrogelator 1; (B) hydrogelator 2. Optical images of the scratch-wound assay to assess the effects of 2T in the media on wound closure; optical images of HeLa cells on the surface (C) 0 h; and (D) 20 h after the creation of wound in the presence of 2T (by adding 500 μM of 2T in the media). (E) The time-dependent course of the digestions of hydrogelators of 2T, 2C, 2A and 2G by proteinase K.
Besides biocompatibility, biostability is also an essential requisite for a biomaterial. Thus, we examine the stability of hydrogelators 2 by incubating them with proteinase K, a powerful protease that catalyzes the hydrolysis of a wide range of peptidic substrates.27,28 As shown in Figure 4E, more than 85 % of 2T and 2G and 95 % of 2C and 2A remain intact after 24 hrs of the incubation with proteinase K, indicating that hydrogelators 2 have excellent resistance to enzymatic digestion.
To further confirm that the incorporation of glycoside at the C-terminal of the peptides enhances the biostabilities of our hydrogelators to resist the proteinase K digestion, we have synthesized another molecule (thymine-FRGD-glycoside, 1T′) by the conjugating thymine, tetrapeptide (FRGD), and D-glucosamine together, and examine its biostabilities by treating with proteinase K. We found that 1T′ self-assembles to form nanofibers (25 nm in diameter) and affords a hydrogel at the concentration of 3.0 wt%. After the addition of proteinase K, almost 100%, more than 60%, and almost 50% of 1T′ remains at 4 hours, 12 hours, and 24 hours, respectively (Figure S8). Without the conjugation of glycoside, thymine-FRGD, like thymine-FF,14 hydrolyzes completely in 4 hours upon the same treatment. This result further confirms the advantage of the conjugation of glycosides.
Despite the rapid progress in the design and synthesis of peptidic supramolecular hydrogels from L-version amino acids,3,29 the inherent susceptibility L-peptides towards proteolytic digestion in vivo has reduced their efficacy and limited their scope of applications when long-term bioavailability is required.30 The replacement of L-amino acids to D-amino acids or β-amino acids easily reduces the proteolytic digestion, but leads to the loss of the bioactivity of peptides. Many efforts have been focused on designing and synthesizing different peptide molecules from D-amino acids or β-amino acids to mimic the structures and functions of peptides or proteins for prolonged or controlled bioavailability,28,31 but the utilization of such unnatural amino acids raises certain safety concerns and limit their in vivo applications.32 Since glycosylation is a strategy, used by cells, for enhancing the stability of proteins without comprising functions,33 the incorporation of glycoside to the C-terminal of amino acid/peptide would be an advantageous approach for developing biostable and multifunctional hydrogels for applications that require long-term biostability.
To further explore the interbase interaction between the hydrogelators and nucleic acids, an attractive feature of these hydrogelators, we investigate whether these hydrogelators facilitate the delivery of nucleic acids into live cells and examine the subcellular distribution of the delivered nucleic acids. Using fluorescein-labeled single strand oligonucleotide (FAM-A10), which contains the same sequence (A10) as that used in both circular dichroism (CD) and rheology studies, we incubate HeLa cells with 1T and FAM-A10. After 24 hrs of the incubation, we remove the culture medium, wash the cells by PBS buffer, and take fluorescent images. As shown in Figure 5, with the assistance of 1T, the green fluorescence is in both the cytosols and the nuclei of the HeLa cells, indicating the presence of FAM-A10 in HeLa cells. The bright green spots overlay with the fluorescence of SYTO 85, a nuclear staining dye, further confirming the FAM-A10 enters the nuclei. In the control experiment (i.e., without using 1T), green fluorescence is absent from the cytosols and nuclei of the HeLa cells, indicating that it is 1T to interact with and to deliver the oligonucleotide into live cells. Moreover, the replacement of 1T with 1G or 1A fails to deliver the FAM-A10 into the cells, confirming that the matched interbase interactions are critical for the delivery. This result is the first example of the use of nucleobase-amino acid-glycoside conjugate as a new neutral, biocompatible and biostable hydrogelator that allows the delivery of oligonucleotides into human cells. Although recent years have also witnessed intensive research activities with the development of various non-viral vectors for gene delivery, including cationic lipids and polymers, these synthetic vectors have suffered from low gene-transfer efficiency, toxicity, and in vivo instability.34 These limitations necessitate the development of new biocompatible carriers. Thus, these neutral and non-toxic small molecular hydrogelators that act as a successful scaffold for the delivery of oligonucleotide may lead to the development of new non-viral vectors for both in vitro and in vivo applications.
Figure 5.
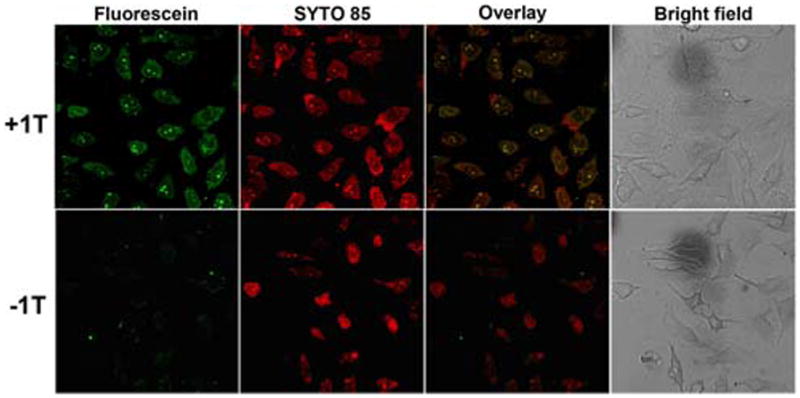
Fluorescence and bright field microscopy images show subcellular distribution of A10, which is labeled with fluorescien dye (green). Cell nuclei were stained with SYTO 85 (orange). (Top) 500 μM 1T and 1 μM FAM-A10 incubated with HeLa cells for 24 hrs. (Bottom) 1 μM FAM-A10 incubated with HeLa cells for 24 hrs.
Conclusion
In conclusion, we have demonstrated that the integration of nucleobase, amino acid, and glycoside, the life's three fundamental building blocks, generates a new type of hydrogelators that self-assemble in water to afford ordered nanostructures and supramolecular hydrogels with multifunctional properties, such as biocompatibilities and biostabilities. Besides like the hydrogelators of nucleopeptides14 to exhibit the excellent cell compatibility, these hydrogelators are able to bind and deliver nucleic acids. This feature are particularly useful and warrants further exploration by incorporating different biofunctional peptides or molecular recognition motifs to achieve nucleic acids condensation, blocking metabolism, endosomal escape, nuclear localisation and receptor targeting.35 Comparing to the hydrogelators of glycosyl-nucleoside-lipid,19,23 the inclusion of a peptide imparts more diverse functions to the hydrogelators than a lipid do. The recent work on sugar-amino acid-nucleoside36 as potential glycosyltransferase inhibitors, in fact, support the notion that the integration of sugar, amino acid, nucleobase into hydrogelators will lead to multifunctional and bioactive soft materials. So this work not only introduces a facile way to expand the current repertoire of building blocks for generating supramolecular assemblies from biomolecules, but also promises more functional supramolecular hydrogels37 for a variety of potential applications, including tissue engineering, drug delivery, and gene delivery.
Supplementary Material
Figure 1.

Optical images of the hydrogels of 1T (pH 7.0), 2T (pH 8.5), 2C (pH 7.5), 1A (pH 5.0), 2A (pH 5.0), 1G (pH 4.0) and 2G (pH 4.0) and the solution of 1C (pH 7.0). All are at 3.0 wt%.
Acknowledgments
This work was partially supported by NIH, NSF (DMR 0820492), a HFSP grant (RGP0056/2008), and start-up grant from Brandeis University. The images were taken at Brandeis EM facilities.
Footnotes
ASSOCIATED CONTENT
Synthesis of hydrogelators 1A, 1T, 1G, 2A, 2T, 2G, 2C, and compound 1C, CD spectra, rheological measurements, and cell viability tests. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zhang J. Annu Rev Biomed Eng. 2000;2:9–29. doi: 10.1146/annurev.bioeng.2.1.9. [DOI] [PubMed] [Google Scholar]; Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]; Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]; Guan ZB, Roland JT, Bai JZ, Ma SX, McIntire TM, Nguyen M. J Am Chem Soc. 2004;126:2058–2065. doi: 10.1021/ja039127p. [DOI] [PubMed] [Google Scholar]; Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345–1360. [Google Scholar]; Cui HG, Webber MJ, Stupp SI. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nagarkar RP, Hule RA, Pochan DJ, Schneider JP. Biopolymers. 2010;94:141–155. doi: 10.1002/bip.21332. [DOI] [PubMed] [Google Scholar]; Su J, Hu BH, Lowe WL, Kaufman DB, Messersmith PB. Biomaterials. 2010;31:308–314. doi: 10.1016/j.biomaterials.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T. Nature. 2010;463:339–343. doi: 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]
- 2.Heeres A, van der Pol C, Stuart MCA, Friggeri A, Feringa BL, van Esch J. J Am Chem Soc. 2003;125:14252–14253. doi: 10.1021/ja036954h. [DOI] [PubMed] [Google Scholar]; Horkay F, Basser PJ, Hecht AM, Geissler E. J Chem Phys. 2008;128:135103. doi: 10.1063/1.2884350. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kato K, Schneider HJ. Eur J Org Chem. 2009:1042–1047. [Google Scholar]
- 3.Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]
- 4.Chan G, Mooney DJ. Trends Biotechnol. 2008;26:382–392. doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 6.Jayawarna V, Ali M, Jowitt TA, Miller AE, Saiani A, Gough JE, Ulijn RV. Adv Mater. 2006;18:611–614. [Google Scholar]; Salick DA, Kretsinger JK, Pochan DJ, Schneider JP. J Am Chem Soc. 2007;129:14793–14799. doi: 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chow LW, Wang LJ, Kaufman DB, Stupp SI. Biomaterials. 2010;31:6154–6161. doi: 10.1016/j.biomaterials.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Garty S, Kimelman-Bleich N, Hayouka Z, Cohn D, Friedler A, Pelled G, Gazit D. Biomacromolecules. 2010;11:1516–1526. doi: 10.1021/bm100157s. [DOI] [PubMed] [Google Scholar]; Liu HJ, Hu YH, Wang HM, Wang JY, Kong DL, Wang L, Chen LY, Yang ZM. Soft Matter. 2011;7:5430–5436. [Google Scholar]
- 7.Schwartz JJ, Zhang SG. Curr Opin Mol Ther. 2000;2:162–167. [PubMed] [Google Scholar]; Zhao F, Ma ML, Xu B. Chem Soc Rev. 2009;38:883–891. doi: 10.1039/b806410p. [DOI] [PubMed] [Google Scholar]; Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576–13577. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]; Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. J Am Chem Soc. 2010;132:17707–17709. doi: 10.1021/ja109269v. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang HM, Yang CH, Wang L, Kong DL, Zhang YJ, Yang ZM. Chem Commun. 2011;47:4439–4441. doi: 10.1039/c1cc10506j. [DOI] [PubMed] [Google Scholar]
- 8.Terech P, Weiss RG. Chem Rev. 1997;97:3133–3159. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]; Jung JH, Amaike M, Shinkai S. Chem Commun. 2000:2343–2344. [Google Scholar]
- 9.Kiyonaka S, Sada K, Yoshimura I, Shinkai S, Kato N, Hamachi I. Nat Mater. 2004;3:58–64. doi: 10.1038/nmat1034. [DOI] [PubMed] [Google Scholar]; Komatsu H, Matsumoto S, Tamaru S, Kaneko K, Ikeda M, Hamachi I. J Am Chem Soc. 2009;131:5580–5585. doi: 10.1021/ja8098239. [DOI] [PubMed] [Google Scholar]
- 10.Seeman NC. Mol Biotechnol. 2007;37:246–257. doi: 10.1007/s12033-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aldaye FA, Senapedis WT, Silver PA, Way JC. J Am Chem Soc. 2010;132:14727–14729. doi: 10.1021/ja105431h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grogan MJ, Pratt MR, Marcaurelle LA, Bertozzi CR. Annu Rev Biochem. 2002;71:593–634. doi: 10.1146/annurev.biochem.71.110601.135334. [DOI] [PubMed] [Google Scholar]
- 13.Azzam ME, Algranat ID. Proc Natl Acad Sci U S A. 1973;70:3866–3869. doi: 10.1073/pnas.70.12.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]; Roviello GN, Benedetti E, Pedone C, Bucci EM. Amino Acids. 2010;39:45–57. doi: 10.1007/s00726-010-0567-6. [DOI] [PubMed] [Google Scholar]
- 14.Li X, K Y, Lin HC, Gao Y, Shi J, Xu B. Angew Chem Int Ed. 2011 doi: 10.1002/anie.201103641. [DOI] [Google Scholar]
- 15.Gorbitz CH. Chem Eur J. 2001;7:5153–5159. doi: 10.1002/1521-3765(20011203)7:23<5153::aid-chem5153>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 16.Porcheddu A, Giacomelli G, Piredda I, Carta M, Nieddu G. Eur J Org Chem. 2008:5786–5797. [Google Scholar]
- 17.Yang ZM, Liang GL, Ma ML, Abbah AS, Lu WW, Xu B. Chem Commun. 2007:843–845. doi: 10.1039/b616563j. [DOI] [PubMed] [Google Scholar]
- 18.Frado LL, Craig R. J Mol Biol. 1992;223:391–397. doi: 10.1016/0022-2836(92)90659-8. [DOI] [PubMed] [Google Scholar]
- 19.Godeau G, Bernard J, Staedel C, Barthelemy P. Chem Commun. 2009:5127–5129. doi: 10.1039/b906212b. [DOI] [PubMed] [Google Scholar]
- 20.Nakashima N, Ando R, Muramatsu T, Kunitake T. Langmuir. 1994;10:232–234. [Google Scholar]
- 21.Schenning A, Kilbinger AFM, Biscarini F, Cavallini M, Cooper HJ, Derrick PJ, Feast WJ, Lazzaroni R, Leclere P, McDonell LA, Meijer EW, Meskers SCJ. J Am Chem Soc. 2002;124:1269–1275. doi: 10.1021/ja0113403. [DOI] [PubMed] [Google Scholar]; Percec V, Smidrkal J, Peterca M, Mitchell CM, Nummelin S, Dulcey AE, Sienkowska MJ, Heiney PA. Chem Eur J. 2007;13:3989–4007. doi: 10.1002/chem.200601582. [DOI] [PubMed] [Google Scholar]; Stals PJM, Smulders MMJ, Martin-Rapun R, Palmans ARA, Meijer EW. Chem Eur J. 2009;15:2071–2080. doi: 10.1002/chem.200802196. [DOI] [PubMed] [Google Scholar]; Peterca M, Imam MR, Ahn CH, Balagurusamy VSK, Wilson DA, Rosen BM, Percec V. J Am Chem Soc. 2011;133:2311–2328. doi: 10.1021/ja110753s. [DOI] [PubMed] [Google Scholar]
- 22.Dong GM, Zhang LR, Zhang LH. Helv Chim Acta. 2003;86:3516–3524. [Google Scholar]; Zhang GS, Guan Z, Zhang LR, Min JM, Zhang LH. Bioorg Med Chem. 2003;11:3273–3278. doi: 10.1016/s0968-0896(03)00278-5. [DOI] [PubMed] [Google Scholar]
- 23.Arigon J, Prata CAH, Grinstaff MW, Barthelemy P. Bi-oconjugate Chem. 2005;16:864–872. doi: 10.1021/bc050029y. [DOI] [PubMed] [Google Scholar]
- 24.Pashuck ET, Cui HG, Stupp SI. J Am Chem Soc. 2010;132:6041–6046. doi: 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang HM, Wang ZH, Song DH, Wang JY, Gao J, Wang L, Kong DL, Yang ZM. Nanotechnology. 2010;21:155602. doi: 10.1088/0957-4484/21/15/155602. [DOI] [PubMed] [Google Scholar]; Zelzer M, Ulijn RV. Chem Soc Rev. 2010;39:3351–3357. doi: 10.1039/c0cs00035c. [DOI] [PubMed] [Google Scholar]
- 25.Gottarelli G, Lena S, Masiero S, Pieraccini S, Spada GP. Chirality. 2008;20:471–485. doi: 10.1002/chir.20459. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez LG, Wu X, Guan JL. In: Cell Migration: Developmental Methods and Protocols. Guan JL, editor. Vol. 294. Humana Press Inc.; To-towa, NJ: 2004. pp. 23–29. [Google Scholar]
- 27.Bromme D, Peters K, Fink S, Fittkau S. Arch Biochem Bio-phys. 1986;244:439–446. doi: 10.1016/0003-9861(86)90611-9. [DOI] [PubMed] [Google Scholar]
- 28.Liang GL, Yang ZM, Zhang RJ, Li LH, Fan YJ, Kuang Y, Gao Y, Wang T, Lu WW, Xu B. Langmuir. 2009;25:8419–8422. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Kuang Y, Gao YA, Xu B. Langmuir. 2011;27:529–537. doi: 10.1021/la1020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jun HW, Yuwono V, Paramonov SE, Hartgerink JD. Adv Mater. 2005;17:2612–2617. [Google Scholar]
- 31.Yang ZM, Liang GL, Xu B. Chem Commun. 2006:738–740. doi: 10.1039/b516133a. [DOI] [PubMed] [Google Scholar]; Yang ZM, Liang GL, Ma ML, Gao Y, Xu B. Small. 2007;3:558–562. doi: 10.1002/smll.200700015. [DOI] [PubMed] [Google Scholar]
- 32.Benevenga NJ, Steele RD. Annu Rev Nutr. 1984;4:157–181. doi: 10.1146/annurev.nu.04.070184.001105. [DOI] [PubMed] [Google Scholar]; Fuchs SA, Berger R, Klomp LWJ, de Koning TJ. Mol Genet Metab. 2005;85:168–180. doi: 10.1016/j.ymgme.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Culyba EK, Price JL, Hanson SR, Dhar A, Wong CH, Gruebele M, Powers ET, Kelly JW. Science. 2011;331:571–575. doi: 10.1126/science.1198461. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sola RJ, Griebenow K. J Pharm Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chesnoy S, Huang L. Annu Rev Biophys Biomolec Struct. 2000;29:27–47. doi: 10.1146/annurev.biophys.29.1.27. [DOI] [PubMed] [Google Scholar]; Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]; Segura T, Shea LD. Ann Rev Mater Res. 2001;31:25–46. [Google Scholar]; Zhao XB, P F, Yaseen M, Lu JR. Annu Rep Prog Chem, Sect C: Phys Chem. 2010;106:305. [Google Scholar]
- 35.Bloomfield VA. Curr Opin Struct Biol. 1996;6:334–341. doi: 10.1016/s0959-440x(96)80052-2. [DOI] [PubMed] [Google Scholar]; Gottschalk S, Sparrow JT, Hauer J, Mims MP, Leland FE, Woo SLC, Smith LC. Gene Ther. 1996;3:448–457. [PubMed] [Google Scholar]; Adami RC, Collard WT, Gupta SA, Kwok KY, Bonadio J, Rice KG. J Pharm Sci. 1998;87:678–683. doi: 10.1021/js9800477. [DOI] [PubMed] [Google Scholar]; McKenzie DL, Collard WT, Rice KG. J Pept Res. 1999;54:311–318. doi: 10.1034/j.1399-3011.1999.00104.x. [DOI] [PubMed] [Google Scholar]; Plank C, Tang MX, Wolfe AR, Szoka FC. Hum Gene Ther. 1999;10:2272–2272. doi: 10.1089/10430349950019101. [DOI] [PubMed] [Google Scholar]; Wagner E. Adv Drug Deliv Rev. 1999;38:279–289. doi: 10.1016/s0169-409x(99)00033-2. [DOI] [PubMed] [Google Scholar]; Schwarze SR, Dowdy SF. Trends Pharmacol Sci. 2000;21:45–48. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]; Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]; Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]; Trehin R, Merkle HP. Eur J Pharm Biopharm. 2004;58:209–223. doi: 10.1016/j.ejpb.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 36.Vembaiyan K, Pearcey JA, Bhasin M, Lowary TL, Zou W. Bioorg Med Chem. 2011;19:58–66. doi: 10.1016/j.bmc.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 37.Mitra S, Gaur U, Ghosh PC, Maitra AN. J Control Release. 2001;74:317–323. doi: 10.1016/s0168-3659(01)00342-x. [DOI] [PubMed] [Google Scholar]; Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; Chen J, McNeil AJ. J Am Chem Soc. 2008;130:16496–16497. doi: 10.1021/ja807651a. [DOI] [PubMed] [Google Scholar]; Schneider HJ, Strongin RM. Acc Chem Res. 2009;42:1489–1500. doi: 10.1021/ar800274u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tam AYY, Wong KMC, Yam VWW. J Am Chem Soc. 2009;131:6253–6260. doi: 10.1021/ja900895x. [DOI] [PubMed] [Google Scholar]; Weiss RG. Langmuir. 2009;25:8369–8369. doi: 10.1021/la901621x. [DOI] [PubMed] [Google Scholar]; Steed JW. Chem Commun. 2011;47:1379–1383. doi: 10.1039/c0cc03293j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.